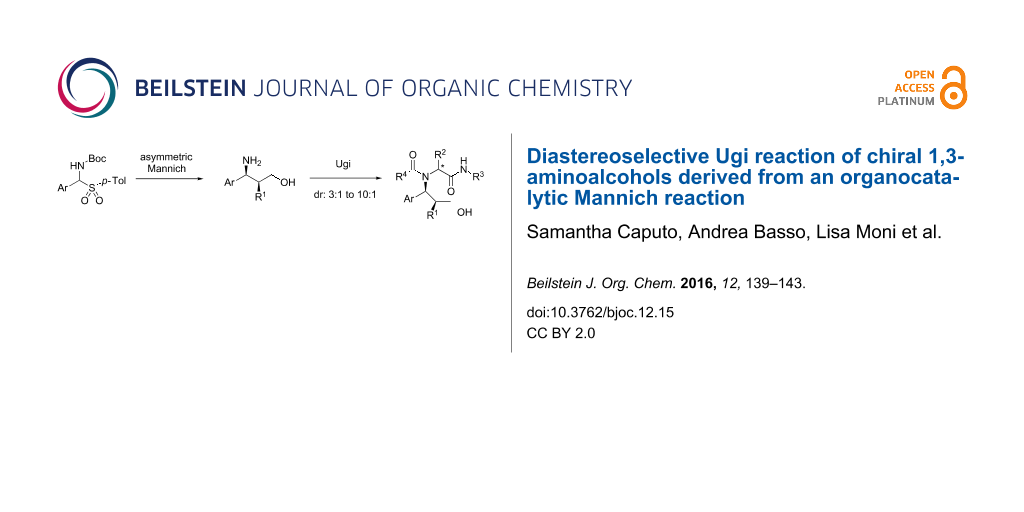
Abstract
Enantiomerically pure β-aminoalcohols, produced through an organocatalytic Mannich reaction, were subjected to an Ugi multicomponent reaction under classical or Lewis acid-promoted conditions with diastereoselectivities ranging from moderate to good. This approach represents a step-economical path to enantiomerically pure, polyfunctionalized peptidomimetics endowed with three stereogenic centers, allowing the introduction of five diversity inputs.

Graphical Abstract
Findings
Isocyanide-based multicomponent reactions [1-3], such as the Ugi reaction, were demonstrated to be very useful in the rapid assembly of complex drug candidates [4], introducing three to four diversity inputs. Furthermore, a nearly limitless variety of heterocycles can be accessed through post-condensation transformations [5-7], adding only one to two steps to the synthetic sequence. However, the main drawback of the Ugi reaction is the poor stereochemical control that is typically achieved [8,9], which hampers its utilization in the diversity-oriented or target-oriented synthesis of complex chiral peptidomimetics. No efficient asymmetric catalytic classic Ugi reaction has been reported to date (whereas some success was obtained on simpler variants) [10-12]. On the other hand, diastereoselective reactions using at least one chiral component are troublesome. Chiral isocyanides and chiral carboxylic acids invariably afford nearly 1:1 mixtures. α-Chiral aldehydes have a high tendency to racemize/epimerize [13,14] and additionally, no report of valuable diastereocontrol by them has appeared so far. Successful examples of diastereoselective Ugi reactions have been reported only with chiral amines [15-19] or with chiral cyclic imines (Ugi–Joullié reaction) [20-23], although in the latter case, racemization/epimerization can again be an issue in special cases [24]. However, the use of amines as chiral auxiliaries has been seldom exploited in peptidomimetic synthesis [16,25,26] because the need to remove the auxiliary reduces the number of diversity inputs and increases the number of synthetic steps.
From the point of view of atom- and step-economy, the use of chiral amines that are retained in the final products will be more valuable [27]. In this case they are not "chiral auxiliaries" and are not removed after the multicomponent reaction, and they contribute to the diversity of the final products. However, the usefulness of this approach relies on an efficient and diversity-oriented preparation of the required amines in high enantiomeric excess.
