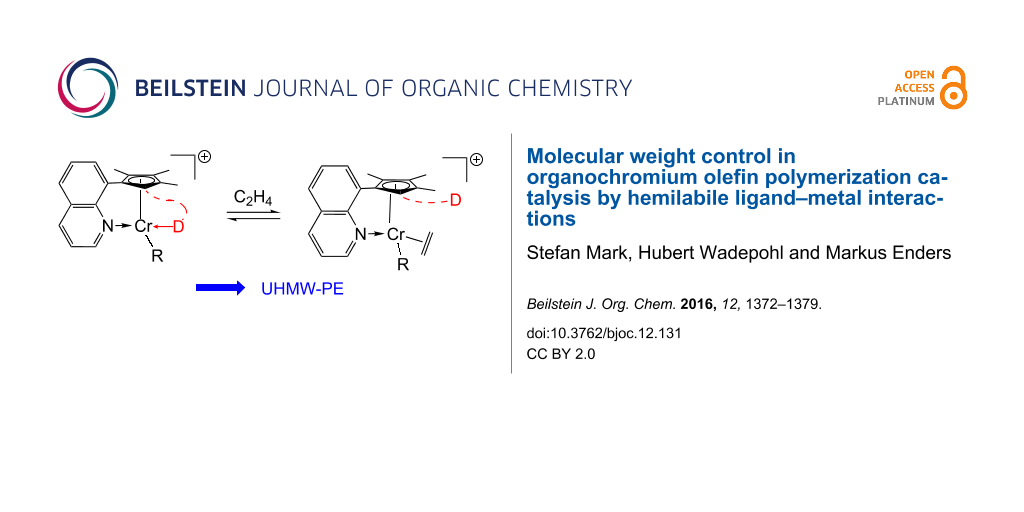
Abstract
A series of Cr(III) complexes based on quinoline-cyclopentadienyl ligands with additional hemilabile side arms were prepared and used as single-site catalyst precursors for ethylene polymerization. The additional donor functions interact with the metal centers only after activation with the co-catalyst. Evidence for this comes from DFT-calculations and from the differing behavior of the complexes in ethylene polymerization. All complexes investigated show very high catalytic activity and the additional side arm minimizes chain-transfer reactions, leading to increase of molecular weights of the resulting polymers.

Graphical Abstract
Introduction
Chelate ligands with both, a strongly coordinating moiety and a weakly coordinating donor function allow the stabilization of vacant coordination sites at metal centers and may act as placeholders for external substrates. The concept of hemilabile ligands has been introduced in 1979 [1] and has been applied for the development of improved transition metal catalysts [2-7]. The donor–acceptor interaction of the hemilabile moiety with the metal center should be weak enough to allow the displacement by a substrate, which should itself be transformed during the catalytic reaction. The bonding ability of internal or external stabilizing ligands in relation to the substrate plays a crucial role. In contrast to external donors, a special feature of a hemilabile donor is the fact that only one stabilizing ligand per metal center is available. Consequently, a large excess of external donors (e.g., solvent molecules, substrates, additives, etc.) may displace a relatively strong intramolecular donor function. Several examples for olefin polymerization catalysts with hemilabile ligands are known and the impact of the hemilabile group on the polymerization behavior can be immense [8-12]. Examples are the switching from polymerization to trimerization selectivity [13] or the suppression of chain termination by weak interactions with fluorine substituents [14-17]. The interaction with fluorine atoms from fluorinated borate anions have also shown to play a role in olefin polymerization [18-24].
Cyclopentadienyl (Cp)-based chromium complexes exhibit very good ethylene polymerization properties, when the coordination sphere of the chromium center is completed by an additional ligand. Improved stability and hence polymer productivities are obtained when the donor is tethered to the Cp ring [25-36]. However, the tethered donor usually does not act as a hemilabile ligand as it remains coordinated during the catalytic process. Many cyclopentadienyl (Cp) ligands where an additional neutral donor function is covalently bonded have been reported [37-39]. Some of those donors bind strongly, others weakly, to a particular transition metal ion. Examples, which are related to the work described here, are Cp ligands with olefinic [40-52] or with a nitrile side arm [53,54].
We have recently described how external modifiers combined with Cp-chromium polymerization catalysts influence the chain-termination process and hence the molecular weight of the produced polyethylene [55]. This paper describes our results with covalently linked modifiers and their influence on the ethylene polymerization behavior. There is neither experimental nor theoretical evidence for a substantial beta-H elimination or beta-H transfer in such catalyst systems so that chain termination is dominated by chain-transfer reactions to the aluminum based co-catalysts [56,57]. Consequently, any component, which modulates the interaction of Al–alkyls with the catalyst center, can influence the molecular weight of the resulting polyethylene. The concept of the present investigation is sketched in Scheme 1. The pre-catalysts feature covalently bonded neutral donor functions (D) which do not interact with the coordinatively saturated chromium centers. The coordination ability of the chosen donors D range from very weak (organofluorine) to medium (olefinic or aryl, respectively) and strong (nitrile). Activation with methylaluminoxane (MAO) leads to the formation of monomethyl complexes and the active species is a cationic alkylchromium complex with one remaining „vacant“ coordination site. This site can bind one of the following donors: ethylene, internal labile donor D, alkylaluminium compound, etc. If the internal donor binds, the cationic chromium center is stabilized but ethylene can displace the donor and insert into the chromium alkyl bond leading to polyethylene. In the presence of aluminum alkyls like trimethylaluminum chain termination may occur by addition of AlMe3 to the cationic chromium alkyl species. A simultaneous coordination of the internal donor D and alkylaluminum is not possible or at least very unlikely so that interaction of the hemilabile donor D can suppress chain transfer. Another possibility is that the donor D directly interacts with AlMe3, which also reduces the chain-transfer rate. Both types of interaction lead to an increase in molecular weight of the polyethylene.
![[1860-5397-12-131-i1]](/bjoc/content/inline/1860-5397-12-131-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Influencing catalyst stability, olefin coordination and chain-transfer reactions by hemilabile donor functions.
Scheme 1: Influencing catalyst stability, olefin coordination and chain-transfer reactions by hemilabile dono...
We have synthesized a number of ligands as presented in Scheme 2 and evaluated the interaction of the donor units with the Lewis acidic chromium center by DFT methods. The synthesized complexes where then tested in ethylene polymerization in order to evaluate the influence of the hemilabile donor in terms of catalyst activity and molecular weight of the polymer.
![[1860-5397-12-131-i2]](/bjoc/content/inline/1860-5397-12-131-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of ligands L1–L8 and chromium complexes 1–8.
Scheme 2: Synthesis of ligands L1–L8 and chromium complexes 1–8.
