Abstract
The synthesis of novel tetrahydroquinolines (THQ) and dihydroquinolines (DHQ) are reported using three practical, scalable synthetic approaches to access highly lipophilic analogues bearing a 6-iodo substituent, each with a different means of cyclisation. A versatile and stable quinolin-2-one intermediate was identified, which could be reduced to the corresponding THQ with borane reagents, or to the DHQ with diisobutylaluminium hydride via a novel elimination that is more favourable at higher temperatures. Coupling these strongly electron-donating scaffolds to electron-accepting moieties caused the resulting structures to exhibit strong fluorescence.

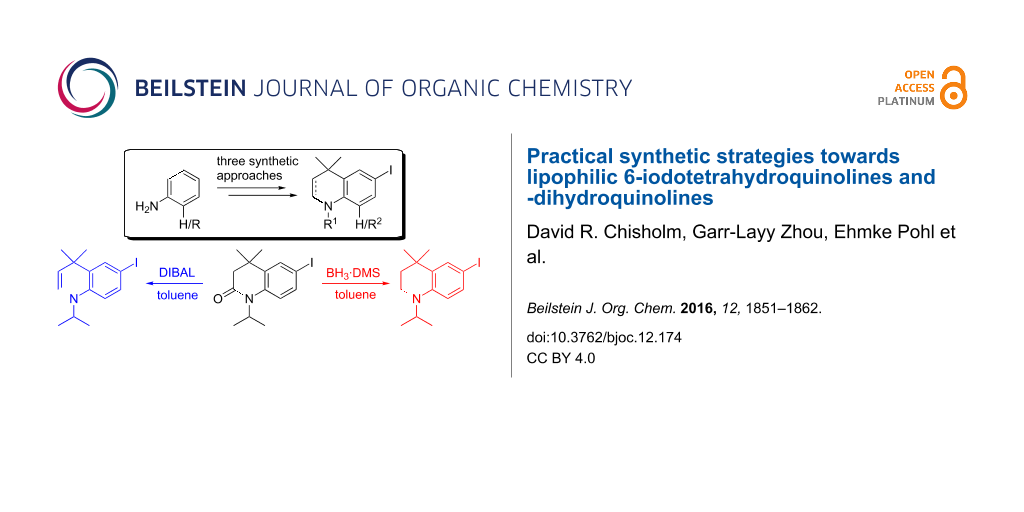
Graphical Abstract
Introduction
Tetrahydroquinolines (THQ) and dihydroquinolines (DHQ) are heterocyclic scaffolds that are ubiquitous in natural products, therapeutics, fluorophores and dyes [1]. Both are structures of great versatility, and their physical and chemical properties can be finely tuned using synthetic chemistry. Methods for their synthesis have been well studied, and range from the classic Skraup–Doebner–von Miller syntheses, to catalytic and asymmetric methods [2-4].
We required a straightforward synthesis of THQs and DHQs for the synthesis of a library of biocompatible fluorophores with the potential to be used in fluorescence microscopy applications. The desired intermediates were to be highly lipophilic, derivatisable and easy to synthesise on both small and multigram scales. A scaffold of general type 1 (Figure 1) was chosen as a synthetic target that could be N-alkylated with bulky alkyl groups for increased lipophilicity. An iodo substituent in the para-position relative to nitrogen (the 6-position) was required for further functionalisation by typical cross-coupling reactions.
![[1860-5397-12-174-1]](/bjoc/content/figures/1860-5397-12-174-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Tetrahydroquinoline (THQ) and dihydroquinoline (DHQ) scaffolds to be synthesised.
Figure 1: Tetrahydroquinoline (THQ) and dihydroquinoline (DHQ) scaffolds to be synthesised.
THQ scaffolds similar to 1 are known in the literature, and have been typically prepared by two approaches. The first involves the alkylation of the starting aniline with the inexpensive 3,3-dimethylallyl bromide/chloride, followed by cyclisation mediated by Lewis acids, or under acidic conditions [5,6]. A second, analogous approach involves an initial acylation with the commercially available 3,3-dimethylacryloyl chloride, followed by Friedel–Crafts cyclisations that also use Lewis acids [7]. The resultant quinolin-2-one is then reduced using strong hydride reducing agents such as LiAlH4 [8]. Similar THQs have also been prepared by a reductive Beckmann rearrangement of an oxime using diisobutylaluminium hydride (DIBAL) [9]. In contrast to this variety, the corresponding 1,4-DHQ of scaffold 1 has not been synthesised to our knowledge. Similar DHQs are typically prepared by methods such as ring-closing metathesis, or by controlled Birch-type reductions of the corresponding quinoline [10-12].
Very few instances of iodine-containing THQs or DHQs exist in the literature [13]. An initial investigation with the corresponding bromides indicated that these were unreactive towards many cross-coupling methodologies, presumably due to the highly electron-rich arene [14]. Reactions such as the Sonogashira coupling, for example, often required high catalyst loadings, phosphine additives and strong heating or microwave conditions in order to effect coupling with most acetylenes. Accordingly, it was anticipated that the aryl iodides would be more versatile due to their greater propensity to undergo oxidative additions.
Therefore, in this paper we report herein the development of facile, scalable and practical syntheses for 6-iodo-THQ and 6-iodo-DHQ scaffolds that can be employed in a variety of cross-coupling and derivatisation reactions. Three synthetic routes are described, each of which involves a different means of cyclisation to form the heterocyclic structure. We also describe a novel formation of the 1,4-DHQ of scaffold 1; a reaction that appears to be favoured both by the use of hydride reducing agents that produce stable tetrahedral intermediates and the high temperature collapse of these intermediates.
Results and Discussion
The aim of this investigation was to synthesise THQs and DHQs of the general scaffold 1 for the use in a variety of cross-coupling reactions, thus necessitating the presence of a halogen or pseudohalogen in the para-position relative to nitrogen. Most of the relevant literature focuses on the synthesis of the corresponding aryl bromides or aryl chlorides, but initial investigation with 6-bromo-THQs indicated that these were remarkably unreactive in cross-coupling reactions [14], and a reliable and scalable synthesis of the likely more reactive iodides was, therefore, sought.
We first considered the retrosynthetic analysis shown in Scheme 1, which requires cyclisation from the alkylated N-allylaniline 4. The use of polyphosphoric acid (PPA) in cyclisation reactions is well known in the literature, and has been used to form a large variety of ring structures [15]. Iodination of 3 could then be realised by utilising the rich iodination literature available [16,17].
![[1860-5397-12-174-i1]](/bjoc/content/inline/1860-5397-12-174-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed retrosynthesis scheme to access N-isopropyl-THQ 2.