Chiral aminoalcohols can be ideal substrates for diastereoselective Ugi reactions: the additional hydroxy group can both help in modulating diastereoselectivity and be employed for post-condensation transformations in order to add further fragments or to form heterocyclic structures. We have previously developed some syntheses of heterocycles through Ugi reactions with 1,2-aminoalcohols followed by nucleophilic substitutions [28], whereas chiral 1,2-aminoalcohols have been proved by Nenajdenko and co-workers to be able to induce good levels of diastereoselectivity in the Ugi reaction [17].
Our attention was drawn by 1,3-aminoalcohols of general formula 5 (Scheme 1), which can be obtained by List's organocatalytic Mannich-type reaction of aldehydes with N-Boc imines 2 and catalytic L-proline [29,30], followed by reduction of 3 and cleavage of the Boc group.
![[1860-5397-12-15-i1]](/bjoc/content/inline/1860-5397-12-15-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
This short and straightforward synthesis allows the introduction of 2 diversity inputs (R1 and Ar), whereas stereochemical diversity can also be explored using D-proline or different, anti-selective organocatalysts.
We prepared two known carbamoyl sulfones 1 [30,31] and transformed them without isolation of intermediates into a series of five Boc-protected β-aminoalcohols 4a–e (Scheme 2). Using caesium carbonate, carbamoyl sulfones were converted into the corresponding N-Boc-protected imines 2 that were immediately submitted to List's organocatalytic Mannich reaction [29,30]. The resulting aldehydes 3 were not isolated (also in view of their known stereochemical lability) but directly reduced to alcohols 4 [32,33]. Purification was carried out through chromatography and, in some cases, by additional crystallization, affording these key intermediates in high ee and de (syn relative configuration, see Supporting Information File 1).
![[1860-5397-12-15-i2]](/bjoc/content/inline/1860-5397-12-15-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Boc-protected aminoalcohols used as inputs in a diastereoselective Ugi reaction.
Scheme 2: Boc-protected aminoalcohols used as inputs in a diastereoselective Ugi reaction.
The tert-butyl urethane was then deblocked with trifluoroacetic acid. Neutralization and extraction afforded crude aminoalcohols 5a–e, that were directly employed in the Ugi reaction. We first optimized this step using isobutyraldehyde, 5-chloro-2-thiophenecarboxylic acid and cyclohexyl isocyanide, to give the two diastereomers of compound 6a (Table 1).
Table 1: Optimization of the synthesis of 6a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-15-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Temp. | Time (h) | Solvent (M) | Lewis acid (equiv) | Yielda | drb |
|---|---|---|---|---|---|---|
| 1 | 25 °C | 12 | MeOH (0.4) | none | 65% | 72:28 |
| 2 | 25 °C | 48 | MeOH (0.1) | none | 72% | 72:28 |
| 3 | 25 °C | 12 | CF3CH2OH (0.1) | none | 65% | 77:23 |
| 4 | 0 °C | 12 | CF3CH2OH (0.1) | none | 61% | 83:17 |
| 5 | −38 °C | 48 | THF (0.1) | ZnCl2·Et2O (1.0) | 55% | 89:11 |
| 6 | −38 °C | 48 | THF (0.1) | ZnCl2 (1.0) | 63% | 73:27 |
| 7 | −38 °C | 48 | THF (0.1) | ZnCl2 (1.5) | 60% | 80:20 |
| 8 | −38 °C | 48 | THF (0.1) | ZnBr2 (1.0) | 82% | 91:9 |
| 9 | −38 °C | 48 | THF (0.1) | ZnI2 (1.0) | 71% | 88:12 |
| 10 | −38 °C | 48 | THF (0.1) | CuBr2 (1.0) | no react. | – |
| 11 | −38 °C | 48 | THF (0.1) | Cu(OTf)2 (1.0) | no react. | – |
| 12 | −38 °C | 48 | THF (0.1) | MgCl2 (1.0) | no react. | – |
| 13 | −38 °C | 48 | THF (0.1) | MgBr2·Et2O (1.0) | no react. | – |
| 14 | −38 °C | 48 | THF (0.1) | Yb(OTf)3 (0.2) | no react. | – |
aOverall yield from aminoalcohol. bRelative configuration not yet determined.