Results and Discussion
It is well known that early transition metal single-site polymerization catalysts can interact with organofluorine groups [15,17,20,22]. In FI-type catalysts the interaction of fluorine substituents with the catalyst center leads to a suppression of chain termination so that living olefin polymerization is possible [15]. The experimental verification of such an interaction was demonstrated by NMR of an MAO activated complex [17]. Consequently, we envisaged the synthesis of ligands where an organofluorine substituent is connected by a side arm with suitable length. In addition to that we choose side arms with olefinic groups, a benzyl unit and a stronger nitrile donor group, respectively. The synthesis of the new ligand derivatives and the corresponding Cr complexes follows known procedures (Scheme 2) [29,58]. The key step for the introduction of the hemilabile donor function is the electrophilic attack of a chlorosilane derivative at quinolyl-functionalized cyclopentadienides (CpQ). We used the trimethyl CpQ derivative as this leads to a single acidic proton in the ligands L1–L8. By this procedure we could introduce side arms with fluorine (L2–L4) or olefinic donor groups (L5, L6) as well as benzyl (L7) or nitrile (L8) moieties. Deprotonation with potassium hydride and subsequent reaction with chromium trichloride leads to the chromium complexes as green-blue solids in yields ranging from 21% (3) to 81% (6).
The ligand L3 as well as the pre-catalysts 4–8 were studied by single crystal X-ray analysis. A selection of molecular structures is presented in Figure 1 and details of the structure determination are presented in Table S1 (see Supporting Information File 1). Due to the rigid and predefined geometry of the CpQ ligand, the coordination environment around the chromium centers is very similar in all cases and in line with previously published structures of such complexes [29,59,60].
![[1860-5397-12-131-1]](/bjoc/content/figures/1860-5397-12-131-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Solid-state molecular structures of selected complexes 4, 7 and 8 (left, middle and right, respectively). Probability level: 50%. Hydrogen atoms and carbon labels are omitted for clarity.
Figure 1: Solid-state molecular structures of selected complexes 4, 7 and 8 (left, middle and right, respecti...
As the metal center in the pre-catalysts 1–8 is coordinatively saturated, the additional side arm functionality cannot interact with the metal. However, after activation with a co-catalyst, the complexes become cationic with a vacant coordination site so that the side arms could interact with the metal centers. Crystals of an activated complex could not be obtained. Only very few examples are known where X-ray diffraction data could be obtained from the active form of chromium polymerization catalysts [61-63]. However, DFT calculation is well suitable for studying such interactions. By such methods it is not only possible to estimate the binding energy but also to compare it with that of ethylene or interaction with solvent molecules like toluene.
All DFT calculations were performed with the B3LYP functional and the 6-311g* basis set. This theoretical level has shown to reproduce well paramagnetic NMR shifts in such compounds [58,64,65]. As we compare only relative energies of the complexes, the errors in the absolute energy values will compensate considerably. As a model for the activated catalysts we calculated the cationic monomethyl complexes 1a+–8a+. The complexes with ligands with a suitable geometry for intramolecular coordination indeed showed minima structures (as shown by the absence of imaginary frequencies) where the functionalized side-arms interact with the metal center (complexes 3a+–8a+, see Figure 2, for the analogous complexes 1a+ and 2a+, respectively, no reasonable minima structures were obtained). We did not consider the conformers where the growing chain is on the “other” site (i.e., oriented in the direction of the Si substituent). From earlier theoretical work on related chromium complexes we know that the chain can move easily from one site to the other so that the hemilabile donor may interact easily [57].
![[1860-5397-12-131-2]](/bjoc/content/figures/1860-5397-12-131-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) Calculated structures of cationic chromium complexes. Chromium–donor distances [Å]: 3a+ [2.21], 4a+ [2.24], 5a+ [2.53/2.51], 6a+ [2.75/2.46], 7a+ [2.58/3.17], 8a+ [2.06]. b) Displacement of hemilabile donor by ethylene under formation of the ethylene complex 4b+. (Energies for the formation of the analogous ethylene complexes 3b+ [−1.3], 5b+ [−4.5], 6b+ [+2.9], 7b+ [−2.9], 8b+ [+15.2].)
Figure 2: a) Calculated structures of cationic chromium complexes. Chromium–donor distances [Å]: 3a+ [2.21], ...
The Cr–F distances in 3a+ and 4a+ are with 2.24 Å and 2.21 Å, respectively, in the range of non-covalent Cr–F interactions as exemplified by a cationic chromium complex with BF4− anion (2.37 Å) [66] and a typical covalent Cr–F bond (~1.95 Å) [67].
According to calculations the distances of the two carbon atoms of the coordinating unit to the Cr centers in 5a+, 6a+ and 7a+ are approximately 2.5 Å (shortest of the two Cr–C distances). No X-ray data are available for olefin complexes of chromium in oxidation state +3 whereas solid state molecular structures of Cr0 and Cr1+ complexes have been reported with C–Cr distances of 2.1 Å–2.2 Å (Cr1+) [68,69] and 2.3 Å–2.4 Å (Cr0) [70,71], respectively.
As an alternative to the intramolecular coordination of the hemilabile donor to the vacant coordination site at the chromium center, a dimerization of the cationic methylchromium fragment could occur and examples of such dicationic dimers have been reported [72-75]. We were able to identify local minimum structures of dimers of compounds 3a+–8a+ but the calculated energies lie considerably higher compared to monomeric forms. Consequently, the saturation by the weak hemilabile donor is energetically preferred over dimerization. Another possible interaction is the coordination of Al–alkyls to the chromium centers. This has been addressed in detail in our previous work, where we could show that the energies of such adducts are similar compared to chromium complexes with olefin coordination [55,57]. For complex 8a+, however, the situation is different: the interaction energy of the nitrile group with the chromium center leads to an energy gain which is 15.2 kcal mol−1 higher compared to the energy of the corresponding ethylene complex. Consequently, the ethylene can hardly displace the nitrile. However, addition of Al–alkyls leads to a strong interaction of the Al center with the nitrile group so that ethylene can coordinate. Related to this behavior is a report of an acetonitrile-stabilized chromium complex which upon activation with MAO leads to a highly active catalyst and even the addition of up to 4 equivalents of acetonitrile to the catalyst solution did not lead to lower catalyst activities [76].