Scheme 1: Proposed retrosynthesis scheme to access N-isopropyl-THQ 2.
Isopropylation of the inexpensive o-toluidine (5) can be conducted by simply adding NaOH and 2-iodopropane to a refluxing solution of the aniline (Scheme 2). While this straightforward procedure was well suited to larger scale, some bis-alkylation was difficult to avoid. However, the desired product 6 can be isolated using basic alumina chromatography. Reductive amination with acetone, mediated by AcOH and NaOAc was also pursued as an alternative strategy [18]. While being much easier to purify due to the elimination of the bis-alkylation product, the yield of this approach was much more variable at larger scales.
![[1860-5397-12-174-i2]](/bjoc/content/inline/1860-5397-12-174-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of THQ 3 by initial N-alkylations, followed by PPA-mediated cyclisation.
Scheme 2: Synthesis of THQ 3 by initial N-alkylations, followed by PPA-mediated cyclisation.
Further N-alkylation with 3,3-dimethylallyl bromide was conducted using K2CO3. The resulting alkylated compound 4 was easily isolated by chromatography in good yield, however, the reaction was found to be difficult to force to completion, presumably due to steric hindrance. Cyclisation of 4 to the THQ 3 was realised by heating a PPA mixture of 4 to 120 °C.
Iodination of 3 using a variety of reagents including I2/HgO [19], ICl/AcOH, N-iodosuccinimide/TFA [20], KI/KIO3/AcOH [21], and N-chlorosuccinimide/NaI/AcOH [22], all gave poor yields (0–30%) according to GC–MS analysis, and the iodinated product 2 was difficult to isolate by chromatography. In light of these results, bromination was conducted with the aim of synthesising the corresponding aryl iodide 2 by halogen exchange (Scheme 3).
![[1860-5397-12-174-i3]](/bjoc/content/inline/1860-5397-12-174-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Bromination of 3 and attempted halogen exchange of the intermediate 7.
Scheme 3: Bromination of 3 and attempted halogen exchange of the intermediate 7.
After careful optimisation of the reaction conditions, the bromide 7 was exclusively obtained by addition of 0.95 equivalents of bromine to a solution of 3 in chloroform maintained at −60 °C. However, halogen exchange under literature conditions was sluggish and even after 3 days only around 30% of the starting material had converted [23].
It was theorised that the low reactivity of 3 towards electrophilic aromatic iodination was caused by distortion of the THQ ring structure due to the need to minimise steric interactions between the N-iPr group and the neighbouring methyl group. This may result in poorer orbital overlap between the nitrogen lone pair and the aromatic π-system, thus reducing the sp2 character of the nitrogen, and therefore lowering the reactivity of the system towards electrophilic aromatic substitution. An alternative, analogous synthesis was accordingly devised, in which the unsubstituted THQ 10 was targeted, as outlined in Scheme 4 [24].
![[1860-5397-12-174-i4]](/bjoc/content/inline/1860-5397-12-174-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of THQ 10, by initial aza-Michael addition, followed by formation of the tertiary alcohol 9, which was then cyclised with H2SO4.
Scheme 4: Synthesis of THQ 10, by initial aza-Michael addition, followed by formation of the tertiary alcohol ...
Reports of aza-Michael additions occurring in water indicated that the desired 8 could be synthesised under mild conditions, and we therefore decided to adapt these published conditions to larger scale work [25,26]. Initial attempts involving a 1:1 molar mixture of o-toluidine (5) and methyl vinyl ketone (MVK) indicated that only around 60% of the starting aniline had converted, particularly at larger scales (>1 g). Two equivalents of MVK were required to effect full conversion, however, under these conditions the bis-adduct was formed in around 6–10% and this was difficult to remove by chromatography or distillation. Using one equivalent lowered the yield, but minimised bis-adduct formation, which allowed facile purification by short path distillation on larger scales. Compound 8 was functionalised to the tertiary alcohol 9 by a Grignard reaction with MeMgBr, which could be directly cyclised without purification to 10 by heating a DCM solution with a nominal amount of concentrated sulphuric acid. This procedure was much more straightforward when compared to the previous PPA reaction, and required a much less laborious work-up.
The increased efficiency of this reaction can be attributed to improved orbital overlap. Protonation of the hydroxy group of 9 under the reaction conditions likely leads to the corresponding tertiary carbocation under equilibrium. The nitrogen lone pair can then assist the electrophilic cyclisation reaction, augmented by improved orbital overlap with the aromatic π-system, which in the case of 9 would be improved over 4 due to the lack of a second alkyl substituent and diminished steric repulsion that likely causes rotation of the nitrogen lone pair out of conjugation with the aromatic ring.
While the increased availability of the nitrogen lone pair appears to assist cyclisation in this system, iodination of 10 was still relatively low yielding, particularly with acidic iodination methods (presumably due to protonation and resultant deactivation of the nitrogen). Slightly higher yields were obtained with pyridine–iodine [16], however, the conversion was also low and isolation of the iodinated product was often difficult.
Despite the relative ease, the overall yield of this second route was considered too low to be a reliable source of THQs of the general scaffold 1. However, we were encouraged by the simplicity and ease of the H2SO4-mediated cyclisation reaction, and a third synthetic route was therefore devised that would employ this methodology.
A common approach in the literature towards similar THQs involves an initial acylation of the starting aniline using 3,3-dimethylacryloyl chloride, followed by a high temperature cyclisation employing Lewis acids such as AlCl3 [7,27]. However, initial experiments indicated that under these cyclisation conditions, significant degradation and de-iodination occurred with iodinated intermediates. It was anticipated that the milder H2SO4 conditions could affect the cyclisation without these side reactions. To probe this, commercially available 4-iodo-2-methylaniline (11) was employed as the starting material in order to circumvent the low-yielding iodination step. This starting material is also readily synthesised from o-toluidine (5) using a pyridine–iodine iodination [16].
Initial acylation of 11 with 3,3-dimethylacryloyl chloride and pyridine provided 12 in good yield (Scheme 5). The acid-catalysed reaction with 12 was predicted to proceed via initial formation of the corresponding tertiary alcohol involving a Markovnikov addition, before cyclisation as with 10. Indeed, cyclisation product 13 (see Supporting Information File 1 for crystal structure) was produced cleanly in a 67% yield, which was easily separated chromatographically from the remaining starting material. However, the reaction occurred at a significantly slower rate than with 9; particularly on larger reaction scales where complete conversion of the starting material required long reaction times (>48 h). The significantly slower rate is likely due to the reduced availability of the nitrogen lone pair in 12. Favourably, however, no signs of de-iodination or degradation were observed under these cyclisation conditions. Cyclisation product 13 was readily reduced to the iodinated THQ 14 (see Supporting Information File 1 for crystal structure) using borane·dimethyl sulphide complex. A slight molar excess of the reducing agent causes de-iodination to give 10 in small quantities.
