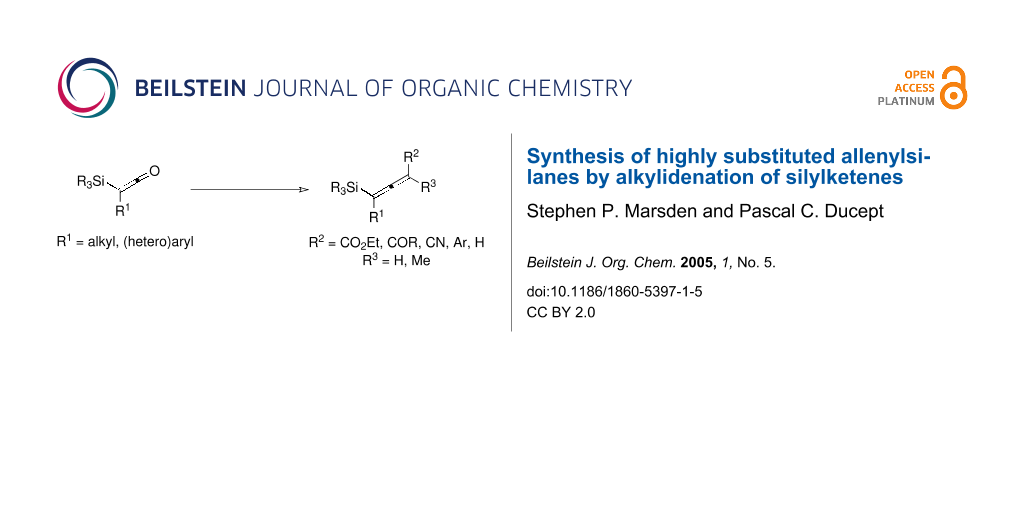
Abstract
Background
Allenylsilanes are useful intermediates in organic synthesis. An attractive, convergent but little used approach for their synthesis is the alkylidenation of stable silylketenes. Reactions thus far have been limited to the use of unsubstituted silylketenes (or equivalents) with stabilised or semi-stabilised ylides only. The current study explores the reactions of substituted ketenes prepared through rhodium(II)-mediated rearrangement of silylated diazoketones.
Results
A range of novel 1,3-disubstituted and 1,3,3-trisubstituted allenylsilanes were prepared using stabilised and semi-stabilised ylides. Alkylidenation with non-stabilised phosphorus ylides was not viable, but the use of titanium-based methylenating reagents was successful, allowing access to 1-substituted allenylsilanes.
Conclusion
Many novel allenylsilanes may be accessed by alkylidenation of substituted silylketenes. Importantly, for the first time, simple methylenation of silylketenes has been achieved using titanium carbenoid-based reagents.

Graphical Abstract
Introduction
Allenylsilanes are versatile intermediates for organic synthesis.[1,2] They have two main modes of reactivity: firstly, as propargyl anion equivalents in thermal [3,4] or Lewis acid-mediated [5,6] addition to carbonyls, acetals and imines, and secondly as three-carbon partners in [3+2] annulation reactions. Thus, reaction with aldehydes,[7] imines/iminiums,[7,8] enones [9-11] and nitrosyl cations [12] leads to dihydrofurans, dihydropyrroles, cyclopentenes and isoxazoles respectively.[13] In most cases the silicon is retained in the final product and can be used as a handle for further synthetic elaboration.
Amongst the myriad methods to prepare allenylsilanes,[1,14] an attractive disconnection is to consider a Wittig-type alkylidenation of a silylketene, Figure 1.
![[1860-5397-1-5-1]](/bjoc/content/figures/1860-5397-1-5-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Alkylidenation approach to the synthesis of allenylsilanes.
Figure 1: Alkylidenation approach to the synthesis of allenylsilanes.