Polymerization results
The ethylene polymerization behavior of all new chromium complexes has been evaluated and compared with the performance of the known derivative 1 as well as with zirconocene dichloride (Cp2ZrCl2). The results are summarized in Table 1 and the exact procedure is described in the experimental part (Supporting Information File 1). The pre-catalysts were activated with PMAO, which is a non-hydrolytically prepared variant of MAO from the company Akzo Nobel (also called PMAO-IP for “polymeric MAO-improved properties”) [77]. All polymerizations were conducted at atmospheric ethylene pressure. The catalytic activities are very high for all derivatives ranging from 1400 to 3900 g polyethylene per mmol catalyst per hour. The lowest activities are obtained with the derivatives with fluorine substituents (complexes 2–4, entries 2–4 in Table 1) whereas all other complexes show considerably higher activities in the range from 2800–3900 g (PE) mmol−1 (cat) h−1. Polymerization under only 1 bar of ethylene pressure may lead to artifacts coming from limitations of ethylene transport into the solution and this may lead to biased turnover numbers. More interesting in terms of the concept of this work is the molecular weight of the polymers. The derivative 1 leads to a molecular weight of 530 000 g mol−1. Introduction of the 3,5-bis(trifluoromethyl)phenyl group lowers the molecular weight drastically to 90 000 g mol−1. In this derivative the electron-withdrawing CF3 groups cannot coordinate to the cationic Cr center in the active catalyst form. Apparently the electron withdrawing C6H3(CF3)2 substituent leads to lower molecular weight. However, when the fluoro substituents are able to coordinate (complexes 3 and 4, respectively) the polymer molecular weight is much higher compared to the results obtained with 2 and slightly higher compared to 1. When the side-arm functionalities possess better donor properties the molecular weight increases considerably up to the UHMW-PE range (entries 5–8, Table 1). With pre-catalysts 5 or 6 it is also possible that the side arm (vinyl or allyl side arm, respectively) is incorporated into the polymer but we cannot verify this by our experimental data.
Table 1: Results of the ethylene-polymerization tests with complexes 1–8 and Cp2ZrCl2 as catalyst precursors.
| entrya | catalyst | Ncat [μmol] | activity [g·mmol−1·h−1] |
Mwb
[103 g·mol−1] |
PE [g] | polym.- time [min] | [Mw/Mn] | degr. of cryst.c [%] | Tmc [°C] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 4.42 | 3240 | 530 | 2.87 | 12 | 3.1 | 65 | 135.5 |
| 2 | 2 | 6.37 | 1590 | 90 | 3.37 | 20 | 3.2 | –d | 132.5 |
| 3 | 3 | 6.88 | 1450 | 660 | 2.32 | 14 | 2.2 | 62 | 133.0 |
| 4 | 4 | 7.82 | 1910 | 610 | 4.48 | 18 | 3.5 | 66 | 135.5 |
| 5 | 5 | 4.30 | 3130 | 900 | 3.37 | 15 | 3.5 | 59 | 135.5 |
| 6 | 6 | 7.68 | 2780 | 1 070 | 4.99 | 14 | 4.0 | 56 | 132.5 |
| 7 | 7 | 3.76 | 3940 | 1 140 | 2.96 | 12 | 3.0 | 54 | 133.5 |
| 8 | 8 | 4.14 | 3560 | 1 450 | 2.46 | 10 | 4.9 | 64 | 133.0 |
| 9 | Cp2ZrCl2 | 10.30 | 2330 | 600 | 4.78 | 12 | 2.4 | 57 | 132.5 |
aStandard conditions: co-catalyst: PMAO (7% solution in toluene), Al:Cr = 1000:1, room temperature, 150 mL of toluene, atmospheric pressure, all reactions were performed with identical flasks and stirring bars. bGPC-measurements. cDSC measurements, for details see Supporting Information File 1. dThe determined crystallinity was unexpectedly high which could be due to artifacts. Therefore, this value is not tabulated.
Figure 3 shows the effect on molecular weight of the produced polyethylene when comparing the known pre-catalyst derivative 1 with the new derivative 8. As mentioned in the Introduction, the dominating chain termination pathway in olefin polymerization with Cp-chromium catalysts is chain transfer to aluminum alkyls and our results clearly show that this process can be suppressed efficiently by using the donor functions in the side arms of the silyl substituents. It has already been shown that external modifiers are also able to suppress chain termination. However, much higher amounts of such modifiers are necessary in order to give considerable effects [55,78].
![[1860-5397-12-131-3]](/bjoc/content/figures/1860-5397-12-131-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Comparison of GPC traces of polyethylene produced by 1/PMAO and 8/PMAO respectively (entries 1 and 8 in Table 1).
Figure 3: Comparison of GPC traces of polyethylene produced by 1/PMAO and 8/PMAO respectively (entries 1 and ...
Conclusion
We have shown that weak donor groups, which are covalently bound to the metal complex can efficiently modulate chain termination processes in chromium-catalyzed ethylene polymerization catalysis. These hemilabile donors are able to protect coordinatively unsaturated metal centers against coordination of aluminum alkyls and hence reduce chain transfer to aluminum. On the other hand, the donors are weak enough in order to be displaced by ethylene monomers so that the insertion polymerization can proceed with high turnover numbers. With this concept, it is possible to tune the catalyst behavior in terms of the molecular weight they produce without lowering their very high catalytic activity.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 319.1 KB | Download |
References
-
Jeffrey, J. C.; Rauchfuss, T. B. Inorg. Chem. 1979, 18, 2658–2666. doi:10.1021/ic50200a004
Return to citation in text: [1] -
Slone, C. S.; Weinberger, D. A.; Mirkin, C. A. The Transition Metal Coordination Chemistry of Hemilabile Ligands. Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: New York, 1999; Vol. 48, pp 233–350.