![[1860-5397-12-174-i5]](/bjoc/content/inline/1860-5397-12-174-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of THQ 14 by initial acylation, cyclisation with H2SO4 and reduction with borane·dimethyl sulphide complex.
Scheme 5: Synthesis of THQ 14 by initial acylation, cyclisation with H2SO4 and reduction with borane·dimethyl...
N-Alkylation with highly lipophilic alkyl groups such as an isopropyl was next considered (Scheme 6). The quinolin-2-one 13 was predicted to be a more amenable alkylation partner than the THQ 14 due to the likely lower pKa of the amide proton. To assess this, 13 was reacted with NaH and 2-iodopropane in DMF at 80 °C overnight. However, 13 displayed a marked lack of reactivity towards isopropylation, and only around 40% of the starting material 13 was converted. Interestingly, the major product from the reaction was the O-iPr imine 15b as indicated by a low field 1H chemical shift of the isopropyl proton (5.39 ppm), and later confirmed by X-ray crystallography (the crystal structure is shown in Supporting Information File 1). The N-iPr product 15a was isolated in only 3% yield. This, therefore, indicates that the electrophile reacts faster with the oxide anion of 13. Repeating the reaction with 15-crown-5 to limit the effect of the sodium cation did not appreciably effect the product distribution. It would, therefore, appear that steric hindrance from the neighbouring methyl group is in fact the main determinant of the product distribution.
![[1860-5397-12-174-i6]](/bjoc/content/inline/1860-5397-12-174-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Alkylation of 14 was also marked by a general lack of reactivity; the methylated 16 was isolated in only a 43% yield. Ethylation was possible, albeit in much lower yield and only negligible amounts (1–2%) of the iPr adduct were isolated. Alkylation using silver(I) oxide was also possible, but with similarly low yields.
The ortho-methyl group had originally been incorporated as protection to block the ortho-amino centre against oxidation [14], and to increase lipophilicity. However, because the methyl group appeared likely to be the cause for the lack of reactivity of 13 and 14 towards alkylation, a synthesis of the corresponding quinolin-2-one derived from 4-iodoaniline (17) was pursued in order to increase the ease of alkylation.
The initial acylation of 17 (commercially available, or easily synthesised using literature methods [16]) proceeded well to give 18, and this could easily be applied to larger scale (>25 g) synthesis. The acid-catalysed cyclisation again was much slower than for the synthesis of 10, and at large scale in particular; the reaction could take at least 48 hours for completion. An alternative large-scale protocol was, therefore, investigated. In contrast to the high temperature Lewis acid-mediated cyclisations reported in the literature [7], it was found that simply stirring a DCM solution of 18 with 1.5 equivalents of AlCl3 at room temperature for 2.5 hours gave the quinolin-2-one 19 in excellent yield after recrystallisation from EtOH (see Supporting Information File 1 for the crystal structure). While facile, the reaction was highly dependent both on the number of equivalents of AlCl3 used and the reaction time. Leaving the reaction mixture to stir with 1.5 equivalents of AlCl3 for longer periods (>3 h) resulted in minor de-iodination, whereas less than 2 hours gave incomplete cyclisation. Using 2 equivalents of AlCl3 causes rapid cyclisation after 1 hour, but the de-iodinated form was also present. Using 1.25 equivalents appeared to inhibit de-iodination completely, but the cyclisation reaction tended to plateau before full completion.
Without the ortho-methyl, preference for isopropylation switched to N-alkylation, providing 50% of 20a and 26% of the O-isopropylimine 20b; a mixture that could be separated by chromatography. Full conversion of the starting material was difficult to achieve and appeared to plateau at around 75%. However, the reaction was easily conducted and both products isolated on larger scale (30 g). Encouragingly, the iodine substituent was stable throughout the optimised synthesis of the quinolin-2-one intermediate (Scheme 7).
![[1860-5397-12-174-i7]](/bjoc/content/inline/1860-5397-12-174-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Facile route for the synthesis of 20a.
Scheme 7: Facile route for the synthesis of 20a.
Reduction to the THQ 21 was easily conducted with borane·dimethyl sulphide complex as described previously, in excellent yield (Scheme 8). In addition, upon trialling different conditions for the reduction, LiAlH4 was found to afford a small amount (ca. 5%) of 1,4-DHQ 22 in addition to 21. Ab initio simulation of 22 (Figure 2) indicated that the enamine functionality causes a flattening of the dihydroquinoline ring to give a more planar, quinoline-like structure. We were intrigued by the result of these stereoelectronic and conformational effects in terms of the chromophoric properties of the resulting systems and an improved synthesis of 22 was therefore pursued.
![[1860-5397-12-174-i8]](/bjoc/content/inline/1860-5397-12-174-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of THQ 21 and DHQ 22 using borane·dimethyl sulphide complex or DIBAL, respectively.
Scheme 8: Synthesis of THQ 21 and DHQ 22 using borane·dimethyl sulphide complex or DIBAL, respectively.
![[1860-5397-12-174-2]](/bjoc/content/figures/1860-5397-12-174-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Simulated structure of 22 indicates a flattened quinoline-like structure. Hartree–Fock calculations (3-21G*) were conducted using Spartan’10, and visualised using UCSF Chimera 1.11 [28,29].
Figure 2: Simulated structure of 22 indicates a flattened quinoline-like structure. Hartree–Fock calculations...
In light of its formation by LiAlH4, we anticipated that production of 22 would be favoured if the intermediate tetrahedral complex in the reduction reaction was more stable. An elimination reaction could then operate to give 22, either during aqueous work-up, or in situ collapse of the intermediate complex and subsequent reduction of the resulting iminium ion via elimination. As a Lewis acidic reducing agent, it was anticipated that DIBAL could be better suited to the reduction of the electron-rich amide of 20a, and DIBAL is also known to produce relatively stable tetrahedral intermediates [30]. Indeed, initial experiments using 1 equivalent of DIBAL in THF at reflux, with an acidic work-up, returned a 35% yield of 22 by 1H NMR, along with 15% of 21, and unreacted starting material.