This convergent approach potentially allows for tremendous variation in substitution pattern at both termini of the allenylsilane, yet has been little exploited thus far. Ruden first demonstrated that the stabilised phosphorane carbethoxymethylenetriphenylphosphorane underwent Wittig condensation with trimethylsilylketene at sub-ambient temperature in high yield, but found that non-stabilised phosphoranes led to complex mixtures of products.[15] Other workers later extended this chemistry to include a wider range of stabilised phosphoranes, but attempts to promote the reaction with semi-stabilised ylides such as benzylidenetriphenylphosphorane were unsuccessful unless bis(trialkylsilyl)ketenes were used as substrates.[16,17] Thus, only 3-substituted and 3,3-disubstituted allenylsilanes have thus far been accessed by alkylidenation of silylketenes, whilst no reports of the successful introduction of non-stabilised ylide equivalents have been forthcoming.
A second impediment to the generalisation of the approach has been the paucity of methods for preparing substituted silylketenes.[18,19] Methods based upon the thermolysis of siloxyalkynes [20] and dehydrohalogenation of substituted α-silyl acid halides [21,22] have not found general application. We recently reported a mild and functional group tolerant approach to substituted silylketenes based upon a rhodium-mediated formal Wolff rearrangement of silylated diazoketones.[23] Related photolytic approaches also hold some promise.[24-27] These methods allow access for the first time to a wide range of substituted silylketenes which will allow the chemistry of these fascinating persistent ketenes [18] to be better delineated.
In this paper we outline the synthesis of 1,3-disubstituted and 1,3,3-trisubstituted allenylsilanes by the condensation of stabilised and semi-stabilised ylides with substituted silylketenes, and report for the first time the methylenation of silylketenes to give 1-substituted allenylsilanes using titanium-based methodology.
Results and Discussion
Our investigations began with the preparation of substituted silylketenes 1 as substrates for the alkylidenation chemistry. This was carried out under our previously reported conditions for rhodium(II) octanoate-mediated rearrangement of silyl diazoketones 2,[23] which in turn were prepared by C-silylation of the parent diazoketones 3 with triethylsilyl triflate,[28] Scheme 1, Table 1. It should be noted that while the alkyl-substituted silylketenes are relatively stable and show little decomposition at room temperature over several days, the (hetero)aromatic-substituted silylketenes are much less robust and should be used quickly or stored in a freezer.
![[1860-5397-1-5-i1]](/bjoc/content/inline/1860-5397-1-5-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of substituted silylketenes 1
Scheme 1: Synthesis of substituted silylketenes 1
With the requisite silylketenes in hand, attention turned to their reaction with the carboethoxy-stabilised phosphoranes 4 and 5. At the outset, it was by no means certain that these would react efficiently with substituted silylketenes 1 since it is well documented that nucleophiles attack silylketenes anti to the silicon, [29]ie the phosphoranes would be approaching from the same side as the R1-substituent. Since in all previous examples this substituent has been a hydrogen atom, the extension to bulkier substituents could not be taken for granted. In the event, however, we were pleased to find that in nearly all cases the desired allenylsilanes were formed in moderate to excellent yield, Scheme 2, Table 2.