When the reaction was carried out under the classical conditions (using methanol as the solvent), only a moderate diastereoselectivity was achieved (Table 1, entry 1), which could be increased by changing the solvent to trifluoroethanol, especially effective at 0 °C. Considering the recent work by Nenajdenko et al. [17], we explored the usage of Lewis acids in an aprotic solvent in order to further improve the diastereoselectivity. We had anticipated that the binding of the Lewis acid to the free alcohol, followed by intramolecular activation of the aldehyde, would establish a cyclic transition state, thereby enabling better stereocontrol. It is indeed well-known that the Ugi reaction does not proceed in aprotic solvents such as THF at low temperature, and therefore the background, uncatalyzed reaction should not interfere. As shown in Table 1, the best results were achieved by using 1 equiv of zinc bromide (Table 1, entry 8), affording a 10:1 diastereomeric ratio and an excellent overall yield. Other zinc-based catalysts were less efficient, whereas most of the other tested Lewis acids failed to promote the reaction at all. The use of Lewis acids in methanol or trifluoroethanol afforded lower yields with no improvement of diastereoselection. It is worth noting that a 10:1 diastereoselectivity is considered excellent for isocyanide-based multicomponent reactions, due to the very low steric biases of isocyanides.
We then moved on to establish the scope of the method, varying the Boc-protected aminoalcohol, the carboxylic acid and the isocyanide (see Table 2). For a comparison, we performed all Ugi reactions either under Lewis acid-promoted conditions, or under the classical Ugi conditions (MeOH, rt). The stereochemical results were found to vary remarkably from case to case. While in some instances (products 6b–d) the activation with ZnBr2 brought about an increase of diastereoselectivity, in other combinations of substrates, the outcome was similar (products 6h and 6j) or even better using the "classical" conditions (products 6e, 6f, 6i). However, in all cases, the two diastereomers could be easily separated and the ratio was typically, with few exceptions, around 3:1 to 5:1. As far as the isolated yields were concerned, the Lewis acid-promoted reaction is typically less efficient, especially with aromatic isocyanides or aldehydes (compounds 6e, 6f, 6g). The relative configuration of the major adduct has not yet been unambiguously determined. However, TLC, HPLC, polarimetric and NMR analogies suggest that the main diastereomer was always the same, with one notable exception: product 6f obtained in the absence of Lewis acid. In this case, it was necessary to carry out the reaction in THF/iPrOH because the isocyanide was poorly soluble in MeOH, and thus the unexpected diastereoselectivity inversion might be due to the different solvent and not to the structure of isocyanide.