This result highlighted an apparent lack of reactivity in THF and the reaction was, therefore, monitored in THF, DCM and toluene using in situ FTIR spectroscopy (ReactIR). The amide carbonyl stretch of 20a was followed by IR during the addition of one equivalent of DIBAL at rt (see Supporting Information File 1 for details). The reactions in DCM and toluene indicated a rapid reduction of the amide carbonyl stretch. In contrast, the reaction in THF displayed a lower initial addition rate, presumably due to competitive coordination of DIBAL by the solvent, and appeared to plateau at a low conversion. A further addition of 1 equivalent of DIBAL did further consume the amide (shown by a further loss of the C=O stretch by IR), however, again, progress appeared to plateau. This indicated that the reaction with the amide group was markedly faster in a non-polar solvent, and further work suggested that the yield of 22 was also increased under such conditions. Addition of DIBAL at lower temperatures (0, −40, and −78 °C) was found to significantly slow the reaction in all solvents.
Further investigation also highlighted the interesting role of the temperature of the reaction mixture prior to the addition of DIBAL on the product distribution. Increased NMR yields of DHQ 22 relative to THQ 21 were recorded (as shown in Table 1), as the temperature of the 20a solution was increased prior to DIBAL addition. This trend indicates that 22 may be formed from in situ fragmentation of the intermediate DIBAL complex followed by an elimination process that is favoured at higher temperature, as postulated in Scheme 9.
Table 1: Temperature and solvent effects from the addition of DIBAL to 20a.a
| Solvent | Temp. (°C) | 20a (%) | 21 (%) | 22 (%) |
|---|---|---|---|---|
| pentane | rt | 26 | 19 | 55 |
| cyclohexane | 60 | 2 | 14 | 79 |
| cyclohexane | 81 | 0 | 15 | 85 |
| toluene | 111 | 10 | 7 | 83 |
| xylenes | 140 | 13 | 4 | 83 |
aOne equivalent of DIBAL (1.0 M in cyclohexane) added to preheated solution of 20a (ca. 0.25 mmol, in 4 mL), stirred rapidly for 1 h, quenched with 20% aq w/v NaOH, extracted in EtOAc, washed with H2O and brine, dried (MgSO4) and evaporated. Yields of 21 and 22 estimated from 1H NMR analysis of the crude mixture.
![[1860-5397-12-174-i9]](/bjoc/content/inline/1860-5397-12-174-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Postulated mechanism for the formation of 22 using DIBAL.
Scheme 9: Postulated mechanism for the formation of 22 using DIBAL.
Chromatographic separation of compounds 21 and 22 on silica resulted in co-elution; however, the two compounds could be separated on neutral alumina using a non-polar eluent. Selectivity for 22 on larger scales was slightly lower, though the DHQ was still isolated in a satisfactory 66% yield on one gram scale. Unlike borane, LiAlH4 and similar reagents, DIBAL did not cause de-iodination, even with larger excesses (up to 2 equivalents).
There are few references to this type of reaction in the literature, however, to our knowledge this is the only such reaction reported with an aromatic amide [31]. Organolithium reagents have been used to synthesise the analogous alkylated DHQ compounds [27].
Compounds 21 and 22 were found to undergo slow degradation (over 2–4 weeks), as indicated by the observation that solutions in CDCl3 slowly turned pink (22 in particular). However, these compounds did not require special handling or precautions, and could be further derivatised without issue. In contrast, 20a was stable indefinitely when stored neat, or in solution at room temperature.
This interesting reaction was conducted with a variety of quinolin-2-ones bearing differing alkyl substituents in order to ascertain whether steric effects promote collapse of the intermediate aluminium complex via fragmentation/elimination, and to assess the generality of this approach. Table 2 highlights the effect of the presence of N-alkyl groups, and an aryl group in the benzylic position of the quinolin-2-one on the synthesis of the corresponding DHQs using the DIBAL reaction developed for the synthesis of 22.
Table 2: DIBAL reductions of quinolin-2-ones 23a–e using the optimised method to synthesise 22.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-174-i11.svg?max-width=637&scale=1.0)
|
|||||
| Quinolin-2-one | R1 | R2 | 23 (%) | 24 (%) | 25 (%) |
|---|---|---|---|---|---|
| 23a | Ph | H | 0 | 100 | 0 |
| 23b | Ph | Me | 0 | 38 | 62 |
| 23c | H | H | 12 | 88 | 0 |
| 23d | H | Et | 0b | 63b | 21b |
| 23e | H | Bn | 0c | 35c | Tracec |
aDIBAL (1.0 M in toluene, 1.2 equivalents or 2.0 equivalents for 23a and 23c) added to a refluxing solution of quinolin-2-one 23a–e (0.5–3.0 mmol) in toluene stirred rapidly at reflux until TLC analysis indicated completion. Then quenched with 20% aq w/v NaOH, extracted with EtOAc, washed with H2O and brine, dried (MgSO4) and evaporated. Yields of 23a–e, 24a–e and 25a–e were estimated from 1H NMR analysis of the crude mixture (see Supporting Information File 1). bA ring-opened ethylaniline (9%) and another byproduct were also present (7%). cA ring-opened benzylaniline was also present (61%).
The absence of an N-alkyl group (23a and 23c) was found to completely disfavour the formation of the corresponding DHQ. Conducting these reactions with 1.2 equivalents of DIBAL resulted in low conversion (30–40%) to the corresponding THQs (24a and 24c), presumably due to competitive deprotonation of the amide proton by DIBAL. When repeated with 2.0 equivalents, 24a and 24c were cleanly synthesised in 100% and 88% yields according to 1H NMR analysis of the crude mixtures.
Addition of an N-alkyl group to supplement the steric effect of the benzylic phenyl (N–Me, 23b) resulted in the reaction favouring the production of the DHQ 25b (62%) over the THQ 24b (38%). Retaining the N-alkyl group whilst removing the steric effect of this benzylic phenyl group (23d) now switched the preference of the reaction towards the THQ 24d (63%) compared to DHQ 25d (21%). In contrast to the clean conversions of the other quinolin-2-ones, crude 1H NMR analysis of the DIBAL reduction of 23d indicated a variety of side products, chief among which was the corresponding ring opened N–ethylaniline (9%). Indeed, when moving to the N–benzylquinolin-2-one 23e, the major product was that of the ring opened N-benzylaniline (61%) and was favoured over the THQ 24e (3%) and only trace indications of DHQ 25e were apparent.
While these results indicated that this reaction was not completely general, comparison of the NMR yields began to provide further mechanistic insight. Clearly, Table 2 shows that an N-alkyl substituent is required in order to encourage formation of the DHQ. The presence of larger N-substituents (23d,e) did little to improve the yield of the corresponding DHQ over 23b, thus indicating that the presence of an alkyl group simply retards reduction of the imine to the THQ, such that the competing elimination process can operate.