![[1860-5397-1-5-i2]](/bjoc/content/inline/1860-5397-1-5-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of substituted silylketenes with ester-stabilised phosphoranes
Scheme 2: Reaction of substituted silylketenes with ester-stabilised phosphoranes
Table 2: Reaction of substituted silylketenes with ester-stabilised phosphoranes
| Entry | Ketene | Ylide | Temp (°C) | t (h) | Solvent | Yield 6/7 (8) |
|---|---|---|---|---|---|---|
| 1 | 1a | 4 | 80 | 24 | PhH | 54% |
| 2 | 1a | 5 | rt | 3 | CH2Cl2 | 60% |
| 3 | 1b | 4 | 110 | 24 | toluene | 45% |
| 4 | 1b | 5 | reflux | 24 | CH2Cl2 | 77% |
| 5 | 1c | 4 | 80 | 24 | PhH | 60% |
| 6 | 1c | 5 | rt | 6 | CH2Cl2 | 81% |
| 7 | 1d | 4 | 110 | 48 | toluene | 22%a |
| 8 | 1d | 5 | 80 | 48 | toluene | 78% |
| 9 | 1e | 4 | 80 | 24 | PhH | 55% (7%) |
| 10 | 1f | 4 | 60 | 5 | CH2Cl2 | 44% (3%) |
| 11 | 1h | 4 | rt | 6 | CH2Cl2 | 0% (57%) |
| 12 | 1h | 4 | 50 | 1 | CH2Cl2 | 7% (23%) |
| 13 | 1i | 4 | rt | 10 | CH2Cl2 | 0% (67%) |
| 14 | 1i | 5 | rt | 2 | CH2Cl2 | 98% |
| 15 | 1j | 4 | 80 | 12 | PhH | 74% (19%) |
a 60% of starting material recovered
As expected, reactions with the more substituted ylide 4 were significantly slower than those with the parent ylide 5 (compare reaction temperatures and times, entries 1, 3 and 5 versus entries 2, 4 and 6). Increasing the steric bulk of the ketene substituent also slows the reaction, in line with our expectations, exemplified by the reactions of cyclohexyl-substituted ketene 1d (entries 7 and 8, compared with entries 1–6). In almost all cases, if reactions were left for extended periods beyond consumption of starting material, small quantities of desilylated allenes were observed. With alkyl-substituted ketenes this could be avoided by careful reaction monitoring, but was unavoidable in the (somewhat sluggish) reaction of ylie 4 with aromatic substituted ketenes 1e/f, while in the case of furyl and thiophenyl-substituted ketenes 1h and 1i, these were the sole or predominant products of reaction. It appears that the presence of the (hetero)aryl group labilises the silicon to cleavage under the reaction conditions, likely reflecting the silyl group acting as an electrofuge with the (hetero)aryl substituent stabilising incipient anionic character at the adjacent carbon.
We next investigated the reaction of silylketenes 1 with other ylides (Scheme 3). Pleasingly, ylides stabilised by alternative electron-withdrawing groups such as cyano (9), acetyl (10) and benzoyl (11) were also successfully employed in allenylsilane formation. Likewise, the semi-stabilised aromatic-substituted ylides 12 and 13 also gave good yields of the allenylsilanes. The incorporation of the 4-bromophenyl group from 13 is particularly interesting since the bromide provides a handle for further functionalisation either of the allenylsilane or of the products of its subsequent reaction.
![[1860-5397-1-5-i3]](/bjoc/content/inline/1860-5397-1-5-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of silylketenes with various ylides
Scheme 3: Reaction of silylketenes with various ylides
Table 3: Reaction of silylketenes with various ylides
| Entry | Ketene | Ylide | R2 | Temp (°C) | t (h) | Solvent | Allene | Yield |
|---|---|---|---|---|---|---|---|---|
| 1 | 1a | 9 | CN | rt | 1 | CH2Cl2 | 14a | 53% |
| 2 | 1b | 9 | CN | rt | 16 | CH2Cl2 | 14b | 94% |
| 3 | 1a | 10 | COMe | rt | 8 | CH2Cl2 | 15a | 52% |
| 4 | 1a | 11 | COPh | 50 | 5 | CH2Cl2 | 16a | 62% |
| 5 | 1b | 11 | COPh | 60 | 24 | CH2Cl2 | 16b | 62% |
| 6 | 1a | 12 | Ph | rt | 0.5 | THF | 17a | 65% |
| 7 | 1b | 12 | Ph | rt | 0.5 | THF | 17b | 68% |
| 8 | 1g | 12 | Ph | rt | 0.5 | THF | 17g | 58% |
| 9 | 1b | 13 | 4-BrPh | rt | 0.5 | THF | 18b | 53% |
a the TBDMS ketene was employed in place of the TES ketene
Finally, the reactions of the non-stabilised ylides Ph3P=CHSPh, Ph3P=CHOMe and Ph3P=CH2 were examined. In all cases, rapid decomposition of the silylketenes to complex mixtures of unidentified products was observed, even at sub-ambient temperatures. One possible mechanism proposed for the decomposition of unsubstituted silylketenes with reactive ylides involves deprotonation by the basic reagent to generate ynolate anions. Clearly, since such a pathway cannot operate with the substituted ketenes used here other mechanisms are at play, which may include nucleophilic desilylation or ketene oligomerisation initiated by nucleophilic addition to the ketene carbonyl group. If the former pathway were occurring, then increasing the size of the silyl substituent ought to retard this pathway, but reaction of the tert-butyldimethylsilyl analogue of 1b with Ph3P=CH2 also gave no observable allenylsilane product. The lack of identifiable products from these reactions precludes further speculation on the mechanism of decomposition at this stage.