Return to citation in text: [1] -
Lindner, E.; Speidel, R.; Fawzi, R.; Hiller, W. Chem. Ber. 1990, 123, 2255–2260. doi:10.1002/cber.19901231205
Return to citation in text: [1] -
Bader, A.; Lindner, E. Coord. Chem. Rev. 1991, 108, 27–110. doi:10.1016/0010-8545(91)80013-4
Return to citation in text: [1] -
Okuda, J. Comments Inorg. Chem. 1994, 16, 185–205. doi:10.1080/02603599408035859
Return to citation in text: [1] -
Braunstein, P.; Naud, F. Angew. Chem., Int. Ed. 2001, 40, 680–699. doi:10.1002/1521-3773(20010216)40:4<680::AID-ANIE6800>3.0.CO;2-0
Return to citation in text: [1] -
Weng, Z.; Teo, S.; Hor, T. S. A. Acc. Chem. Res. 2007, 40, 676–684. doi:10.1021/ar600003h
Return to citation in text: [1] -
Wang, C.; Ma, Z.; Sun, X.-L.; Gao, Y.; Guo, Y.-H.; Tang, Y.; Shi, L.-P. Organometallics 2006, 25, 3259–3266. doi:10.1021/om060062j
Return to citation in text: [1] -
Tshuva, E. Y.; Groysman, S.; Goldberg, I.; Kol, M.; Goldschmidt, Z. Organometallics 2002, 21, 662–670. doi:10.1021/om010493w
Return to citation in text: [1] -
Qian, Y.; Huang, J.; Bala, M. D.; Lian, B.; Zhang, H.; Zhang, H. Chem. Rev. 2003, 103, 2633–2690. doi:10.1021/cr020002x
Return to citation in text: [1] -
Flores, J. C.; Chien, J. C. W.; Rausch, M. D. Organometallics 1994, 13, 4140–4142. doi:10.1021/om00023a006
Return to citation in text: [1] -
Müller, C.; Lilge, D.; Kristen, M. O.; Jutzi, P. Angew. Chem., Int. Ed. 2000, 39, 789–792. doi:10.1002/(SICI)1521-3773(20000218)39:4<789::AID-ANIE789>3.0.CO;2-F
Return to citation in text: [1] -
Deckers, P. J. W.; Hessen, B.; Teuben, J. H. Angew. Chem., Int. Ed. 2001, 40, 2516–2519. doi:10.1002/1521-3773(20010702)40:13<2516::AID-ANIE2516>3.0.CO;2-V
Return to citation in text: [1] -
Chan, M. C. W.; Kui, S. C. F.; Cole, J. M.; McIntyre, G. J.; Matsui, S.; Zhu, N.; Tam, K.-H. Chem. – Eur. J. 2006, 12, 2607–2619. doi:10.1002/chem.200501054
Return to citation in text: [1] -
Mitani, M.; Mohri, J.-i.; Yoshida, Y.; Saito, J.; Ishii, S.; Tsuru, K.; Matsui, S.; Furuyama, R.; Nakano, T.; Tanaka, H.; Kojoh, S.-i.; Matsugi, T.; Kashiwa, N.; Fujita, T. J. Am. Chem. Soc. 2002, 124, 3327–3336. doi:10.1021/ja0117581
Return to citation in text: [1] [2] [3] -
Chan, M. C. W. Chem. – Asian J. 2008, 3, 18–27. doi:10.1002/asia.200700226
Return to citation in text: [1] -
Bryliakov, K. P.; Talsi, E. P.; Möller, H. M.; Baier, M. C.; Mecking, S. Organometallics 2010, 29, 4428–4430. doi:10.1021/om100729y
Return to citation in text: [1] [2] [3] -
Ruwwe, J.; Erker, G.; Fröhlich, R. Angew. Chem., Int. Ed. Engl. 1996, 35, 80–82. doi:10.1002/anie.199600801
Return to citation in text: [1] -
Sun, Y.; Spence, R. E. v. H.; Piers, W. E.; Parvez, M.; Yap, G. P. A. J. Am. Chem. Soc. 1997, 119, 5132–5143. doi:10.1021/ja970140h
Return to citation in text: [1] -
Karl, J.; Erker, G.; Fröhlich, R. J. Am. Chem. Soc. 1997, 119, 11165–11173. doi:10.1021/ja971720h
Return to citation in text: [1] [2] -
Horton, A. D.; Orpen, A. G. Organometallics 1991, 10, 3910–3918. doi:10.1021/om00057a024
Return to citation in text: [1] -
Siedle, A. R.; Newmark, R. A.; Lamanna, W. M.; Huffman, J. C. Organometallics 1993, 12, 1491–1492. doi:10.1021/om00029a002
Return to citation in text: [1] [2] -
Yang, X.; Stern, C. L.; Marks, T. J. J. Am. Chem. Soc. 1994, 116, 10015–10031. doi:10.1021/ja00101a022
Return to citation in text: [1] -
Chen, E. Y.-X.; Marks, T. J. Chem. Rev. 2000, 100, 1391–1434. doi:10.1021/cr980462j
Return to citation in text: [1] -
Thomas, B. J.; Noh, S. K.; Schulte, G. K.; Sendlinger, S. C.; Theopold, K. H. J. Am. Chem. Soc. 1991, 113, 893–902. doi:10.1021/ja00003a024
Return to citation in text: [1] -
Liang, Y.; Yap, G. P. A.; Rheingold, A. L.; Theopold, K. H. Organometallics 1996, 15, 5284–5286. doi:10.1021/om960774h
Return to citation in text: [1] -
White, P. A.; Calabrese, J.; Theopold, K. H. Organometallics 1996, 15, 5473–5475. doi:10.1021/om960697q
Return to citation in text: [1] -
Döhring, A.; Göhre, J.; Jolly, P. W.; Kryger, B.; Rust, J.; Verhovnik, G. P. J. Organometallics 2000, 19, 388–402. doi:10.1021/om990643r
Return to citation in text: [1] -
Enders, M.; Fernández, P.; Ludwig, G.; Pritzkow, H. Organometallics 2001, 20, 5005–5007. doi:10.1021/om010753+
Return to citation in text: [1] [2] [3] -
Ikeda, H.; Monoi, T.; Ogata, K.; Yasuda, H. Macromol. Chem. Phys. 2001, 202, 1806–1811. doi:10.1002/1521-3935(20010601)202:9<1806::AID-MACP1806>3.0.CO;2-A
Return to citation in text: [1] -
Mani, G.; Gabbaï, F. P. Angew. Chem. 2004, 116, 2313–2316. doi:10.1002/ange.200353040
Return to citation in text: [1] -
Zhang, H.; Ma, J.; Qian, Y.; Huang, J. Organometallics 2004, 23, 5681–5688. doi:10.1021/om049731o
Return to citation in text: [1] -
Randoll, S.; Jones, P. G.; Tamm, M. Organometallics 2008, 27, 3232–3239. doi:10.1021/om800145z
Return to citation in text: [1] -
Zhang, L.; Gao, W.; Tao, X.; Wu, Q.; Mu, Y.; Ye, L. Organometallics 2011, 30, 433–440. doi:10.1021/om1005935
Return to citation in text: [1] -
Sieb, D.; Baker, R. W.; Wadepohl, H.; Enders, M. Organometallics 2012, 31, 7368–7374. doi:10.1021/om300582j
Return to citation in text: [1] -
Romano, D.; Ronca, S.; Rastogi, S. Macromol. Rapid Commun. 2015, 36, 327–331. doi:10.1002/marc.201400514
Return to citation in text: [1] -
Siemeling, U. Chem. Rev. 2000, 100, 1495–1526. doi:10.1021/cr990287m
Return to citation in text: [1] -
Butenschön, H. Chem. Rev. 2000, 100, 1527–1564. doi:10.1021/cr940265u
Return to citation in text: [1] -
Müller, C.; Vos, D.; Jutzi, P. J. Organomet. Chem. 2000, 600, 127–143. doi:10.1016/S0022-328X(00)00060-7