Table 2: Scope of the synthesis of Ugi adducts 6.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-15-i4.svg?max-width=637&scale=1.0)
|
||||||||
| Prod. | Ar | R1 | R2 | R3 | R4 | Cond.a | Yieldb | dr |
|---|---|---|---|---|---|---|---|---|
| 6a | Ph | Me | iPr | cy-Hex | 5-Cl-2-thienyl | A | 82% | 91:9 |
| B | 72% | 72:28 | ||||||
| 6b | Ph | Me | iPr | n-C5H11 | 5-Cl-2-thienyl | A | 53% | 85:15 |
| B | 82% | 73:27 | ||||||
| 6c | 2-BnOC6H4 | Me | iPr | cyclohexyl | 5-Cl-2-thienyl | A | 31% | 82:18 |
| B | 40% | 64:36 | ||||||
| 6d | Ph | Me | iPr | cyclohexyl | Et | A | 55% | 79:21 |
| B | 82% | 74:26 | ||||||
| 6e | Ph | Me | iPr | 2,6-di-MeC6H3 | 5-Cl-2-thienyl | A | 55% | 65:35 |
| B | 95% | 86:14 | ||||||
| 6f | Ph | Me | iPr | 4-(BnOCO)-C6H4 | 5-Cl-2-thienyl | A | <5% | n.d. |
| C | 60% | 35:65 | ||||||
| 6g | Ph | Me | Ph | n-C5H11 | 5-Cl-2-thienyl | A | <20% | n.d. |
| B | 79% | 57:43 | ||||||
| 6h | Ph | iPr | iPr | cyclohexyl | 5-Cl-2-thienyl | A | 40% | 78:22 |
| B | 70% | 77:23 | ||||||
| 6i | 2-BnOC6H4 | iPr | iPr | cyclohexyl | 5-Cl-2-thienyl | A | 30% | 64:36 |
| B | 54% | 69:31 | ||||||
| 6j | 2-BnOC6H4 | Bn | iPr | t-Bu | CbzNH-CH2 | A | 48% | 80:20 |
| B | 77% | 81:19 | ||||||
aOverall yield from Boc aminoalcohols 4. bRelative configuration not yet determined. A: THF, −38 °C, 1 equiv of ZnBr2; B: MeOH, 25 °C; C: iPrOH/THF 2:1, 25 °C. All reactions carried out for 48 h at 0.1 M concentration of aminoalcohol with 1.00 equiv of aminoalcohol 5, 1.05 equiv of aldehyde, 1.2 equiv of carboxylic acid and isocyanide and 100 mg of powdered 3 Å molecular sieves per mmol of aminoalcohol.
The synthetic route from carbamoyl sulfones 1 to peptidomimetics 6 is quite short: intermediate purification was carried out only at the level of the Boc-protected aminoalcohols 4 and of the final products 6. Thus, this method offers an operationally simple route to enantiomerically pure complex structures like 6, introducing up to five diversity inputs and controlling three stereogenic centers (also thanks to the final chromatography).
Compounds 6 are endowed with several functionalities that can be exploited for post-Ugi cyclization steps or as a handle for attaching further fragments: the primary alcohol and the secondary amide (which are present in all products), a protected phenol (for compounds 6c, 6i, 6j), and a protected amine (6j). Studies towards this goal are in progress and will be reported in due course.
Supporting Information
| Supporting Information File 1: General remarks, experimental procedures and characterization data; 1H and 13C NMR spectra of new compounds 4 and 6 (major isomer only). | ||
| Format: PDF | Size: 4.6 MB | Download |
References
-
van der Heijden, G.; Ruijter, E.; Orru, R. V. A. Synlett 2013, 666–685. doi:10.1055/s-0032-1318222
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Riva, R. The Passerini Reaction. In Multicomponent Reactions; Müller, T. J. J., Ed.; Science of Synthesis, Vol. 1; Thieme: Stuttgart, Germany, 2013; pp 327–414.
Return to citation in text: [1] -
Wessjohann, L. A.; Kaluderovic, G. N.; Neves Filho, R. A. W.; Morejon, M. C.; Lemanski, G.; Ziegler, T. Further Components Carboxylic Acids and Amine (Ugi Reaction). In Multicomponent Reactions; Müller, T. J. J., Ed.; Science of Synthesis, Vol. 1; Thieme: Stuttgart, Germany, 2013; pp 415–495.