The confirmation that non-stabilised ylides such as methylenetriphenylphosphorane were unsuitable reaction partners for condensation with even substituted silylketenes was disappointing, since the 1-substituted allenylsilanes 19 that would be thus formed are useful synthetic intermediates. Our attention therefore turned to the application of milder (and specifically less basic/nucleophilic) methylenating reagents. We first investigated the Lombardo reagent,[30] and were pleased to find that a modest yield of allenylsilane 19b was obtained from this reaction (Scheme 4).
![[1860-5397-1-5-i4]](/bjoc/content/inline/1860-5397-1-5-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Methylenation of silylketene 1b with the Lombardo reagent
Scheme 4: Methylenation of silylketene 1b with the Lombardo reagent
Although the Lombardo reaction had several disadvantages (including the lengthy preparation of the reagent and the heterogeneous nature of the reaction medium) which led us to discount it as a long term solution to the problem, it gave us the encouragement to screen other titanium carbenoid-based methylenating reagents. The Petasis reagent (dimethyltitanocene) [31,32] was our next choice, and we were pleased to find that it afforded modest to excellent yields of the allenylsilanes in reaction with most of the ketene substrates examined (Scheme 5, Table 4).
![[1860-5397-1-5-i5]](/bjoc/content/inline/1860-5397-1-5-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Methylenation of silylketenes with the Petasis reagent
Scheme 5: Methylenation of silylketenes with the Petasis reagent
Table 4: Methylenation of silylketenes with the Petasis reagent
| Entry | Ketene | Time (h) | Yield |
|---|---|---|---|
| 1 | 1b | 6 | 78% |
| 2 | 1b* | 2 | 79% |
| 3 | 1c | 5 | 74% |
| 4 | 1d | 26 | 82% |
| 5 | 1e | 5 | 38% |
a the TBDMS ketene was employed in place of the TES ketene
The low yield with phenyl-substituted ketene 1e is likely attributable to the instability of the ketene at the elevated temperatures for the Petasis reaction. Consistent with this, attempted reaction of the heteroaryl-substituted ketenes 1h/j failed to return any allenylsilane product. We have not as yet examined the use of the more reactive Tebbe reagent which may function effectively at lower temperatures and hence offer a solution to this current limitation. Additionally, it will be of future interest to investigate the chromium-based Takai procedures for olefination with higher alkylidene reagents,[33] since this has the potential to deliver alkyl-substituted allenylsilanes.
In summary, we have further developed the reaction of stabilised and semi-stabilised ylides with silylketenes to include, for the first time, carbon-substituted silylketenes leading to a range of novel 1,3-disubstituted and 1,3,3-trisubstituted allenylsilanes. Additionally, we have demonstrated for the first time that titanium carbenoids can be used to methylenate silylketenes, providing the first access to 1-substituted allenylsilanes from these substrates. Given the broad range of useful chemistry undertaken by allenylsilanes, we expect that these new highly functionalised members of the class will find broad synthetic utility. Our own studies on their application in heterocyclic synthesis will be disclosed in due course.
Description of Supporting Information
Supporting Information features copies of 1H nmr spectra of silylated diazoketones 2 and silylketenes 1, plus 1H and 13C nmr spectra of allenylsilanes 6, 7, and 14–19.
References
-
Pornet, J. Product Subclass 32: Allenylsilanes. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 669–683.
