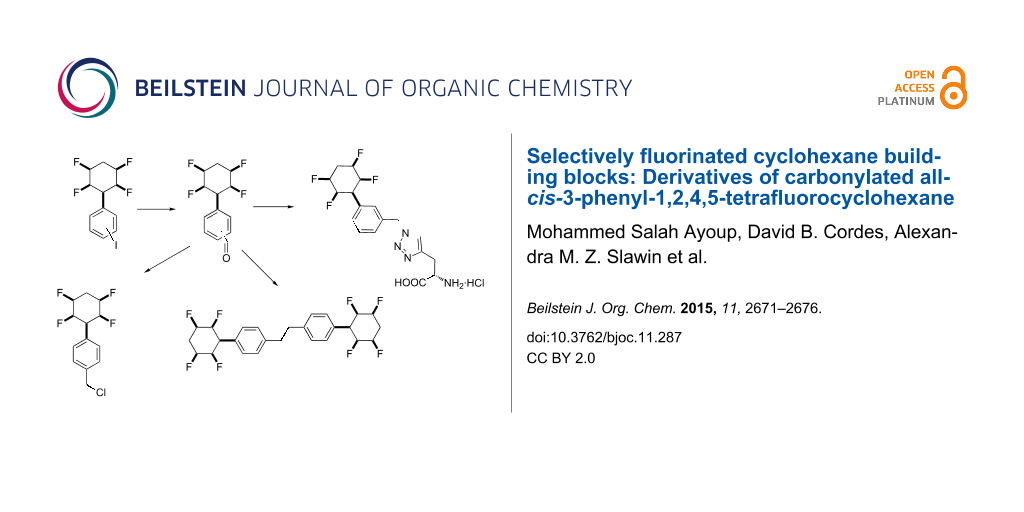
Abstract
Palladium catalysed carbonylation reactions using the meta- and para-iodo derivatives of all-cis-3-phenyl-1,2,4,5-tetrafluorocyclohexane (4) are illustrated as the start point for a variety of functional group interconversions. The resultant benzaldehyde and benzoic acids offer novel building blocks for further derivatisation and facilitate the incorporation of the facially polarised all-cis-1,2,4,5-tetrafluorocyclohexane motif into more advanced molecular scaffolds.

Graphical Abstract
Introduction
Selectively fluorinated building blocks have proven invaluable in drug discovery [1] and agrochemical research programmes [2]. Most such building blocks possess aryl/heteroaryl-F, aryl/heteroaryl-(X)CF3 or variants thereof, whereas selectively fluorinated aliphatics are much rarer. We have had a focus on introducing selectively fluorinated cyclohexane rings, to complement aromatic building blocks. In this context we have recently prepared the all-cis-tetrafluorocyclohexanes 2 and 3 [3,4] and also all-cis-hexafluorocyclohexane (1) [5] as shown in Figure 1. In the case of the hexafluorocyclohexane, the ring maintains a chair conformation and therefore there are three C–F bonds orientated triaxial, on one face of the ring. This gives rise to a large dipole moment (6.2 D) and a molecule which is among the most polar aliphatics known in organic chemistry. The all-cis-tetrafluorocyclohexanes 2 and 3 are also facially polarised because in the chair conformation, they always have two 1,3-diaxial C–F bonds on one face of the ring, and this results in ring systems also with large dipole moments (4.9 D for 2 and 5.2 D for 3). The nature of these cyclohexanes, where the two faces are oppositely polarised, presents a unique property for use in pharmaceuticals and agrochemicals discovery programmes.
![[1860-5397-11-287-1]](/bjoc/content/figures/1860-5397-11-287-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: All cis-hexafluoro- 1 and tetrafluorocyclohexanes 2 and 3 result in facially polarised ring motifs with an electrostatically negative (δ –ve) fluorine face and an electrostatically positive (δ +ve) hydrogen face.
Figure 1: All cis-hexafluoro- 1 and tetrafluorocyclohexanes 2 and 3 result in facially polarised ring motifs ...
Compounds 1–3 are unfunctionalised and not amenable to derivatisation, and therefore we started to explore the chemistry of all-cis-3-phenyl-1,2,4,5-tetrafluorocyclohexane (4) [6], to access derivatives carrying this ring motif. In that context it has been demonstrated that 4 can be elaborated in a relatively straightforward manner by mainstream reactions of electrophilic aromatic substitution [7]. This extended to the synthesis of cyclohexane substituted (S)-L-phenylalanines with orthogonal protecting groups suitable for their incorporation into peptides [8]. In this paper it is demonstrated that palladium mediated carbonylation of the aryl iodinated derivatives 5–7 forms the basis of a diversity of new products which may prove attractive as building blocks for structure activity studies in bioactive research projects.