Return to citation in text: [1] -
Hulme, C.; Ayaz, M.; Martinez Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry: Applications in Small Molecule Drug Discovery. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W.; Hamley, P., Eds.; Wiley: Weinheim, Germany, 2015; pp 145–187. doi:10.1002/9781118771723.ch6
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Riva, R. Synthesis of Heterocycles Through Classical Ugi and Passerini Reactions Followed by Secondary Transformations Involving One or Two Additional Functional Groups. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A.; Ruitjer, E., Eds.; Topics in Heterocyclic Chemistry, Vol. 23; Springer: Berlin, Germany, 2010; pp 1–39. doi:10.1007/7081_2009_23
Return to citation in text: [1] -
Sharma, U. K.; Sharma, N.; Vachhani, D. D.; Van der Eycken, E. V. Chem. Soc. Rev. 2015, 44, 1836–1860. doi:10.1039/C4CS00253A
Return to citation in text: [1] -
Ayaz, M.; De Moliner, F.; Dietrich, J.; Hulme, C. Applications of Isocyanides in IMCRs for the Rapid Generation of Molecular Diversity. In Isocyanide Chemistry: Applications in Synthesis and Material Science; Nenajdenko, V. G., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp 335–384. doi:10.1002/9783527652532.ch10
Return to citation in text: [1] -
van Berkel, S. S.; Bögels, B. G. M.; Wijdeven, M. A.; Westermann, B.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2012, 3543–3559. doi:10.1002/ejoc.201200030
Return to citation in text: [1] -
de Graaff, C.; Ruijter, E.; Orru, R. V. A. Chem. Soc. Rev. 2012, 41, 3969–4009. doi:10.1039/c2cs15361k
Return to citation in text: [1] -
Zhao, W.; Huang, L.; Guan, Y.; Wulff, W. D. Angew. Chem., Int. Ed. 2014, 53, 3436–3441. doi:10.1002/anie.201310491
Return to citation in text: [1] -
Yue, T.; Wang, M.-X.; Wang, D.-X.; Masson, G.; Zhu, J. Angew. Chem., Int. Ed. 2009, 48, 6717–6721. doi:10.1002/anie.200902385
Return to citation in text: [1] -
Su, Y.; Bouma, M. J.; Alcaraz, L.; Stocks, M.; Furber, M.; Masson, G.; Zhu, J. Chem. – Eur. J. 2012, 18, 12624–12627. doi:10.1002/chem.201202174
Return to citation in text: [1] -
Kelly, G. L.; Lawrie, K. W. M.; Morgan, P.; Willis, C. L. Tetrahedron Lett. 2000, 41, 8001–8005. doi:10.1016/S0040-4039(00)01389-7
Return to citation in text: [1] -
Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. J. Org. Chem. 2015, 80, 3411–3428. doi:10.1021/jo502829j
Return to citation in text: [1] -
Ross, G. F.; Herdtweck, E.; Ugi, I. Tetrahedron 2002, 58, 6127–6133. doi:10.1016/S0040-4020(02)00484-2
Return to citation in text: [1] -
Hebach, C.; Kazmaier, U. Chem. Commun. 2003, 596–597. doi:10.1039/b210952b
Return to citation in text: [1] [2] -
Nenajdenko, V. G.; Gulevich, A. V.; Chernichenko, K. Yu.; Sokolova, N. V.; Balenkova, E. S. Mendeleev Commun. 2011, 21, 245–246. doi:10.1016/j.mencom.2011.09.004
Return to citation in text: [1] [2] [3] -
Basso, A.; Banfi, L.; Riva, R.; Guanti, G. J. Org. Chem. 2005, 70, 575–579. doi:10.1021/jo048389m