Return to citation in text: [1] [2] -
Masse, C. E.; Panek, J. S. Chem. Rev. 1995, 95, 1293–1316. doi:10.1021/cr00037a008
Return to citation in text: [1] -
Jian, J.; Smith, D. T.; Weinreb, S. M. J. Org. Chem. 1995, 60, 5366–5367. doi:10.1021/jo00122a001
Return to citation in text: [1] -
Weinreb, S. M.; Smith, D. T.; Jin, J. Synthesis 1998, 509–521. doi:10.1055/s-1998-5920
Return to citation in text: [1] -
Danheiser, R. L.; Carini, D. J. J. Org. Chem. 1980, 45, 3925–3927. doi:10.1021/jo01307a044
Return to citation in text: [1] -
Danheiser, R. L.; Carini, D. J.; Kwasigroch, C. A. J. Org. Chem. 1986, 51, 3870–3878. doi:10.1021/jo00370a023
Return to citation in text: [1] -
Danheiser, R. L.; Kwasigroch, C. A.; Tsai, Y.-M. J. Am. Chem. Soc. 1985, 107, 7233–7235. doi:10.1021/ja00310a109
Return to citation in text: [1] [2] -
Daidouji, K.; Fuchibe, K.; Akiyama, T. Org. Lett. 2005, 7, 1051–1053. doi:10.1021/ol047343c
Return to citation in text: [1] -
Danheiser, R. L.; Carini, D. J.; Basak, A. J. Am. Chem. Soc. 1981, 103, 1604–1606. doi:10.1021/ja00396a071
Return to citation in text: [1] -
Danheiser, R. L.; Carini, D. J.; Fink, D. M.; Basak, A. Tetrahedron 1983, 39, 935–947. doi:10.1016/S0040-4020(01)88592-6
Return to citation in text: [1] -
Danheiser, R. L.; Fink, D. M. Tetrahedron Lett. 1985, 26, 2513–2516. doi:10.1016/S0040-4039(00)98824-5
Return to citation in text: [1] -
Danheiser, R. L.; Becker, D. A. Heterocycles 1987, 25, 277–281.
Return to citation in text: [1] -
Yadav, V. K.; Sriramurthy, V. Org. Lett. 2004, 6, 4495–4498. doi:10.1021/ol047991w
Annulations leading to cyclohexenes are known, see reference 11.
Return to citation in text: [1] -
Danheiser, R. L.; Tsai, Y.-M.; Fink, D. M. Org. Synth. 1987, 66, 1–4.
Return to citation in text: [1] -
Ruden, R. A. J. Org. Chem. 1974, 39, 3607–3608. doi:10.1021/jo00938a041
Return to citation in text: [1] -
Orlov, V. Y.; Lebedev, S. A.; Ponomarev, S. V.; Lutzenko, I. F. Zh. Obshch. Khim. 1975, 45, 708–9.
Return to citation in text: [1] -
Kita, Y.; Tsuzuki, Y.; Kitagaki, S.; Akai, S. Chem. Pharm. Bull. 1994, 42, 233–236.
Return to citation in text: [1] -
Pommier, A.; Kocienski, P.; Pons, J.-M. J. Chem. Soc., Perkin Trans. 1 1998, 2105–2118. doi:10.1039/a801254g
Return to citation in text: [1] [2] -
Pons, J.-M.; Kocienski, P. Product Subclass 31: Silylketenes. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 657–668.