Results and Discussion
A key reaction for this programme involved the carbonylation of aryl iodides 5, 6 and 7 which are derived by HIO4 treatment of all-cis-3-phenyl-1,2,4,5-tetrafluorocyclohexane (4). The ortho isomer 5 was separated from the meta and para isomers 6 and 7, the latter of which were recovered as a mixture as previously reported [8]. Palladium catalysed carboxylation [9] was explored with aryl iodides 5 or 6/7 as a mixture, and these gave the corresponding ethyl esters 8 and 9/10, respectively. The meta and para esters 9 and 10 were easily separated by chromatography. Hydrolysis of esters 8–10 using trifluoroacetic acid (TFA) or 6 M HCl in dioxane [10] gave the corresponding benzoic acids 11, 12, and 13 as illustrated in Scheme 1.
![[1860-5397-11-287-i1]](/bjoc/content/inline/1860-5397-11-287-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of benzoic acids 11–13; i. HIO4·2H2O (50%), AcOH, H2SO4, I2, H2O, 70 °C 24 h, 92%.; ii. Pd(OAc)2, Ph3P,Et3N, EtOH, CO (1 atm), 80 °C, 16 h. iii. HCl (6 M), 1,4-dioxane, 70 °C, 24 h or TFA/H2O (9:1), 100 °C, 24 h.
Scheme 1: Preparation of benzoic acids 11–13; i. HIO4·2H2O (50%), AcOH, H2SO4, I2, H2O, 70 °C 24 h, 92%.; ii....
Direct palladium catalysed formylation of aryl iodides 6/7 as a mixture was explored using Pd(PPh3)4, Bu4SnH and CO [11]. This generated benzaldehyde derivatives 14 and 15 in a 1:3 ratio, respectively, which could be separated by column chromatography. The reaction is illustrated in Scheme 2. The structure of aldehyde 15 was confirmed by X-ray structure analysis and is shown in Figure 2.
![[1860-5397-11-287-i2]](/bjoc/content/inline/1860-5397-11-287-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of benzaldehyde derivatives 14 and 15: i. Pd(PPh3)4, Bu3SnH, THF, CO (1 atm), 50 °C, 2–3 h, 70%.
Scheme 2: Synthesis of benzaldehyde derivatives 14 and 15: i. Pd(PPh3)4, Bu3SnH, THF, CO (1 atm), 50 °C, 2–3 ...
![[1860-5397-11-287-2]](/bjoc/content/figures/1860-5397-11-287-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structure of aldehyde 15. CCDC number 1432193.
Figure 2: X-ray structure of aldehyde 15. CCDC number 1432193.
Transformations of benzaldehyde 15 were explored. For example McMurry coupling [12] of 15 gave a mixture of stilbene isomers 16. Direct hydrogenation of the mixture afford dihydrostilbene 17 as a crystalline solid, a structure which was confirmed by X-ray crystallography and is shown in the inset in Scheme 3. Aldehyde 15 could also be converted to styrene 18 in good yield using the Simmons–Smith method [13] as illustrated in Scheme 3.
![[1860-5397-11-287-i3]](/bjoc/content/inline/1860-5397-11-287-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Olefination reactions of 15 and the X-ray structure of 17 (CCDC number 1432194): i. Zn, TiCl4, THF, 0 °C to reflux, 12 h; ii. Pd/C, H2, EtOAc, 20 °C, 16 h, 76% over 2 steps. iii. Zn, CH2I2, TiCl4, THF, 0 °C to 20 °C, 12 h; 79%.
Scheme 3: Olefination reactions of 15 and the X-ray structure of 17 (CCDC number 1432194): i. Zn, TiCl4, THF,...
For further elaboration, aldehyde 15 was reduced with NaBH4 in good yield to generate the corresponding benzyl alcohol 19 [14]. Iodination of 19 to generate 20 with HI in chloroform [15] proved superior (95% yield) to the more classical Appel protocol which gave low yields in our hands. Nucleophilic substitution with the resultant benzyl iodide 20 using azide gave the corresponding benzyl azide 21 in good yield [16]. Chlorination to generate 23 was accomplished by treatment with mesyl chloride/Et3N in a one pot protocol, presumably via mesylate 22, as illustrated in Scheme 4 [17].
![[1860-5397-11-287-i4]](/bjoc/content/inline/1860-5397-11-287-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions from aldehyde 15: i. NaBH4, THF, 20 °C, 1 h, 98%.; ii. HI (57%), CHCl3, 30 h, 95%; iii. Bu4NN3, acetone/H2O (4:1), 20 °C, 3 h, 94%; iv. MsCl, Et3N, DCM, −78 °C to 20 °C, 85%.
Scheme 4: Reactions from aldehyde 15: i. NaBH4, THF, 20 °C, 1 h, 98%.; ii. HI (57%), CHCl3, 30 h, 95%; iii. Bu...