A clear increase in the yield of the DHQ is also observed upon addition of a steric influence in the benzylic position of the quinolin-2-one (20a/23b). Given the high temperature, it may be that 1,3-diaxial steric interactions between the benzylic substituent(s) and the intermediary iminium ion are such that a conformation needs to be adopted that both allows and indeed, favours the proposed elimination pathway, with the expelled DIBAL-derived aluminate likely functioning as the base.
With optimised syntheses of 21 and 22 in hand, the reactivity of their iodo substituents towards cross-coupling reactions was compared to that of the corresponding aryl bromides. Table 3 shows an example where 21 and 6-bromo-THQ 26 (synthesised using the optimised conditions for 21) were reacted under typical Suzuki conditions with boronic ester 27. Analysis of the crude 1H NMR spectra for the reactions indicated full conversion of the 6-iodo-THQ 21, but moderate conversion (56%) in the case of the 6-bromo-THQ 26. This highlights the greater propensity for 21 to undergo oxidative addition, due to the more reactive iodo-substituent. Further analysis indicated that biaryl 28 strongly absorbs light at 360 nm, and when excited at this wavelength exhibited a red-shifted, intense, emission peaking at 450 nm (Figure 3), thus highlighting the chromophoric effect of attaching a conjugated electron acceptor to the strongly electron donating THQ moiety.
Table 3: Comparing the reactivity of 6-iodo-THQ 21 and 6-bromo-THQ 26 in a typical Suzuki reaction with boronic ester 27 to give biaryl 28.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-174-i12.svg?max-width=637&scale=1.0)
|
|||
| THQ | Halide | Conversion of starting materiala | Isolated yield of 28 |
|---|---|---|---|
| 21 | I | 100% | 68% |
| 26 | Br | 56% | N/Ab |
aConversion of the starting THQs 21/26 to the coupling product 28 was estimated by comparing the integrals of the CH2 adjacent to the nitrogen (the 2-position) in the crude 1H NMR spectrum. bNot isolated.
![[1860-5397-12-174-3]](/bjoc/content/figures/1860-5397-12-174-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Combined, normalised absorption and emission spectra of 28 in chloroform. Absorption spectrum was recorded at 10 μM. Emission spectrum was recorded at 100 nM, with excitation at 360 nm.
Figure 3: Combined, normalised absorption and emission spectra of 28 in chloroform. Absorption spectrum was r...
To enable a wider range of cross-coupling reactions and to compare their 3-dimensional structures, the corresponding boronic esters 29 and 30 were prepared from 21 and 22 using typical Miyaura borylation conditions (Scheme 10) [32]. Both were highly crystalline compounds, and comparison of the two crystal structures (Figure 4) highlighted the more planar structure of 30 as a result of the enamine function, as predicted by ab initio methods in Figure 2. Furthermore, TLC samples of 30 were found to be fluorescent under a UV lamp, and an absorbance/emission analysis was accordingly conducted in diethyl ether and chloroform.
![[1860-5397-12-174-i10]](/bjoc/content/inline/1860-5397-12-174-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Miyaura borylation of 21 and 22 to give crystalline boronic esters 29 and 30.
Scheme 10: Miyaura borylation of 21 and 22 to give crystalline boronic esters 29 and 30.
![[1860-5397-12-174-4]](/bjoc/content/figures/1860-5397-12-174-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Comparison of the crystal structures of 29 (left) and 30 (right) as viewed along the plane of the aromatic ring, with the pinacolate ester groups removed for clarity. DHQ 30 exhibits a more planar structure. Compounds 29 and 30 were visualised using Olex2 [33].
Figure 4: Comparison of the crystal structures of 29 (left) and 30 (right) as viewed along the plane of the a...
Figure 5 shows the combined, normalised absorbance and emission profiles in diethyl ether at 10 μM and 100 nM, respectively. In diethyl ether, 30 exhibited a major absorbance at 318 nm, and when excited at this wavelength, exhibited an emission that peaks at 370 nm. A similar maximal absorbance was observed in chloroform (323 nm), and excitation at this wavelength produced an emission that was significantly lower in intensity, and mildly red shifted (to 376 nm). This behaviour indicates that 1,4-DHQ structures of this type may function as more effective donor chromophores in dyes and fluorophores than the corresponding THQs due to improved orbital overlap between the nitrogen lone pair and the aromatic π-system by virtue of the more planar ring structure.
![[1860-5397-12-174-5]](/bjoc/content/figures/1860-5397-12-174-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Combined, normalised absorption and emission spectra of 30 in diethyl ether. Absorption spectrum was recorded at 10 μM. Emission spectrum was recorded at 100 nM, with excitation at 318 nm.
Figure 5: Combined, normalised absorption and emission spectra of 30 in diethyl ether. Absorption spectrum wa...
Conclusion
In conclusion, we have explored a wide range of methods for the synthesis of highly lipophilic tetrahydroquinolines and dihydroquinolines. Their iodo-substituents are reactive substrates for direct cross coupling using the plethora of cross-coupling methodologies in the literature. Iodoquinolin-2-ones 13, 19 and 20a, are stable intermediates, and we have developed straightforward, practical and scalable procedures towards their synthesis. We have also demonstrated the novel synthesis of dihydroquinoline 22, by what appears to be a collapse of the intermediate DIBAL complex that is favoured at higher temperatures. Further mechanistic insight into this interesting reaction was garnered by varying the structure of the quinolin-2-one starting material, and indicated that increased steric bulk in the benzylic position and an N-alkyl substituent improves selectivity for the DHQ over the corresponding THQ. Comparison of the reactivity of 6-iodo-THQ 21 and 6-bromo-THQ 26 in a typical Suzuki coupling reaction showed significantly improved conversion in the case of the iodide. The resulting biaryl 28 was found to be highly fluorescent, highlighting the suitability of general structure 1 as an electron donor for the design of charge transfer fluorophores. Finally, crystallographic analysis of the boronic esters 29 and 30 highlighted a subtle flattening of the DHQ structure when compared to the saturated THQ; a structural characteristic that was found to cause fluorescence in 30, indicating that the DHQ may be a more effective electron donor than the THQ. We are currently utilising the optimised syntheses of these interesting compounds in a range of novel fluorophores, and their applications will be communicated in due course.