Return to citation in text: [1] -
Sakurai, H.; Shirahata, A.; Sasaki, K.; Hosomi, A. Synthesis 1979, 740–741. doi:10.1055/s-1979-28823
Return to citation in text: [1] -
Danheiser, R. L.; Sard, H. J. Org. Chem. 1980, 45, 4810–4812. doi:10.1021/jo01311a065
Return to citation in text: [1] -
Brady, W. T.; Cheng, T. C. J. Organomet. Chem. 1977, 137, 287–292. doi:10.1016/S0022-328X(00)81435-7
Return to citation in text: [1] -
Marsden, S. P.; Pang, W.-K. Chem. Commun. 1999, 1199–1200. doi:10.1039/a902549i
Return to citation in text: [1] [2] -
Maas, G.; Brückmann, R. J. Org. Chem. 1985, 50, 2801–2802. doi:10.1021/jo00215a049
Return to citation in text: [1] -
Brückmann, R.; Schneider, K.; Maas, G. Tetrahedron 1989, 45, 5517–5530. doi:10.1016/S0040-4020(01)89498-9
Return to citation in text: [1] -
Loebach, J. L.; Bennett, D. M.; Danheiser, R. L. J. Am. Chem. Soc. 1998, 120, 9690–9691. doi:10.1021/ja982101r
Return to citation in text: [1] -
Loebach, J. L.; Bennett, D. M.; Danheiser, R. L. J. Org. Chem. 1998, 63, 8380–8389. doi:10.1021/jo981289u
Return to citation in text: [1] -
Allspach, T.; Gümbel, H.; Regitz, M. J. Organomet. Chem. 1985, 290, 33–39. doi:10.1016/0022-328X(85)80146-7
Return to citation in text: [1] -
Baigrie, L. M.; Seiklay, H. R.; Tidwell, T. T. J. Am. Chem. Soc. 1985, 107, 5391–5396. doi:10.1021/ja00305a012
Return to citation in text: [1] -
Lombardo, L. Org. Synth. 1987, 65, 81–85.
Return to citation in text: [1] -
Petasis, N. A.; Lu, S.-P.; Bzowej, E. I.; Fu, D. K.; Staszewski, J. P.; Akritopoulou-Zanze, I.; Patane, M. A.; Hu, Y.-H. Pure Appl. Chem. 1996, 68, 667–670.
Return to citation in text: [1] -
Payack, J. F.; Hughes, D. L.; Cai, D.; Cottrell, I. F.; Verhoeven, T. R. Org. Synth. 1991, 79, 19–22.
Return to citation in text: [1] -
Fürstner, A. Chem. Rev. 1999, 99, 991–1045. doi:10.1021/cr9703360
Return to citation in text: [1]
| 28. | Allspach, T.; Gümbel, H.; Regitz, M. J. Organomet. Chem. 1985, 290, 33–39. doi:10.1016/0022-328X(85)80146-7 |
| 18. | Pommier, A.; Kocienski, P.; Pons, J.-M. J. Chem. Soc., Perkin Trans. 1 1998, 2105–2118. doi:10.1039/a801254g |
| 23. | Marsden, S. P.; Pang, W.-K. Chem. Commun. 1999, 1199–1200. doi:10.1039/a902549i |
| 1. | Pornet, J. Product Subclass 32: Allenylsilanes. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 669–683. |
| 2. | Masse, C. E.; Panek, J. S. Chem. Rev. 1995, 95, 1293–1316. doi:10.1021/cr00037a008 |
| 7. | Danheiser, R. L.; Kwasigroch, C. A.; Tsai, Y.-M. J. Am. Chem. Soc. 1985, 107, 7233–7235. doi:10.1021/ja00310a109 |
| 8. | Daidouji, K.; Fuchibe, K.; Akiyama, T. Org. Lett. 2005, 7, 1051–1053. doi:10.1021/ol047343c |
| 23. | Marsden, S. P.; Pang, W.-K. Chem. Commun. 1999, 1199–1200. doi:10.1039/a902549i |
| 7. | Danheiser, R. L.; Kwasigroch, C. A.; Tsai, Y.-M. J. Am. Chem. Soc. 1985, 107, 7233–7235. doi:10.1021/ja00310a109 |