Reduction of benzyl azide 21 to generate amine 27 as its hydrochloride salt was achieved by a Staudinger reduction in good yield [18]. Azide 21 was also amenable to a ‘click’ reaction, in this case with the acetylenic protected amino acid 24 [19]. This gave the 1,2,3-triazol linked adduct 25 which could be fully deprotected to generate the free amino acid hydrochloride 26 as illustrated in Scheme 5.
![[1860-5397-11-287-i5]](/bjoc/content/inline/1860-5397-11-287-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reactions of benzyl azide 21; i. 24, Cu(OAc)2, Na ascorbate, t-BuOH, H2O, 20 °C, 16 h, 72%; ii. HCl (6 M), 1,4-dioxane, 80 °C, 48 h, 96%; iii. Ph3P, THF/H2O (10:1), then HCl, rt, 1 h, 74%.
Scheme 5: Reactions of benzyl azide 21; i. 24, Cu(OAc)2, Na ascorbate, t-BuOH, H2O, 20 °C, 16 h, 72%; ii. HCl...
The reactions described above were equally amenable to the meta-benzaldehyde 14, as illustrated in Scheme 6.
![[1860-5397-11-287-i6]](/bjoc/content/inline/1860-5397-11-287-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactions of aldehyde 14: i. NaBH4, THF, rt, 1 h, 98%.; ii. HI (57%), CHCl3, 30 h, 94%; iii. Bu4NN3, acetone/H2O (4:1), rt, 3 h, 91%; iv. 24, Cu(OAc)2, Na ascorbate, t-BuOH, H2O, rt, 12 h, 81%; v. HCl (6 M), 1,4-dioxane, 70 °C, 48 h, 94%.
Scheme 6: Reactions of aldehyde 14: i. NaBH4, THF, rt, 1 h, 98%.; ii. HI (57%), CHCl3, 30 h, 94%; iii. Bu4NN3...
Conclusion
In summary it is demonstrated that the all-cis-1,2,4,5-tetrafluorocyclohexane motif has been incorporated in a range of products prepared from aryl iodides 5–7. These derivatives derive from aryl carboxylation or carbonylation, and complement those that can be prepared directly by electrophilic aromatic substitution of phenyl derivative 4. This chemistry should more readily facilitate the exploration of the properties and potential of the all-cis-1,2,4,5-tetrafluorocyclohexane motif in a diversity of research programmes.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 3.5 MB | Download |
Acknowledgements
MA thanks the Egyptian Government for a Scholarship under the Channel Scheme and we are grateful to the support of Professor L. Fathey of the University of Alexandria, Department of Chemistry, Egypt. DO'H thanks the Royal Society for a Wolfson Research Merit Award and we also thank the EPSRC Mass Spectrometry Service at Swansea University, UK for analyses.
References
-
Wang, J.; Sánchez-Rosolló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014
Return to citation in text: [1] -
Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2011, 47, 8265–8267. doi:10.1039/C1CC13016A
Return to citation in text: [1] -
Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/C2CC34679F
Return to citation in text: [1] -
Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O’Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232
Return to citation in text: [1] -
Durie, A. J.; Fujiwara, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. J. Org. Chem. 2014, 79, 8228–8233. doi:10.1021/jo501432x
Return to citation in text: [1] -
Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354
Return to citation in text: [1] -
Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2015, 13, 5621–5624. doi:10.1039/C5OB00650C
Return to citation in text: [1] [2] -
Moriello, A. S.; Balas, L.; Ligresti, A.; Cascio, M. G.; Durand, T.; Morea, E.; Ortar, G.; Di Marzo, V. J. Med. Chem. 2006, 49, 2320–2332. doi:10.1021/jm051226l
Return to citation in text: [1] -
Barfoot, C. W.; Harvey, J. E.; Kenworthy, M. N.; Kilburn, J. P.; Ahmed, M.; Taylor, R. J. K. Tetrahedron 2005, 61, 3403–3417. doi:10.1016/j.tet.2004.10.097
Return to citation in text: [1] -
Baillargeon, V. P.; Stille, J. K. J. Am. Chem. Soc. 1986, 108, 452–461. doi:10.1021/ja00263a015
Return to citation in text: [1] -
Ruzié, C.; Karpinska, J.; Kennedy, A. R.; Geerts, Y. H. J. Org. Chem. 2013, 78, 7741–7748. doi:10.1021/jo401134c
