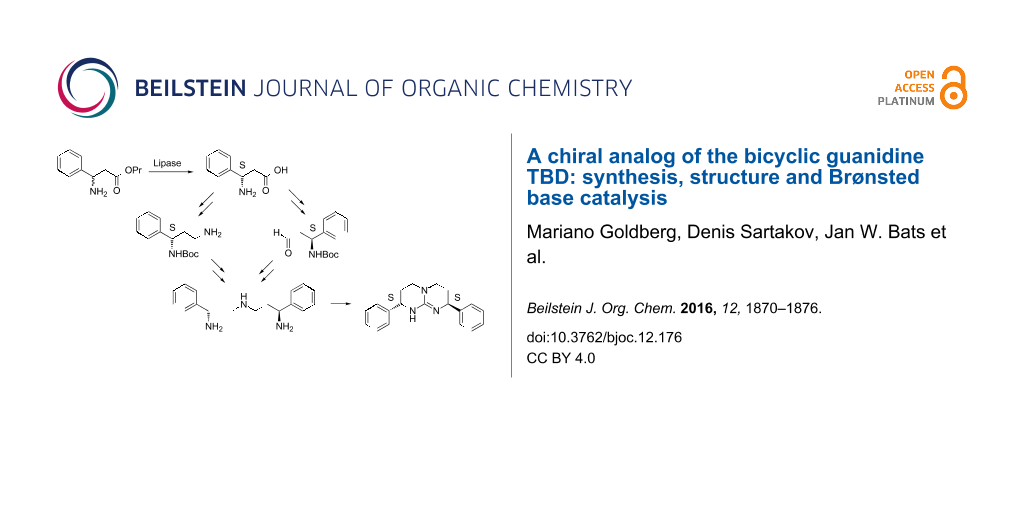
Abstract
Starting from (S)-β-phenylalanine, easily accessible by lipase-catalyzed kinetic resolution, a chiral triamine was assembled by a reductive amination and finally cyclized to form the title compound 10. In the crystals of the guanidinium benzoate salt the six membered rings of 10 adopt conformations close to an envelope with the phenyl substituents in pseudo-axial positions. The unprotonated guanidine 10 catalyzes Diels–Alder reactions of anthrones and maleimides (25–30% ee). It also promotes as a strong Brønsted base the retro-aldol reaction of some cycloadducts with kinetic resolution of the enantiomers. In three cases, the retro-aldol products (48–83% ee) could be recrystallized to high enantiopurity (≥95% ee). The absolute configuration of several compounds is supported by anomalous X-ray diffraction and by chemical correlation.

Graphical Abstract
Introduction
In guanidinium ions charge delocalization is an important factor to stabilize the protonated form. As a result, guanidines are exceptionally strong nitrogen bases. As part of the amino acid arginine, they play an important role in biochemistry, mainly by forming ion pairs. In addition, numerous guanidine derivatives with complex cyclic structures can be found in natural products [1]. Simple guanidines such as tetramethylguanidine have been used as strong Brønsted bases in countless applications [2,3]. The bicylic guanidine 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 1, Figure 1) [4], another important Brønsted base in preparative chemistry, may also act as a powerful nucleophilic catalyst [3]. Substituted analogs of TBD [5], such as the chiral compound 2, have become popular in the field of molecular recognition, however, without being tested as catalysts [5-10]. A first example of enantioselective Michael addition has been reported for guanidine 3, albeit with low selectivity [11]. Compounds 4 and 5, inspired by the structure of ptilomycalin A and related natural products, have been used as chiral phase-transfer catalysts [12,13].
![[1860-5397-12-176-1]](/bjoc/content/figures/1860-5397-12-176-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Bicyclic guanidines with five-membered rings are also known from the alkaloid isoalchornein [14,15]. In subsequent years, synthetic compounds (6–9) of this structural type have been developed as chiral Brønsted bases [16-19] and used for highly enantioselective Strecker [17] and Diels–Alder reactions [19].
Compared to guanidine 7 ring expansion into structure 10 would shift the stereogenic phenyl groups into closer proximity to a hydrogen-bonded guest molecule and thereby might improve the enantioselective control exerted by 10 when used as a catalyst. In this article we describe the synthesis of guanidine 10 together with some initial applications as a chiral Brønsted base.