Return to citation in text: [1] -
Buzinkai, J. F.; Schrock, R. R. Organometallics 1987, 6, 1447–1452. doi:10.1021/om00150a014
Return to citation in text: [1] -
Okuda, J.; Zimmermann, K. H. J. Organomet. Chem. 1988, 344, C1–C4. doi:10.1016/0022-328X(88)80219-5
Return to citation in text: [1] -
Kohl, F. X.; Dickbreder, R.; Jutzi, P.; Müller, G.; Huber, B. Chem. Ber. 1989, 122, 871–878. doi:10.1002/cber.19891220513
Return to citation in text: [1] -
Miguel-Garcia, J. A.; Maitlis, P. M. J. Chem. Soc., Chem. Commun. 1990, 1472–1473. doi:10.1039/c39900001472
Return to citation in text: [1] -
Lehmkuhl, H.; Näser, J.; Mehler, G.; Keil, T.; Danowski, F.; Benn, R.; Mynott, R.; Schroth, G.; Gabor, B.; Krüger, C.; Betz, P. Chem. Ber. 1991, 124, 441–452. doi:10.1002/cber.19911240306
Return to citation in text: [1] -
Ogasa, M.; Mallin, D. T.; Macomber, D. W.; Rausch, M. D.; Rogers, R. D.; Rollins, A. N. J. Organomet. Chem. 1991, 405, 41–52. doi:10.1016/0022-328X(91)83154-V
Return to citation in text: [1] -
Erker, G.; Aul, R. Chem. Ber. 1991, 124, 1301–1310. doi:10.1002/cber.19911240550
Return to citation in text: [1] -
Zimmermann, K. H.; Pilato, R. S.; Horvath, I. T.; Okuda, J. Organometallics 1992, 11, 3935–3937. doi:10.1021/om00060a004
Return to citation in text: [1] -
Okuda, J.; Zimmermann, K. H.; Herdtweck, E. Angew. Chem., Int. Ed. Engl. 1991, 30, 430–431. doi:10.1002/anie.199104301
Return to citation in text: [1] -
Okuda, J.; Zimmermann, K. H. Chem. Ber. 1992, 125, 637–641. doi:10.1002/cber.19921250315
Return to citation in text: [1] -
Alt, H. G.; Jung, S. H.; Thewalt, U. J. Organomet. Chem. 1993, 456, 89–95. doi:10.1016/0022-328X(93)83322-M
Return to citation in text: [1] -
Spence, R. E. v. H.; Piers, W. E. Organometallics 1995, 14, 4617–4624. doi:10.1021/om00010a028
Return to citation in text: [1] -
Galakhov, M. V. Chem. Commun. 1998, 17–32. doi:10.1039/a704886f
Return to citation in text: [1] -
Leong, W. L. J.; Garland, M. V.; Goh, L. Y.; Leong, W. K. Inorg. Chim. Acta 2009, 362, 2089–2092. doi:10.1016/j.ica.2008.09.026
Return to citation in text: [1] -
Pinkas, J.; Gyepes, R.; Kubišta, J.; Horáček, M.; Lamač, M. J. Organomet. Chem. 2011, 696, 2364–2372. doi:10.1016/j.jorganchem.2011.02.032
Return to citation in text: [1] -
Mark, S.; Kurek, A.; Mülhaupt, R.; Xu, R.; Klatt, G.; Köppel, H.; Enders, M. Angew. Chem., Int. Ed. 2010, 49, 8751–8754. doi:10.1002/anie.201003918
Return to citation in text: [1] [2] [3] -
Döhring, A.; Jensen, V. R.; Jolly, P. W.; Thiel, W.; Weber, J. C. Organometallics 2001, 20, 2234–2245. doi:10.1021/om010146m
Return to citation in text: [1] -
Xu, R.; Klatt, G.; Enders, M.; Köppel, H. J. Phys. Chem. A 2012, 116, 1077–1085. doi:10.1021/jp209451p
Return to citation in text: [1] [2] [3] -
Fernández, P.; Pritzkow, H.; Carbó, J. J.; Hofmann, P.; Enders, M. Organometallics 2007, 26, 4402–4412. doi:10.1021/om070173y
Return to citation in text: [1] [2] -
Enders, M.; Fernández, P.; Mihan, S.; Pritzkow, H. J. Organomet. Chem. 2003, 687, 125–130. doi:10.1016/j.jorganchem.2003.07.017
Return to citation in text: [1] -
Enders, M.; Kohl, G.; Pritzkow, H. Organometallics 2004, 23, 3832–3839. doi:10.1021/om030694v
Return to citation in text: [1] -
Bazan, G. C.; Rogers, J. S.; Fang, C. C. Organometallics 2001, 20, 2059–2064. doi:10.1021/om001048o
Return to citation in text: [1] -
MacAdams, L. A.; Buffone, G. P.; Incarvito, C. D.; Rheingold, A. L.; Theopold, K. H. J. Am. Chem. Soc. 2005, 127, 1082–1083. doi:10.1021/ja043877x
Return to citation in text: [1] -
Theopold, K. H. Eur. J. Inorg. Chem. 1998, 15–24. doi:10.1002/(SICI)1099-0682(199801)1998:1<15::AID-EJIC15>3.0.CO;2-M
Return to citation in text: [1] -
Liimatainen, H.; Pennanen, T. O.; Vaara, J. Can. J. Chem. 2009, 87, 954–964. doi:10.1139/V09-045
Return to citation in text: [1] -
Martin, B.; Autschbach, J. Phys. Chem. Chem. Phys. 2016. doi:10.1039/c5cp07667f
Return to citation in text: [1] -
Clérac, R.; Cotton, F. A.; Daniels, L. M.; Dunbar, K. R.; Murillo, C. A.; Pascual, I. Inorg. Chem. 2000, 39, 748–751. doi:10.1021/ic990793u
Return to citation in text: [1] -
Thomas, B. J.; Mitchell, J. F.; Theopold, K. H.; Leafy, J. A. J. Organomet. Chem. 1988, 348, 333–342. doi:10.1016/0022-328X(88)80414-5
Return to citation in text: [1] -
Emrich, R.; Heinemann, O.; Jolly, P. W.; Krüger, C.; Verhovnik, G. P. J. Organometallics 1997, 16, 1511–1513. doi:10.1021/om961044c
Return to citation in text: [1] -
Monillas, W. H.; Yap, G. P. A.; MacAdams, L. A.; Theopold, K. H. J. Am. Chem. Soc. 2007, 129, 8090–8091. doi:10.1021/ja0725549
Return to citation in text: [1] -
Wink, D. J.; Wang, N. F.; Creagan, B. T. Organometallics 1989, 8, 561–562. doi:10.1021/om00104a047
Return to citation in text: [1] -
Fischer, H.; Hofmann, J. Chem. Ber. 1991, 124, 981–988. doi:10.1002/cber.19911240503
Return to citation in text: [1] -
Noh, S. K.; Sendlinger, S. C.; Janiak, C.; Theopold, K. H. J. Am. Chem. Soc. 1989, 111, 9127–9129. doi:10.1021/ja00207a034
Return to citation in text: [1] -
Richeson, D. S.; Mitchell, J. F.; Theopold, K. H. Organometallics 1989, 8, 2570–2577. doi:10.1021/om00113a009
Return to citation in text: [1] -
Messere, R.; Spirlet, M.-R.; Jan, D.; Demonceau, A.; Noels, A. F. Eur. J. Inorg. Chem. 2000, 1151–1153. doi:10.1002/(SICI)1099-0682(200006)2000:6<1151::AID-EJIC1151>3.3.CO;2-R
Return to citation in text: [1] -
Nicoara, C. Neue Cyclopentadienyl ~ N-Donor – Liganden für Chrom-basierte Single-Site-Katalysatoren zur Olefinpolymerisation. Ph.D. Thesis, Universiät Heidelberg, Heidelberg, Germany, 2006.