Experimental
Synthetic chemistry
Reagents were purchased from Sigma-Aldrich, Acros Organics, Alfa-Aesar and Fluorochem and used without further purification unless otherwise stated. 4-Iodo-2-methylaniline and 4-iodoaniline were either purchased from Fluorochem or prepared according to a literature method [16]. Solvents were used as supplied, and dried before use if required with appropriate drying agents or using an Innovative Technologies Inc. Solvent Purification System. Thin-layer chromatography (TLC) was conducted using Merck Millipore silica gel 60G F254 25 glassplates and/or TLC-PET foils of aluminium oxide with fluorescent indicator 254 nm (40 × 80 mm) with visualisation by UV lamp or appropriate staining agents. Flash column chromatography was performed using SiO2 from Sigma-Aldrich (230–400 mesh, 40–63 μm, 60 Å), or activated neutral aluminium oxide (Alumina) from Sigma-Aldrich, and monitored using TLC. NMR spectra were recorded in CDCl3 using Varian VNMRS-700, Varian VNMRS-600, Bruker Avance-400 or Varian Mercury-400 spectrometers operating at ambient probe temperature. NMR peaks are reported as singlet (s), doublet (d), triplet (t), quartet (q), broad (br), septet (sept), combinations thereof, or as a multiplet (m). ESMS was performed by the Durham University departmental service using a TQD (Waters Ltd, UK) mass spectrometer with an Acquity UPLC (Waters Ltd, UK), and accurate mass measurements were obtained using a QtoF Premier mass spectrometer with an Acquity UPLC (Waters Ltd, UK). IR spectra were recorded using a Perkin Elmer FTIR spectrometer. Melting points were obtained using a Gallenkamp melting point apparatus and are uncorrected. Elemental analysis was conducted by the Durham University departmental service using an Exeter Analytical CE-440 analyser. UV–vis spectroscopy was conducted using a CARY100 UV–visible spectrophotometer using the Cary WinUV Scan software 3.00(182). Fluorescence emission spectroscopy was conducted using a Perkin Elmer LS 55 fluorescence spectrometer. All in situ FTIR spectroscopy experiments (ReactIR, Mettler Toledo) were carried out using a ReactIR 15 with MCT detector. Apodization = Happ General. Probe: Prob A DiComp (Diamond) connected via KAgX 9.5 mm × 2 m Fiber (Silver Halide); Sampling 2000–650 at 8 cm−1 resolution; Scan option: auto select, gain 1X.
X-ray crystallography
Single-crystal diffraction experiments were conducted on a Bruker APEX-II CCD diffractometer (13,14, 15b, 29 and 30) and an Xcalibur Sapphire3 diffractomer (19), using Mo Kα radiation. Crystals were cooled using Cryostream (Oxford Cryosystems) open-flow N2 cryostats. The structures were solved within Olex2 by direct methods and refined by full-matrix least squares against F2 of all data, using SHELXTL software [33-37]. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model. CCDC (1433617–1433622) contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/getstructures.
Supporting Information
Supporting information features full synthetic procedures, in situ FTIR data plots, copies of 1H, 13C and 11B NMR spectra, and full crystallographic data including images and CIF files.
| Supporting Information File 1: Full synthetic procedures, in situ FTIR data plots, and copies of 1H, 13C and 11B NMR spectra. | ||
| Format: PDF | Size: 2.7 MB | Download |
| Supporting Information File 2: Crystallographic Information Files of compounds 13, 14, 15b, 19, 29, and 30. | ||
| Format: ZIP | Size: 2.9 MB | Download |
Acknowledgements
DRC thanks the EPSRC, BBSRC and High Force Research Ltd. for doctoral funding. GLZ also thanks the EPSRC and High Force Research Ltd. for doctoral funding. We thank Helen Osborne and Prof. Todd B. Marder for helpful discussions, Dr. Horst Puschmann for general crystallographic guidance and for assistance in the solution of the crystal structure of 19, and Dr. Dmitry S. Yufit for general crystallographic guidance.
References
-
Sridharan, V.; Suryavanshi, P. A.; Menéndez, J. C. Chem. Rev. 2011, 111, 7157–7259. doi:10.1021/cr100307m
Return to citation in text: [1] -
Wang, Z.; Li, S.; Yu, B.; Wu, H.; Wang, Y.; Sun, X. J. Org. Chem. 2012, 77, 8615–8620. doi:10.1021/jo301560w
Return to citation in text: [1] -
Li, H. Y.; Horn, J.; Campbell, A.; House, D.; Nelson, A.; Marsden, S. P. Chem. Commun. 2014, 50, 10222–10224. doi:10.1039/C4CC04940C
Return to citation in text: [1] -
Akiyama, T.; Morita, H.; Fuchibe, K. J. Am. Chem. Soc. 2006, 128, 13070–13071. doi:10.1021/ja064676r
Return to citation in text: [1] -
Cheng, X.; Xia, Y.; Wei, H.; Xu, B.; Zhang, C.; Li, Y.; Qian, G.; Zhang, X.; Li, K.; Li, W. Eur. J. Org. Chem. 2008, 1929–1936. doi:10.1002/ejoc.200701080
Return to citation in text: [1] -
Kamino, S.; Murakami, M.; Tanioka, M.; Shirasaki, Y.; Watanabe, K.; Horigome, J.; Ooyama, Y.; Enomoto, S. Org. Lett. 2014, 16, 258–261. doi:10.1021/ol403262x
Return to citation in text: [1] -
Benbrook, D. M.; Madler, M. M.; Spruce, L. W.; Birckbichler, P. J.; Nelson, E. C.; Subramanian, S.; Weerasekare, G. M.; Gale, J. B.; Patterson, M. K., Jr.; Wang, B.; Wang, W.; Lu, S.; Rowland, T. C.; DiSivestro, P.; Lindamood, C., III; Hill, D. L.; Berlin, K. D. J. Med. Chem. 1997, 40, 3567–3583. doi:10.1021/jm970196m
Return to citation in text: [1] [2] [3] -
Dhar, A.; Liu, S.; Klucik, J.; Berlin, K. D.; Madler, M. M.; Lu, S.; Ivey, R. T.; Zacheis, D.; Brown, C. W.; Nelson, E. C.; Birckbichler, P. J.; Benbrook, D. M. J. Med. Chem. 1999, 42, 3602–3614. doi:10.1021/jm9900974
Return to citation in text: [1] -
Atwal, K. S.; Ferrara, F. N.; Ding, C. Z.; Grover, G. J.; Sleph, P. G.; Dzwonczyk, S.; Baird, A. J.; Normandin, D. E. J. Med. Chem. 1996, 39, 304–313. doi:10.1021/jm950646f
Return to citation in text: [1] -
Sánchez, I.; Pujol, M. D. Synthesis 2006, 1823–1828. doi:10.1055/s-2006-942360
Return to citation in text: [1] -
Birch, A. J.; Lehman, P. G. J. Chem. Soc., Perkin Trans. 1 1973, 2754–2759. doi:10.1039/P19730002754
Return to citation in text: [1] -
Smith, W. H.; Bard, A. J. J. Am. Chem. Soc. 1975, 97, 6491–6495. doi:10.1021/ja00855a034
Return to citation in text: [1] -
Li, Y.-M.; Shen, Y.; Chang, K.-J.; Yang, S.-D. Tetrahedron Lett. 2014, 55, 2119–2122. doi:10.1016/j.tetlet.2014.02.043
Return to citation in text: [1] -
Barnard, J. H.; Zhou, G.-L.; Collings, J. C.; Whiting, A.; Marder, T. B. Durham University, Durham, UK, Unpublished work.