Results and Discussion
Previous syntheses of bicylic guanidines 2 and 6–9 started from enantiomerically pure α-amino acids [6,8,10,16,17] or from their reduction products, chiral α-amino alcohols [9,18]. In contrast, our approach used the racemic ester rac-12 of β-phenylalanine, easily accessible in a Knoevenagel-type condensation of benzaldehyde 11, ammonium acetate and malonic acid, followed by esterification (Scheme 1) [20,21]. A kinetic resolution of the enantiomers was achieved by enzymatic hydrolysis with Amano lipase PS from Burkholderia cepacia [22,23], a method already optimized for technical use [24]. The best results were obtained with methyl tert-butyl ether as a cosolvent [24]. By simple precipitation, batches larger than 15 g of the S-configurated acid 13 could be isolated in 90% yield (45% based on rac-12). In a two-step procedure 13 was converted into amino alcohol 14 without recrystallization in order to keep the enantiomeric excess unchanged. It was determined at this stage to be better than 99%. The S-configuration was assigned to 13 in accord with published data [22-24]. Intermediate 14 was converted into amine 16 by mesylation (79%), reaction with NaN3 (96%) and hydrogenation (84%). Aldehyde 17, also accessible from 14 by oxidation (87%), then could be coupled with amine 16 in a reductive amination to form 18 (58%). After removal of the Boc protecting group (quant.), triamine 19 was reacted with dimethyl trithiocarbonate in refluxing nitromethane. The thiourea intermediate was activated in situ by S-alkylation with MeI. Upon further heating the final cyclization occured forming the iodide salt 10a of S,S-configurated guanidine 10 (67%). The free base could be isolated by extraction with CH2Cl2 from 20 M aqueous NaOH (95%). For experimental details see Supporting Information File 1.
![[1860-5397-12-176-i1]](/bjoc/content/inline/1860-5397-12-176-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of guanidine 10. Conditions: (a) 1 equiv HOOC-CH2-COOH, 2 equiv NH4OAc, EtOH, 78 °C, 5 h, 38%; (b) 10 equiv n-PrOH, 1.5 equiv H2SO4, 97 °C, 4 h, 82%; (c) Amano-Lipase PS (from Burkholderia cepacia), aqueous Na2HPO4 buffer, pH 7.00, 50 °C, 1 h, methyl tert-butyl ether, 50 °C, 24 h, 90% (45% based on rac-12), >99% ee; (d) 2.5 equiv NaBH4, 1.2 equiv I2, THF, 66 °C, 18 h, 86%; (e) 1 equiv Boc2O, 1.2 equiv triethylamine, CH2Cl2, 0 °C, 3 h, 100%; (f) 1.1 equiv MsCl, 1.1 equiv triethylamine, CH2Cl2, 0 °C, 3 h, 79%; (g) 3 equiv NaN3, DMF, 24 °C, 120 h, 96%; (h) 2 equiv SO3*Py, 2.3 equiv pyridine, 4.1 equiv triethylamine, DMSO, CH2Cl2, 0 °C, 10 min, 24 °C, 2 h, 87%; (i) H2, Pd/C, MeOH, overnight, 84%; (k) 1 equiv 17, THF, 48 h, 2 equiv NaBH4; MeOH, 96 h, 58%; (l) 10 equiv TFA, CH2Cl2, 40 °C, 24 h, 100%; (m) 1.3 equiv dimethyl trithiocarbonate, MeNO2, 101 °C, 2 h, 4 equiv AcOH, 2 equiv MeI, 101 °C, 3 h, 67% as iodide salt 10a.
Scheme 1: Synthesis of guanidine 10. Conditions: (a) 1 equiv HOOC-CH2-COOH, 2 equiv NH4OAc, EtOH, 78 °C, 5 h,...
A crystal structure could be obtained from the benzoate salt of 10 (Figure 2). The asymmetric unit contains two cations, two benzoate anions and ethyl acetate as a solvate molecule. Each cation is connected by two N–H···O hydrogen bonds to benzoate ions. The rings of cation 10 adopt a conformation close to an envelope with the phenyl substituents in pseudo-axial positions. The ion pairs and the solvate molecule are also connected by a number of very weak intermolecular C–H···π (phenyl) and C–H···O contacts (see Supporting Information File 2).
![[1860-5397-12-176-2]](/bjoc/content/figures/1860-5397-12-176-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of guanidine 10 as a benzoate salt. Only one of the ion pairs is shown for the sake of clarity.
Figure 2: Crystal structure of guanidine 10 as a benzoate salt. Only one of the ion pairs is shown for the sa...