Return to citation in text: [1] -
Kirillov, E.; Roisnel, T.; Razavi, A.; Carpentier, J.-F. Organometallics 2009, 28, 2401–2409. doi:10.1021/om801196d
Return to citation in text: [1] -
Smith, G. M.; Palmaka, S. W.; Rogers, J. S.; Malpass, D. B. Polyalkylaluminoxane compositions formed by non-hydrolytic means. U.S. Patent US5.831.109, Nov 3, 1998.
Return to citation in text: [1] -
Busico, V.; Cipullo, R.; Cutillo, F.; Friederichs, N.; Ronca, S.; Wang, B. J. Am. Chem. Soc. 2003, 125, 12402–12403. doi:10.1021/ja0372412
Return to citation in text: [1]
| 77. | Smith, G. M.; Palmaka, S. W.; Rogers, J. S.; Malpass, D. B. Polyalkylaluminoxane compositions formed by non-hydrolytic means. U.S. Patent US5.831.109, Nov 3, 1998. |
| 55. | Mark, S.; Kurek, A.; Mülhaupt, R.; Xu, R.; Klatt, G.; Köppel, H.; Enders, M. Angew. Chem., Int. Ed. 2010, 49, 8751–8754. doi:10.1002/anie.201003918 |
| 78. | Busico, V.; Cipullo, R.; Cutillo, F.; Friederichs, N.; Ronca, S.; Wang, B. J. Am. Chem. Soc. 2003, 125, 12402–12403. doi:10.1021/ja0372412 |
| 1. | Jeffrey, J. C.; Rauchfuss, T. B. Inorg. Chem. 1979, 18, 2658–2666. doi:10.1021/ic50200a004 |
| 14. | Chan, M. C. W.; Kui, S. C. F.; Cole, J. M.; McIntyre, G. J.; Matsui, S.; Zhu, N.; Tam, K.-H. Chem. – Eur. J. 2006, 12, 2607–2619. doi:10.1002/chem.200501054 |
| 15. | Mitani, M.; Mohri, J.-i.; Yoshida, Y.; Saito, J.; Ishii, S.; Tsuru, K.; Matsui, S.; Furuyama, R.; Nakano, T.; Tanaka, H.; Kojoh, S.-i.; Matsugi, T.; Kashiwa, N.; Fujita, T. J. Am. Chem. Soc. 2002, 124, 3327–3336. doi:10.1021/ja0117581 |
| 16. | Chan, M. C. W. Chem. – Asian J. 2008, 3, 18–27. doi:10.1002/asia.200700226 |
| 17. | Bryliakov, K. P.; Talsi, E. P.; Möller, H. M.; Baier, M. C.; Mecking, S. Organometallics 2010, 29, 4428–4430. doi:10.1021/om100729y |
| 17. | Bryliakov, K. P.; Talsi, E. P.; Möller, H. M.; Baier, M. C.; Mecking, S. Organometallics 2010, 29, 4428–4430. doi:10.1021/om100729y |
| 13. | Deckers, P. J. W.; Hessen, B.; Teuben, J. H. Angew. Chem., Int. Ed. 2001, 40, 2516–2519. doi:10.1002/1521-3773(20010702)40:13<2516::AID-ANIE2516>3.0.CO;2-V |
| 29. | Enders, M.; Fernández, P.; Ludwig, G.; Pritzkow, H. Organometallics 2001, 20, 5005–5007. doi:10.1021/om010753+ |
| 58. | Fernández, P.; Pritzkow, H.; Carbó, J. J.; Hofmann, P.; Enders, M. Organometallics 2007, 26, 4402–4412. doi:10.1021/om070173y |
| 8. | Wang, C.; Ma, Z.; Sun, X.-L.; Gao, Y.; Guo, Y.-H.; Tang, Y.; Shi, L.-P. Organometallics 2006, 25, 3259–3266. doi:10.1021/om060062j |
| 9. | Tshuva, E. Y.; Groysman, S.; Goldberg, I.; Kol, M.; Goldschmidt, Z. Organometallics 2002, 21, 662–670. doi:10.1021/om010493w |
| 10. | Qian, Y.; Huang, J.; Bala, M. D.; Lian, B.; Zhang, H.; Zhang, H. Chem. Rev. 2003, 103, 2633–2690. doi:10.1021/cr020002x |
| 11. | Flores, J. C.; Chien, J. C. W.; Rausch, M. D. Organometallics 1994, 13, 4140–4142. doi:10.1021/om00023a006 |
| 12. | Müller, C.; Lilge, D.; Kristen, M. O.; Jutzi, P. Angew. Chem., Int. Ed. 2000, 39, 789–792. doi:10.1002/(SICI)1521-3773(20000218)39:4<789::AID-ANIE789>3.0.CO;2-F |
| 15. | Mitani, M.; Mohri, J.-i.; Yoshida, Y.; Saito, J.; Ishii, S.; Tsuru, K.; Matsui, S.; Furuyama, R.; Nakano, T.; Tanaka, H.; Kojoh, S.-i.; Matsugi, T.; Kashiwa, N.; Fujita, T. J. Am. Chem. Soc. 2002, 124, 3327–3336. doi:10.1021/ja0117581 |
| 17. | Bryliakov, K. P.; Talsi, E. P.; Möller, H. M.; Baier, M. C.; Mecking, S. Organometallics 2010, 29, 4428–4430. doi:10.1021/om100729y |
| 20. | Karl, J.; Erker, G.; Fröhlich, R. J. Am. Chem. Soc. 1997, 119, 11165–11173. doi:10.1021/ja971720h |
| 22. | Siedle, A. R.; Newmark, R. A.; Lamanna, W. M.; Huffman, J. C. Organometallics 1993, 12, 1491–1492. doi:10.1021/om00029a002 |
| 2. | Slone, C. S.; Weinberger, D. A.; Mirkin, C. A. The Transition Metal Coordination Chemistry of Hemilabile Ligands. Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: New York, 1999; Vol. 48, pp 233–350. |
| 3. | Lindner, E.; Speidel, R.; Fawzi, R.; Hiller, W. Chem. Ber. 1990, 123, 2255–2260. doi:10.1002/cber.19901231205 |
| 4. | Bader, A.; Lindner, E. Coord. Chem. Rev. 1991, 108, 27–110. doi:10.1016/0010-8545(91)80013-4 |
| 5. | Okuda, J. Comments Inorg. Chem. 1994, 16, 185–205. doi:10.1080/02603599408035859 |