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Chiappe, C.; De Moliner, F.; Riva, R.; Sonaglia, L. Org. Biomol. Chem. 2012, 10, 3819–3829. doi:10.1039/c2ob25060h
Return to citation in text: [1] -
Banfi, L.; Bagno, A.; Basso, A.; De Santis, C.; Riva, R.; Rastrelli, F. Eur. J. Org. Chem. 2013, 2013, 5064–5075. doi:10.1002/ejoc.201300541
Return to citation in text: [1] -
Cerulli, V.; Banfi, L.; Basso, A.; Rocca, V.; Riva, R. Org. Biomol. Chem. 2012, 10, 1255–1274. doi:10.1039/c1ob06632c
Return to citation in text: [1] -
Zhu, D.; Xia, L.; Pan, L.; Li, S.; Chen, R.; Mou, Y.; Chen, X. J. Org. Chem. 2012, 77, 1386–1395. doi:10.1021/jo2021967
Return to citation in text: [1] -
van Rijssel, E. R.; Goumans, T. P. M.; Lodder, G.; Overkleeft, H. S.; van der Marel, G. A.; Codée, J. D. C. Org. Lett. 2013, 15, 3026–3029. doi:10.1021/ol4012053
Return to citation in text: [1] -
Moni, L.; Banfi, L.; Basso, A.; Galatini, A.; Spallarossa, M.; Riva, R. J. Org. Chem. 2014, 79, 339–351. doi:10.1021/jo402527w
Return to citation in text: [1] -
Gulevich, A. V.; Shevchenko, N. E.; Balenkova, E. S.; Röschenthaler, G.-V.; Nenajdenko, V. G. Tetrahedron 2008, 64, 11706–11712. doi:10.1016/j.tet.2008.10.004
Return to citation in text: [1] -
Lamberth, C.; Jeanguenat, A.; Cederbaum, F.; De Mesmaeker, A.; Zeller, M.; Kempf, H.-J.; Zeun, R. Bioorg. Med. Chem. 2008, 16, 1531–1545. doi:10.1016/j.bmc.2007.10.019
Return to citation in text: [1] -
Sandulenko, Y.; Krasavin, M. Chem. Heterocycl. Compd. 2012, 48, 606–612. doi:10.1007/s10593-012-1033-z
Return to citation in text: [1] -
Banfi, L.; Riva, R.; Basso, A. Synlett 2010, 23–41. doi:10.1055/s-0029-1218527
Return to citation in text: [1] -
Yang, J. W.; Stadler, M.; List, B. Angew. Chem., Int. Ed. 2007, 46, 609–611. doi:10.1002/anie.200603188
Return to citation in text: [1] [2] -
Yang, J. W.; Pan, S. C.; List, B. Org. Synth. 2009, 86, 11–17. doi:10.15227/orgsyn.086.0011
Return to citation in text: [1] [2] [3] -
Morana, F.; Basso, A.; Riva, R.; Rocca, V.; Banfi, L. Chem. – Eur. J. 2013, 19, 4563–4569. doi:10.1002/chem.201300023
Return to citation in text: [1] -
Joannesse, C.; Johnston, C. P.; Morrill, L. C.; Woods, P. A.; Kieffer, M.; Nigst, T. A.; Mayr, H.; Lebl, T.; Philp, D.; Bragg, R. A.; Smith, A. D. Chem. – Eur. J. 2012, 18, 2398–2408. doi:10.1002/chem.201102847
Return to citation in text: [1] -
Joannesse, C.; Johnston, C. P.; Concellón, C.; Simal, C.; Philp, D.; Smith, A. D. Angew. Chem., Int. Ed. 2009, 48, 8914–8918. doi:10.1002/anie.200904333
Return to citation in text: [1]
| 32. | Joannesse, C.; Johnston, C. P.; Morrill, L. C.; Woods, P. A.; Kieffer, M.; Nigst, T. A.; Mayr, H.; Lebl, T.; Philp, D.; Bragg, R. A.; Smith, A. D. Chem. – Eur. J. 2012, 18, 2398–2408. doi:10.1002/chem.201102847 |
| 33. | Joannesse, C.; Johnston, C. P.; Concellón, C.; Simal, C.; Philp, D.; Smith, A. D. Angew. Chem., Int. Ed. 2009, 48, 8914–8918. doi:10.1002/anie.200904333 |