Return to citation in text: [1] -
Takai, K.; Hotta, Y.; Oshima, K.; Nozaki, H. Bull. Chem. Soc. Jpn. 1980, 53, 1698–1702. doi:10.1246/bcsj.53.1698
Return to citation in text: [1] -
Johnson, M. R.; Rickborn, B. J. Org. Chem. 1970, 35, 1041–1045. doi:10.1021/jo00829a039
Return to citation in text: [1] -
Bianchini, G.; Aschi, M.; Cavicchio, G.; Crucianelli, M.; Preziuso, S.; Gallina, C.; Nastari, A.; Gavuzzo, E.; Mazza, F. Bioorg. Med. Chem. 2005, 13, 4740–4749. doi:10.1016/j.bmc.2005.04.079
Return to citation in text: [1] -
Campbell-Verduyn, L. S.; Mirfeizi, L.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Chem. Commun. 2009, 2139–2141. doi:10.1039/B822994E
Return to citation in text: [1] -
Amer, A.; Ayoup, M. S.; Khattab, S. N.; Hassan, S. Y.; Langer, V.; Senior, S.; El-Massry, A. M. Carbohydr. Res. 2010, 345, 2474–2484. doi:10.1016/j.carres.2010.08.010
Return to citation in text: [1] -
Wallace, K. J.; Hanes, R.; Anslyn, E.; Morey, J.; Kilway, K. V.; Siegel, J. Synthesis 2005, 2080–2083. doi:10.1055/s-2005-869963
Return to citation in text: [1] -
Kuijpers, B. H. M.; Groothuys, S.; Keereweer, A. R.; Quaedflieg, P. J. L. M.; Blaauw, R. H.; van Delft, F. L.; Rutjes, F. P. J. T. Org. Lett. 2004, 6, 3123–3126. doi:10.1021/ol048841o
Return to citation in text: [1]
| 19. | Kuijpers, B. H. M.; Groothuys, S.; Keereweer, A. R.; Quaedflieg, P. J. L. M.; Blaauw, R. H.; van Delft, F. L.; Rutjes, F. P. J. T. Org. Lett. 2004, 6, 3123–3126. doi:10.1021/ol048841o |
| 17. | Amer, A.; Ayoup, M. S.; Khattab, S. N.; Hassan, S. Y.; Langer, V.; Senior, S.; El-Massry, A. M. Carbohydr. Res. 2010, 345, 2474–2484. doi:10.1016/j.carres.2010.08.010 |
| 18. | Wallace, K. J.; Hanes, R.; Anslyn, E.; Morey, J.; Kilway, K. V.; Siegel, J. Synthesis 2005, 2080–2083. doi:10.1055/s-2005-869963 |
| 1. | Wang, J.; Sánchez-Rosolló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 6. | Durie, A. J.; Fujiwara, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. J. Org. Chem. 2014, 79, 8228–8233. doi:10.1021/jo501432x |
| 15. | Bianchini, G.; Aschi, M.; Cavicchio, G.; Crucianelli, M.; Preziuso, S.; Gallina, C.; Nastari, A.; Gavuzzo, E.; Mazza, F. Bioorg. Med. Chem. 2005, 13, 4740–4749. doi:10.1016/j.bmc.2005.04.079 |
| 5. | Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O’Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232 |
| 16. | Campbell-Verduyn, L. S.; Mirfeizi, L.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Chem. Commun. 2009, 2139–2141. doi:10.1039/B822994E |
| 3. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2011, 47, 8265–8267. doi:10.1039/C1CC13016A |
| 4. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/C2CC34679F |
| 13. | Takai, K.; Hotta, Y.; Oshima, K.; Nozaki, H. Bull. Chem. Soc. Jpn. 1980, 53, 1698–1702. doi:10.1246/bcsj.53.1698 |
| 2. | Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014 |
| 14. | Johnson, M. R.; Rickborn, B. J. Org. Chem. 1970, 35, 1041–1045. doi:10.1021/jo00829a039 |
| 9. | Moriello, A. S.; Balas, L.; Ligresti, A.; Cascio, M. G.; Durand, T.; Morea, E.; Ortar, G.; Di Marzo, V. J. Med. Chem. 2006, 49, 2320–2332. doi:10.1021/jm051226l |
| 11. | Baillargeon, V. P.; Stille, J. K. J. Am. Chem. Soc. 1986, 108, 452–461. doi:10.1021/ja00263a015 |
| 8. | Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2015, 13, 5621–5624. doi:10.1039/C5OB00650C |
| 12. | Ruzié, C.; Karpinska, J.; Kennedy, A. R.; Geerts, Y. H. J. Org. Chem. 2013, 78, 7741–7748. doi:10.1021/jo401134c |
| 8. | Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2015, 13, 5621–5624. doi:10.1039/C5OB00650C |
| 7. | Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354 |
| 10. | Barfoot, C. W.; Harvey, J. E.; Kenworthy, M. N.; Kilburn, J. P.; Ahmed, M.; Taylor, R. J. K. Tetrahedron 2005, 61, 3403–3417. doi:10.1016/j.tet.2004.10.097 |
© 2015 Ayoup et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)