| 6. | Braunstein, P.; Naud, F. Angew. Chem., Int. Ed. 2001, 40, 680–699. doi:10.1002/1521-3773(20010216)40:4<680::AID-ANIE6800>3.0.CO;2-0 |
| 7. | Weng, Z.; Teo, S.; Hor, T. S. A. Acc. Chem. Res. 2007, 40, 676–684. doi:10.1021/ar600003h |
| 15. | Mitani, M.; Mohri, J.-i.; Yoshida, Y.; Saito, J.; Ishii, S.; Tsuru, K.; Matsui, S.; Furuyama, R.; Nakano, T.; Tanaka, H.; Kojoh, S.-i.; Matsugi, T.; Kashiwa, N.; Fujita, T. J. Am. Chem. Soc. 2002, 124, 3327–3336. doi:10.1021/ja0117581 |
| 40. | Buzinkai, J. F.; Schrock, R. R. Organometallics 1987, 6, 1447–1452. doi:10.1021/om00150a014 |
| 41. | Okuda, J.; Zimmermann, K. H. J. Organomet. Chem. 1988, 344, C1–C4. doi:10.1016/0022-328X(88)80219-5 |
| 42. | Kohl, F. X.; Dickbreder, R.; Jutzi, P.; Müller, G.; Huber, B. Chem. Ber. 1989, 122, 871–878. doi:10.1002/cber.19891220513 |
| 43. | Miguel-Garcia, J. A.; Maitlis, P. M. J. Chem. Soc., Chem. Commun. 1990, 1472–1473. doi:10.1039/c39900001472 |
| 44. | Lehmkuhl, H.; Näser, J.; Mehler, G.; Keil, T.; Danowski, F.; Benn, R.; Mynott, R.; Schroth, G.; Gabor, B.; Krüger, C.; Betz, P. Chem. Ber. 1991, 124, 441–452. doi:10.1002/cber.19911240306 |
| 45. | Ogasa, M.; Mallin, D. T.; Macomber, D. W.; Rausch, M. D.; Rogers, R. D.; Rollins, A. N. J. Organomet. Chem. 1991, 405, 41–52. doi:10.1016/0022-328X(91)83154-V |
| 46. | Erker, G.; Aul, R. Chem. Ber. 1991, 124, 1301–1310. doi:10.1002/cber.19911240550 |
| 47. | Zimmermann, K. H.; Pilato, R. S.; Horvath, I. T.; Okuda, J. Organometallics 1992, 11, 3935–3937. doi:10.1021/om00060a004 |
| 48. | Okuda, J.; Zimmermann, K. H.; Herdtweck, E. Angew. Chem., Int. Ed. Engl. 1991, 30, 430–431. doi:10.1002/anie.199104301 |
| 49. | Okuda, J.; Zimmermann, K. H. Chem. Ber. 1992, 125, 637–641. doi:10.1002/cber.19921250315 |
| 50. | Alt, H. G.; Jung, S. H.; Thewalt, U. J. Organomet. Chem. 1993, 456, 89–95. doi:10.1016/0022-328X(93)83322-M |
| 51. | Spence, R. E. v. H.; Piers, W. E. Organometallics 1995, 14, 4617–4624. doi:10.1021/om00010a028 |
| 52. | Galakhov, M. V. Chem. Commun. 1998, 17–32. doi:10.1039/a704886f |
| 55. | Mark, S.; Kurek, A.; Mülhaupt, R.; Xu, R.; Klatt, G.; Köppel, H.; Enders, M. Angew. Chem., Int. Ed. 2010, 49, 8751–8754. doi:10.1002/anie.201003918 |
| 37. | Siemeling, U. Chem. Rev. 2000, 100, 1495–1526. doi:10.1021/cr990287m |
| 38. | Butenschön, H. Chem. Rev. 2000, 100, 1527–1564. doi:10.1021/cr940265u |
| 39. | Müller, C.; Vos, D.; Jutzi, P. J. Organomet. Chem. 2000, 600, 127–143. doi:10.1016/S0022-328X(00)00060-7 |
| 56. | Döhring, A.; Jensen, V. R.; Jolly, P. W.; Thiel, W.; Weber, J. C. Organometallics 2001, 20, 2234–2245. doi:10.1021/om010146m |
| 57. | Xu, R.; Klatt, G.; Enders, M.; Köppel, H. J. Phys. Chem. A 2012, 116, 1077–1085. doi:10.1021/jp209451p |
| 25. | Thomas, B. J.; Noh, S. K.; Schulte, G. K.; Sendlinger, S. C.; Theopold, K. H. J. Am. Chem. Soc. 1991, 113, 893–902. doi:10.1021/ja00003a024 |
| 26. | Liang, Y.; Yap, G. P. A.; Rheingold, A. L.; Theopold, K. H. Organometallics 1996, 15, 5284–5286. doi:10.1021/om960774h |
| 27. | White, P. A.; Calabrese, J.; Theopold, K. H. Organometallics 1996, 15, 5473–5475. doi:10.1021/om960697q |
| 28. | Döhring, A.; Göhre, J.; Jolly, P. W.; Kryger, B.; Rust, J.; Verhovnik, G. P. J. Organometallics 2000, 19, 388–402. doi:10.1021/om990643r |
| 29. | Enders, M.; Fernández, P.; Ludwig, G.; Pritzkow, H. Organometallics 2001, 20, 5005–5007. doi:10.1021/om010753+ |
| 30. | Ikeda, H.; Monoi, T.; Ogata, K.; Yasuda, H. Macromol. Chem. Phys. 2001, 202, 1806–1811. doi:10.1002/1521-3935(20010601)202:9<1806::AID-MACP1806>3.0.CO;2-A |
| 31. | Mani, G.; Gabbaï, F. P. Angew. Chem. 2004, 116, 2313–2316. doi:10.1002/ange.200353040 |
| 32. | Zhang, H.; Ma, J.; Qian, Y.; Huang, J. Organometallics 2004, 23, 5681–5688. doi:10.1021/om049731o |
| 33. | Randoll, S.; Jones, P. G.; Tamm, M. Organometallics 2008, 27, 3232–3239. doi:10.1021/om800145z |
| 34. | Zhang, L.; Gao, W.; Tao, X.; Wu, Q.; Mu, Y.; Ye, L. Organometallics 2011, 30, 433–440. doi:10.1021/om1005935 |
| 35. | Sieb, D.; Baker, R. W.; Wadepohl, H.; Enders, M. Organometallics 2012, 31, 7368–7374. doi:10.1021/om300582j |
| 36. | Romano, D.; Ronca, S.; Rastogi, S. Macromol. Rapid Commun. 2015, 36, 327–331. doi:10.1002/marc.201400514 |