| 17. | Nenajdenko, V. G.; Gulevich, A. V.; Chernichenko, K. Yu.; Sokolova, N. V.; Balenkova, E. S. Mendeleev Commun. 2011, 21, 245–246. doi:10.1016/j.mencom.2011.09.004 |
| 1. | van der Heijden, G.; Ruijter, E.; Orru, R. V. A. Synlett 2013, 666–685. doi:10.1055/s-0032-1318222 |
| 2. | Banfi, L.; Basso, A.; Riva, R. The Passerini Reaction. In Multicomponent Reactions; Müller, T. J. J., Ed.; Science of Synthesis, Vol. 1; Thieme: Stuttgart, Germany, 2013; pp 327–414. |
| 3. | Wessjohann, L. A.; Kaluderovic, G. N.; Neves Filho, R. A. W.; Morejon, M. C.; Lemanski, G.; Ziegler, T. Further Components Carboxylic Acids and Amine (Ugi Reaction). In Multicomponent Reactions; Müller, T. J. J., Ed.; Science of Synthesis, Vol. 1; Thieme: Stuttgart, Germany, 2013; pp 415–495. |
| 10. | Zhao, W.; Huang, L.; Guan, Y.; Wulff, W. D. Angew. Chem., Int. Ed. 2014, 53, 3436–3441. doi:10.1002/anie.201310491 |
| 11. | Yue, T.; Wang, M.-X.; Wang, D.-X.; Masson, G.; Zhu, J. Angew. Chem., Int. Ed. 2009, 48, 6717–6721. doi:10.1002/anie.200902385 |
| 12. | Su, Y.; Bouma, M. J.; Alcaraz, L.; Stocks, M.; Furber, M.; Masson, G.; Zhu, J. Chem. – Eur. J. 2012, 18, 12624–12627. doi:10.1002/chem.201202174 |
| 30. | Yang, J. W.; Pan, S. C.; List, B. Org. Synth. 2009, 86, 11–17. doi:10.15227/orgsyn.086.0011 |
| 31. | Morana, F.; Basso, A.; Riva, R.; Rocca, V.; Banfi, L. Chem. – Eur. J. 2013, 19, 4563–4569. doi:10.1002/chem.201300023 |
| 8. | van Berkel, S. S.; Bögels, B. G. M.; Wijdeven, M. A.; Westermann, B.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2012, 3543–3559. doi:10.1002/ejoc.201200030 |
| 9. | de Graaff, C.; Ruijter, E.; Orru, R. V. A. Chem. Soc. Rev. 2012, 41, 3969–4009. doi:10.1039/c2cs15361k |
| 29. | Yang, J. W.; Stadler, M.; List, B. Angew. Chem., Int. Ed. 2007, 46, 609–611. doi:10.1002/anie.200603188 |
| 30. | Yang, J. W.; Pan, S. C.; List, B. Org. Synth. 2009, 86, 11–17. doi:10.15227/orgsyn.086.0011 |
| 5. | Banfi, L.; Basso, A.; Riva, R. Synthesis of Heterocycles Through Classical Ugi and Passerini Reactions Followed by Secondary Transformations Involving One or Two Additional Functional Groups. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A.; Ruitjer, E., Eds.; Topics in Heterocyclic Chemistry, Vol. 23; Springer: Berlin, Germany, 2010; pp 1–39. doi:10.1007/7081_2009_23 |
| 6. | Sharma, U. K.; Sharma, N.; Vachhani, D. D.; Van der Eycken, E. V. Chem. Soc. Rev. 2015, 44, 1836–1860. doi:10.1039/C4CS00253A |
| 7. | Ayaz, M.; De Moliner, F.; Dietrich, J.; Hulme, C. Applications of Isocyanides in IMCRs for the Rapid Generation of Molecular Diversity. In Isocyanide Chemistry: Applications in Synthesis and Material Science; Nenajdenko, V. G., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp 335–384. doi:10.1002/9783527652532.ch10 |