| 24. | Maas, G.; Brückmann, R. J. Org. Chem. 1985, 50, 2801–2802. doi:10.1021/jo00215a049 |
| 25. | Brückmann, R.; Schneider, K.; Maas, G. Tetrahedron 1989, 45, 5517–5530. doi:10.1016/S0040-4020(01)89498-9 |
| 26. | Loebach, J. L.; Bennett, D. M.; Danheiser, R. L. J. Am. Chem. Soc. 1998, 120, 9690–9691. doi:10.1021/ja982101r |
| 27. | Loebach, J. L.; Bennett, D. M.; Danheiser, R. L. J. Org. Chem. 1998, 63, 8380–8389. doi:10.1021/jo981289u |
| 5. | Danheiser, R. L.; Carini, D. J. J. Org. Chem. 1980, 45, 3925–3927. doi:10.1021/jo01307a044 |
| 6. | Danheiser, R. L.; Carini, D. J.; Kwasigroch, C. A. J. Org. Chem. 1986, 51, 3870–3878. doi:10.1021/jo00370a023 |
| 20. | Sakurai, H.; Shirahata, A.; Sasaki, K.; Hosomi, A. Synthesis 1979, 740–741. doi:10.1055/s-1979-28823 |
| 3. | Jian, J.; Smith, D. T.; Weinreb, S. M. J. Org. Chem. 1995, 60, 5366–5367. doi:10.1021/jo00122a001 |
| 4. | Weinreb, S. M.; Smith, D. T.; Jin, J. Synthesis 1998, 509–521. doi:10.1055/s-1998-5920 |
| 21. | Danheiser, R. L.; Sard, H. J. Org. Chem. 1980, 45, 4810–4812. doi:10.1021/jo01311a065 |
| 22. | Brady, W. T.; Cheng, T. C. J. Organomet. Chem. 1977, 137, 287–292. doi:10.1016/S0022-328X(00)81435-7 |
| 1. | Pornet, J. Product Subclass 32: Allenylsilanes. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 669–683. |
| 14. | Danheiser, R. L.; Tsai, Y.-M.; Fink, D. M. Org. Synth. 1987, 66, 1–4. |
| 16. | Orlov, V. Y.; Lebedev, S. A.; Ponomarev, S. V.; Lutzenko, I. F. Zh. Obshch. Khim. 1975, 45, 708–9. |
| 17. | Kita, Y.; Tsuzuki, Y.; Kitagaki, S.; Akai, S. Chem. Pharm. Bull. 1994, 42, 233–236. |
| 31. | Petasis, N. A.; Lu, S.-P.; Bzowej, E. I.; Fu, D. K.; Staszewski, J. P.; Akritopoulou-Zanze, I.; Patane, M. A.; Hu, Y.-H. Pure Appl. Chem. 1996, 68, 667–670. |
| 32. | Payack, J. F.; Hughes, D. L.; Cai, D.; Cottrell, I. F.; Verhoeven, T. R. Org. Synth. 1991, 79, 19–22. |
| 13. |
Yadav, V. K.; Sriramurthy, V. Org. Lett. 2004, 6, 4495–4498. doi:10.1021/ol047991w
Annulations leading to cyclohexenes are known, see reference 11. |
| 18. | Pommier, A.; Kocienski, P.; Pons, J.-M. J. Chem. Soc., Perkin Trans. 1 1998, 2105–2118. doi:10.1039/a801254g |
| 19. | Pons, J.-M.; Kocienski, P. Product Subclass 31: Silylketenes. In Science of Synthesis; Fleming, I., Ed.; Georg Thieme Verlag: Stuttgart, 2002; Vol. 4, pp 657–668. |
| 29. | Baigrie, L. M.; Seiklay, H. R.; Tidwell, T. T. J. Am. Chem. Soc. 1985, 107, 5391–5396. doi:10.1021/ja00305a012 |
| 9. | Danheiser, R. L.; Carini, D. J.; Basak, A. J. Am. Chem. Soc. 1981, 103, 1604–1606. doi:10.1021/ja00396a071 |
| 10. | Danheiser, R. L.; Carini, D. J.; Fink, D. M.; Basak, A. Tetrahedron 1983, 39, 935–947. doi:10.1016/S0040-4020(01)88592-6 |
| 11. | Danheiser, R. L.; Fink, D. M. Tetrahedron Lett. 1985, 26, 2513–2516. doi:10.1016/S0040-4039(00)98824-5 |
© 2005 Marsden and Ducept; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)