| 18. | Ruwwe, J.; Erker, G.; Fröhlich, R. Angew. Chem., Int. Ed. Engl. 1996, 35, 80–82. doi:10.1002/anie.199600801 |
| 19. | Sun, Y.; Spence, R. E. v. H.; Piers, W. E.; Parvez, M.; Yap, G. P. A. J. Am. Chem. Soc. 1997, 119, 5132–5143. doi:10.1021/ja970140h |
| 20. | Karl, J.; Erker, G.; Fröhlich, R. J. Am. Chem. Soc. 1997, 119, 11165–11173. doi:10.1021/ja971720h |
| 21. | Horton, A. D.; Orpen, A. G. Organometallics 1991, 10, 3910–3918. doi:10.1021/om00057a024 |
| 22. | Siedle, A. R.; Newmark, R. A.; Lamanna, W. M.; Huffman, J. C. Organometallics 1993, 12, 1491–1492. doi:10.1021/om00029a002 |
| 23. | Yang, X.; Stern, C. L.; Marks, T. J. J. Am. Chem. Soc. 1994, 116, 10015–10031. doi:10.1021/ja00101a022 |
| 24. | Chen, E. Y.-X.; Marks, T. J. Chem. Rev. 2000, 100, 1391–1434. doi:10.1021/cr980462j |
| 53. | Leong, W. L. J.; Garland, M. V.; Goh, L. Y.; Leong, W. K. Inorg. Chim. Acta 2009, 362, 2089–2092. doi:10.1016/j.ica.2008.09.026 |
| 54. | Pinkas, J.; Gyepes, R.; Kubišta, J.; Horáček, M.; Lamač, M. J. Organomet. Chem. 2011, 696, 2364–2372. doi:10.1016/j.jorganchem.2011.02.032 |
| 58. | Fernández, P.; Pritzkow, H.; Carbó, J. J.; Hofmann, P.; Enders, M. Organometallics 2007, 26, 4402–4412. doi:10.1021/om070173y |
| 64. | Liimatainen, H.; Pennanen, T. O.; Vaara, J. Can. J. Chem. 2009, 87, 954–964. doi:10.1139/V09-045 |
| 65. | Martin, B.; Autschbach, J. Phys. Chem. Chem. Phys. 2016. doi:10.1039/c5cp07667f |
| 29. | Enders, M.; Fernández, P.; Ludwig, G.; Pritzkow, H. Organometallics 2001, 20, 5005–5007. doi:10.1021/om010753+ |
| 59. | Enders, M.; Fernández, P.; Mihan, S.; Pritzkow, H. J. Organomet. Chem. 2003, 687, 125–130. doi:10.1016/j.jorganchem.2003.07.017 |
| 60. | Enders, M.; Kohl, G.; Pritzkow, H. Organometallics 2004, 23, 3832–3839. doi:10.1021/om030694v |
| 61. | Bazan, G. C.; Rogers, J. S.; Fang, C. C. Organometallics 2001, 20, 2059–2064. doi:10.1021/om001048o |
| 62. | MacAdams, L. A.; Buffone, G. P.; Incarvito, C. D.; Rheingold, A. L.; Theopold, K. H. J. Am. Chem. Soc. 2005, 127, 1082–1083. doi:10.1021/ja043877x |
| 63. | Theopold, K. H. Eur. J. Inorg. Chem. 1998, 15–24. doi:10.1002/(SICI)1099-0682(199801)1998:1<15::AID-EJIC15>3.0.CO;2-M |
| 55. | Mark, S.; Kurek, A.; Mülhaupt, R.; Xu, R.; Klatt, G.; Köppel, H.; Enders, M. Angew. Chem., Int. Ed. 2010, 49, 8751–8754. doi:10.1002/anie.201003918 |
| 57. | Xu, R.; Klatt, G.; Enders, M.; Köppel, H. J. Phys. Chem. A 2012, 116, 1077–1085. doi:10.1021/jp209451p |
| 76. | Kirillov, E.; Roisnel, T.; Razavi, A.; Carpentier, J.-F. Organometallics 2009, 28, 2401–2409. doi:10.1021/om801196d |
| 70. | Wink, D. J.; Wang, N. F.; Creagan, B. T. Organometallics 1989, 8, 561–562. doi:10.1021/om00104a047 |
| 71. | Fischer, H.; Hofmann, J. Chem. Ber. 1991, 124, 981–988. doi:10.1002/cber.19911240503 |
| 72. | Noh, S. K.; Sendlinger, S. C.; Janiak, C.; Theopold, K. H. J. Am. Chem. Soc. 1989, 111, 9127–9129. doi:10.1021/ja00207a034 |
| 73. | Richeson, D. S.; Mitchell, J. F.; Theopold, K. H. Organometallics 1989, 8, 2570–2577. doi:10.1021/om00113a009 |
| 74. | Messere, R.; Spirlet, M.-R.; Jan, D.; Demonceau, A.; Noels, A. F. Eur. J. Inorg. Chem. 2000, 1151–1153. doi:10.1002/(SICI)1099-0682(200006)2000:6<1151::AID-EJIC1151>3.3.CO;2-R |
| 75. | Nicoara, C. Neue Cyclopentadienyl ~ N-Donor – Liganden für Chrom-basierte Single-Site-Katalysatoren zur Olefinpolymerisation. Ph.D. Thesis, Universiät Heidelberg, Heidelberg, Germany, 2006. |
| 67. | Thomas, B. J.; Mitchell, J. F.; Theopold, K. H.; Leafy, J. A. J. Organomet. Chem. 1988, 348, 333–342. doi:10.1016/0022-328X(88)80414-5 |
| 68. | Emrich, R.; Heinemann, O.; Jolly, P. W.; Krüger, C.; Verhovnik, G. P. J. Organometallics 1997, 16, 1511–1513. doi:10.1021/om961044c |
| 69. | Monillas, W. H.; Yap, G. P. A.; MacAdams, L. A.; Theopold, K. H. J. Am. Chem. Soc. 2007, 129, 8090–8091. doi:10.1021/ja0725549 |
| 57. | Xu, R.; Klatt, G.; Enders, M.; Köppel, H. J. Phys. Chem. A 2012, 116, 1077–1085. doi:10.1021/jp209451p |
| 66. | Clérac, R.; Cotton, F. A.; Daniels, L. M.; Dunbar, K. R.; Murillo, C. A.; Pascual, I. Inorg. Chem. 2000, 39, 748–751. doi:10.1021/ic990793u |
© 2016 Mark et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)