Return to citation in text: [1] [2] [3] -
Koo, J. J. Am. Chem. Soc. 1953, 75, 1891–1895. doi:10.1021/ja01104a034
Return to citation in text: [1] -
Monnereau, C.; Blart, E.; Odobel, F. Tetrahedron Lett. 2005, 46, 5421–5423. doi:10.1016/j.tetlet.2005.05.117
Return to citation in text: [1] [2] [3] [4] [5] -
Emmanuvel, L.; Shukla, R. K.; Sudalai, A.; Gurunath, S.; Sivaram, S. Tetrahedron Lett. 2006, 47, 4793–4796. doi:10.1016/j.tetlet.2006.05.062
Return to citation in text: [1] -
Kawade, R. K.; Huple, D. B.; Lin, R.-J.; Liu, R.-S. Chem. Commun. 2015, 51, 6625–6628. doi:10.1039/C5CC01287B
Return to citation in text: [1] -
Tomcufcik, A. S.; Seeger, D. R. J. Org. Chem. 1961, 26, 3351–3356. doi:10.1021/jo01067a079
Return to citation in text: [1] -
Castanet, A.-S.; Colobert, F.; Broutin, P.-E. Tetrahedron Lett. 2002, 43, 5047–5048. doi:10.1016/S0040-4039(02)01010-9
Return to citation in text: [1] -
Adimurthy, S.; Ramachandraiah, G.; Ghosh, P. K.; Bedekar, A. V. Tetrahedron Lett. 2003, 44, 5099–5101. doi:10.1016/S0040-4039(03)01144-4
Return to citation in text: [1] -
Yamamoto, T.; Toyota, K.; Morita, N. Tetrahedron Lett. 2010, 51, 1364–1366. doi:10.1016/j.tetlet.2009.12.125
Return to citation in text: [1] -
Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 14844–14845. doi:10.1021/ja028865v
Return to citation in text: [1] -
Thompson, M. D.; Berlin, K. D. Proc. Okla. Acad. Sci. 1985, 65, 39–43.
Return to citation in text: [1] -
Phippen, C. B. W.; Beattie, J. K.; McErlean, C. S. P. Chem. Commun. 2010, 46, 8234–8236. doi:10.1039/c0cc02502j
Return to citation in text: [1] -
De, K.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. J. Org. Chem. 2009, 74, 6260–6265. doi:10.1021/jo9012699
Return to citation in text: [1] -
Reist, E. J.; Hamlow, H. P.; Junga, I. G.; Silverstein, R. M.; Baker, B. R. J. Org. Chem. 1960, 25, 1368–1378. doi:10.1021/jo01078a021
Return to citation in text: [1] [2] -
Spartan'10; Wavefunction, Inc.: Irvine, CA, 2010.
Return to citation in text: [1] -
Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. J. Comput. Chem. 2004, 25, 1605–1612. doi:10.1002/jcc.20084
Return to citation in text: [1] -
Winterfeldt, E. Synthesis 1975, 617–630. doi:10.1055/s-1975-23856
Return to citation in text: [1] -
Stevens, R. V.; Mehra, R. K.; Zimmerman, R. L. J. Chem. Soc. D 1969, 877–878. doi:10.1039/c29690000877
Return to citation in text: [1] -
Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508–7510. doi:10.1021/jo00128a024
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/S0021889808042726
Return to citation in text: [1] [2] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/S2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/S2053229614024218
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] -
APEX2; SAINT; SADABS; Bruker AXS, Inc.: Madison, WI, 2014.
Return to citation in text: [1]
| 31. | Stevens, R. V.; Mehra, R. K.; Zimmerman, R. L. J. Chem. Soc. D 1969, 877–878. doi:10.1039/c29690000877 |
| 27. | Reist, E. J.; Hamlow, H. P.; Junga, I. G.; Silverstein, R. M.; Baker, B. R. J. Org. Chem. 1960, 25, 1368–1378. doi:10.1021/jo01078a021 |
| 1. | Sridharan, V.; Suryavanshi, P. A.; Menéndez, J. C. Chem. Rev. 2011, 111, 7157–7259. doi:10.1021/cr100307m |
| 8. | Dhar, A.; Liu, S.; Klucik, J.; Berlin, K. D.; Madler, M. M.; Lu, S.; Ivey, R. T.; Zacheis, D.; Brown, C. W.; Nelson, E. C.; Birckbichler, P. J.; Benbrook, D. M. J. Med. Chem. 1999, 42, 3602–3614. doi:10.1021/jm9900974 |
| 20. | Castanet, A.-S.; Colobert, F.; Broutin, P.-E. Tetrahedron Lett. 2002, 43, 5047–5048. doi:10.1016/S0040-4039(02)01010-9 |
| 7. | Benbrook, D. M.; Madler, M. M.; Spruce, L. W.; Birckbichler, P. J.; Nelson, E. C.; Subramanian, S.; Weerasekare, G. M.; Gale, J. B.; Patterson, M. K., Jr.; Wang, B.; Wang, W.; Lu, S.; Rowland, T. C.; DiSivestro, P.; Lindamood, C., III; Hill, D. L.; Berlin, K. D. J. Med. Chem. 1997, 40, 3567–3583. doi:10.1021/jm970196m |
| 21. | Adimurthy, S.; Ramachandraiah, G.; Ghosh, P. K.; Bedekar, A. V. Tetrahedron Lett. 2003, 44, 5099–5101. doi:10.1016/S0040-4039(03)01144-4 |
| 5. | Cheng, X.; Xia, Y.; Wei, H.; Xu, B.; Zhang, C.; Li, Y.; Qian, G.; Zhang, X.; Li, K.; Li, W. Eur. J. Org. Chem. 2008, 1929–1936. doi:10.1002/ejoc.200701080 |
| 6. | Kamino, S.; Murakami, M.; Tanioka, M.; Shirasaki, Y.; Watanabe, K.; Horigome, J.; Ooyama, Y.; Enomoto, S. Org. Lett. 2014, 16, 258–261. doi:10.1021/ol403262x |