| 17. | Nenajdenko, V. G.; Gulevich, A. V.; Chernichenko, K. Yu.; Sokolova, N. V.; Balenkova, E. S. Mendeleev Commun. 2011, 21, 245–246. doi:10.1016/j.mencom.2011.09.004 |
| 4. | Hulme, C.; Ayaz, M.; Martinez Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry: Applications in Small Molecule Drug Discovery. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W.; Hamley, P., Eds.; Wiley: Weinheim, Germany, 2015; pp 145–187. doi:10.1002/9781118771723.ch6 |
| 29. | Yang, J. W.; Stadler, M.; List, B. Angew. Chem., Int. Ed. 2007, 46, 609–611. doi:10.1002/anie.200603188 |
| 30. | Yang, J. W.; Pan, S. C.; List, B. Org. Synth. 2009, 86, 11–17. doi:10.15227/orgsyn.086.0011 |
| 24. | Moni, L.; Banfi, L.; Basso, A.; Galatini, A.; Spallarossa, M.; Riva, R. J. Org. Chem. 2014, 79, 339–351. doi:10.1021/jo402527w |
| 27. | Sandulenko, Y.; Krasavin, M. Chem. Heterocycl. Compd. 2012, 48, 606–612. doi:10.1007/s10593-012-1033-z |
| 20. | Banfi, L.; Bagno, A.; Basso, A.; De Santis, C.; Riva, R.; Rastrelli, F. Eur. J. Org. Chem. 2013, 2013, 5064–5075. doi:10.1002/ejoc.201300541 |
| 21. | Cerulli, V.; Banfi, L.; Basso, A.; Rocca, V.; Riva, R. Org. Biomol. Chem. 2012, 10, 1255–1274. doi:10.1039/c1ob06632c |
| 22. | Zhu, D.; Xia, L.; Pan, L.; Li, S.; Chen, R.; Mou, Y.; Chen, X. J. Org. Chem. 2012, 77, 1386–1395. doi:10.1021/jo2021967 |
| 23. | van Rijssel, E. R.; Goumans, T. P. M.; Lodder, G.; Overkleeft, H. S.; van der Marel, G. A.; Codée, J. D. C. Org. Lett. 2013, 15, 3026–3029. doi:10.1021/ol4012053 |
| 28. | Banfi, L.; Riva, R.; Basso, A. Synlett 2010, 23–41. doi:10.1055/s-0029-1218527 |
| 15. | Ross, G. F.; Herdtweck, E.; Ugi, I. Tetrahedron 2002, 58, 6127–6133. doi:10.1016/S0040-4020(02)00484-2 |
| 16. | Hebach, C.; Kazmaier, U. Chem. Commun. 2003, 596–597. doi:10.1039/b210952b |
| 17. | Nenajdenko, V. G.; Gulevich, A. V.; Chernichenko, K. Yu.; Sokolova, N. V.; Balenkova, E. S. Mendeleev Commun. 2011, 21, 245–246. doi:10.1016/j.mencom.2011.09.004 |
| 18. | Basso, A.; Banfi, L.; Riva, R.; Guanti, G. J. Org. Chem. 2005, 70, 575–579. doi:10.1021/jo048389m |
| 19. | Banfi, L.; Basso, A.; Chiappe, C.; De Moliner, F.; Riva, R.; Sonaglia, L. Org. Biomol. Chem. 2012, 10, 3819–3829. doi:10.1039/c2ob25060h |
| 13. | Kelly, G. L.; Lawrie, K. W. M.; Morgan, P.; Willis, C. L. Tetrahedron Lett. 2000, 41, 8001–8005. doi:10.1016/S0040-4039(00)01389-7 |
| 14. | Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. J. Org. Chem. 2015, 80, 3411–3428. doi:10.1021/jo502829j |
| 16. | Hebach, C.; Kazmaier, U. Chem. Commun. 2003, 596–597. doi:10.1039/b210952b |
| 25. | Gulevich, A. V.; Shevchenko, N. E.; Balenkova, E. S.; Röschenthaler, G.-V.; Nenajdenko, V. G. Tetrahedron 2008, 64, 11706–11712. doi:10.1016/j.tet.2008.10.004 |
| 26. | Lamberth, C.; Jeanguenat, A.; Cederbaum, F.; De Mesmaeker, A.; Zeller, M.; Kempf, H.-J.; Zeun, R. Bioorg. Med. Chem. 2008, 16, 1531–1545. doi:10.1016/j.bmc.2007.10.019 |
© 2016 Caputo et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)