| 18. | Kawade, R. K.; Huple, D. B.; Lin, R.-J.; Liu, R.-S. Chem. Commun. 2015, 51, 6625–6628. doi:10.1039/C5CC01287B |
| 2. | Wang, Z.; Li, S.; Yu, B.; Wu, H.; Wang, Y.; Sun, X. J. Org. Chem. 2012, 77, 8615–8620. doi:10.1021/jo301560w |
| 3. | Li, H. Y.; Horn, J.; Campbell, A.; House, D.; Nelson, A.; Marsden, S. P. Chem. Commun. 2014, 50, 10222–10224. doi:10.1039/C4CC04940C |
| 4. | Akiyama, T.; Morita, H.; Fuchibe, K. J. Am. Chem. Soc. 2006, 128, 13070–13071. doi:10.1021/ja064676r |
| 19. | Tomcufcik, A. S.; Seeger, D. R. J. Org. Chem. 1961, 26, 3351–3356. doi:10.1021/jo01067a079 |
| 14. | Barnard, J. H.; Zhou, G.-L.; Collings, J. C.; Whiting, A.; Marder, T. B. Durham University, Durham, UK, Unpublished work. |
| 16. | Monnereau, C.; Blart, E.; Odobel, F. Tetrahedron Lett. 2005, 46, 5421–5423. doi:10.1016/j.tetlet.2005.05.117 |
| 13. | Li, Y.-M.; Shen, Y.; Chang, K.-J.; Yang, S.-D. Tetrahedron Lett. 2014, 55, 2119–2122. doi:10.1016/j.tetlet.2014.02.043 |
| 16. | Monnereau, C.; Blart, E.; Odobel, F. Tetrahedron Lett. 2005, 46, 5421–5423. doi:10.1016/j.tetlet.2005.05.117 |
| 17. | Emmanuvel, L.; Shukla, R. K.; Sudalai, A.; Gurunath, S.; Sivaram, S. Tetrahedron Lett. 2006, 47, 4793–4796. doi:10.1016/j.tetlet.2006.05.062 |
| 33. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/S0021889808042726 |
| 34. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/S2053273314026370 |
| 35. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/S2053229614024218 |
| 36. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 37. | APEX2; SAINT; SADABS; Bruker AXS, Inc.: Madison, WI, 2014. |
| 10. | Sánchez, I.; Pujol, M. D. Synthesis 2006, 1823–1828. doi:10.1055/s-2006-942360 |
| 11. | Birch, A. J.; Lehman, P. G. J. Chem. Soc., Perkin Trans. 1 1973, 2754–2759. doi:10.1039/P19730002754 |
| 12. | Smith, W. H.; Bard, A. J. J. Am. Chem. Soc. 1975, 97, 6491–6495. doi:10.1021/ja00855a034 |
| 32. | Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508–7510. doi:10.1021/jo00128a024 |
| 9. | Atwal, K. S.; Ferrara, F. N.; Ding, C. Z.; Grover, G. J.; Sleph, P. G.; Dzwonczyk, S.; Baird, A. J.; Normandin, D. E. J. Med. Chem. 1996, 39, 304–313. doi:10.1021/jm950646f |
| 14. | Barnard, J. H.; Zhou, G.-L.; Collings, J. C.; Whiting, A.; Marder, T. B. Durham University, Durham, UK, Unpublished work. |
| 33. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/S0021889808042726 |
| 22. | Yamamoto, T.; Toyota, K.; Morita, N. Tetrahedron Lett. 2010, 51, 1364–1366. doi:10.1016/j.tetlet.2009.12.125 |
| 23. | Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 14844–14845. doi:10.1021/ja028865v |
| 7. | Benbrook, D. M.; Madler, M. M.; Spruce, L. W.; Birckbichler, P. J.; Nelson, E. C.; Subramanian, S.; Weerasekare, G. M.; Gale, J. B.; Patterson, M. K., Jr.; Wang, B.; Wang, W.; Lu, S.; Rowland, T. C.; DiSivestro, P.; Lindamood, C., III; Hill, D. L.; Berlin, K. D. J. Med. Chem. 1997, 40, 3567–3583. doi:10.1021/jm970196m |
| 28. | Spartan'10; Wavefunction, Inc.: Irvine, CA, 2010. |
| 29. | Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. J. Comput. Chem. 2004, 25, 1605–1612. doi:10.1002/jcc.20084 |
| 14. | Barnard, J. H.; Zhou, G.-L.; Collings, J. C.; Whiting, A.; Marder, T. B. Durham University, Durham, UK, Unpublished work. |
| 16. | Monnereau, C.; Blart, E.; Odobel, F. Tetrahedron Lett. 2005, 46, 5421–5423. doi:10.1016/j.tetlet.2005.05.117 |
| 7. | Benbrook, D. M.; Madler, M. M.; Spruce, L. W.; Birckbichler, P. J.; Nelson, E. C.; Subramanian, S.; Weerasekare, G. M.; Gale, J. B.; Patterson, M. K., Jr.; Wang, B.; Wang, W.; Lu, S.; Rowland, T. C.; DiSivestro, P.; Lindamood, C., III; Hill, D. L.; Berlin, K. D. J. Med. Chem. 1997, 40, 3567–3583. doi:10.1021/jm970196m |
| 27. | Reist, E. J.; Hamlow, H. P.; Junga, I. G.; Silverstein, R. M.; Baker, B. R. J. Org. Chem. 1960, 25, 1368–1378. doi:10.1021/jo01078a021 |
| 16. | Monnereau, C.; Blart, E.; Odobel, F. Tetrahedron Lett. 2005, 46, 5421–5423. doi:10.1016/j.tetlet.2005.05.117 |
| 25. | Phippen, C. B. W.; Beattie, J. K.; McErlean, C. S. P. Chem. Commun. 2010, 46, 8234–8236. doi:10.1039/c0cc02502j |
| 26. | De, K.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. J. Org. Chem. 2009, 74, 6260–6265. doi:10.1021/jo9012699 |
| 16. | Monnereau, C.; Blart, E.; Odobel, F. Tetrahedron Lett. 2005, 46, 5421–5423. doi:10.1016/j.tetlet.2005.05.117 |
© 2016 Chisholm et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)