The chiral guanidine 10 was tested as a Brønsted base catalyst to promote the reaction of anthrones 20 or 21 with maleimides 22–24 (Scheme 2) [19,25-28]. Depending on the substituents and the strength of the Brønsted base either Diels–Alder adducts or Michael products may be formed. Weaker bases such as ion-free bisoxazolines [28] or triethylamine are known to induce Diels–Alder adducts selectively whereas strongly basic guanidines such as 8 may also form the Michael products [19] which are the dominant products in polar solvents [25]. Previous mechanistic studies by Koerner and Rickborn [25] have collected strong arguments for a fast concerted [4 + 2]-cycloaddition of the deprotonated anthrone. Michael products were shown to be secondary products of a base-catalyzed retro-aldol reaction and they are not converted backwards into Diels–Alder adducts under such conditions [19,25,26]. Our results shown below give further support to this view. Riant, Kagan and Ricard have demonstrated for the first time that deprotonated anthrones may coordinate to chiral counterions in less polar solvents. Up to 61% ee could be obtained using cinchona alkaloids as catalysts [26]. The subsequent work of Tan and co-workers with guanidine catalyst 8 achieved enantioselectivities as high as 99% ee [19]. In recent years functionalized chiral amines have been successfully used as catalysts for anthrone maleimide cycloadditions [29-35]. In the presence of 10, dichloroanthrone 21 reacted with maleimides 22 and 23 to produce exclusively cycloadducts 26 and 27. In contrast, a mixture of 25 and 28 resulted from the reaction of anthrone 20 and N-phenylmaleimide (22). For the remaining combinations (20 + 23; 20 + 24; 21 + 24) only the Michael products 29–31 could be observed (0.1 equiv of 10, CH2Cl2, −15 °C).
![[1860-5397-12-176-i2]](/bjoc/content/inline/1860-5397-12-176-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of anthrones and N-arylmaleimides catalyzed by guanidine 10. The guanidine deprotonates anthrones 20 or 21 and forms chiral ion pairs. Primary products of the ion pair complexes are the Diels–Alder adducts that may be further transformed in a base catalyzed retro-aldol reaction.
Scheme 2: Reaction of anthrones and N-arylmaleimides catalyzed by guanidine 10. The guanidine deprotonates an...
To determine the enantioselectivity of guanidine 10 we started with the structurally simplest case, the reaction of anthrone 20 and N-phenylmaleimide (22) which finally turned out to be the most complex one (Table 1). The best but still low ee values for cycloadduct 25 were obtained in CHCl3 (37% ee) and CH2Cl2 (34% ee). Much better enantioselectivities of the Michael product 28 were found for reactions in solvents of increased polarity. Assuming that 28 is formed from 25 in a retro-aldol step, one might expect similar ee values for both compounds. This is clearly not the case. In THF the 83% ee for compound 28 strongly deviates from the low value for 25 (6% ee). Obviously, in the reaction of 20 and 22, a Diels–Alder step with low to moderate stereoselectivity is superimposed with a kinetic resolution of enantiomers in the subsequent retro-aldol reaction. Both are catalyzed by the chiral guanidine 10. As a result, the numbers shown in Table 1 are not constant but depend on the relative turnover of each reaction.
Table 1: Reaction of anthrone 20 and N-phenylmaleimide (22) in different solvents.a
| solvent | Diels–Alder product 25 | Michael product 28 | |||
|---|---|---|---|---|---|
| yieldb | eec | yieldb | eec | ||
| toluene | 36% | 22% | 51% | 47% | |
| (CH2Cl)2 | 88% | 6% | 8% | 35% | |
| CHCl3 | 3% | 37% | 91% | 25% | |
| CH2Cl2 | 28% | 34% | 70% | 41% | |
| THF | 51% | 6% | 42% | 83% | |
| CH3CN | 15% | 1% | 53% | 74% | |
aAll reactions were carried out with 0.1 equiv of guanidine 10 for 64 h at −15 °C. bIsolated yields after chromatographic separation. cDetermined by HPLC on a chiral column.
When rac-25 reacted with 0.1 equiv of 10 in THF at −15 °C, at 22% conversion ee values of 18% and 70% were found for compounds 25 and 28. The dominant isomer of Michael product 28 is shown below to be R configurated which corresponds to the slower running peak in Figure 3B. The R isomer of 28 is formed from the S,S isomer of 25 with retention of configuration (Scheme 2). The change from S to R is caused by a change in the CIP priorities of the substituents. Thus the faster running smaller peak in Figure 3A must correspond to the S,S enantiomer of 25. Assuming independent first order rate laws for the opening of the S,S and R,R enantiomers of 25, the best numerical fit is obtained for kS,S/kR,R ≈ 6.5. When both steps, Diels–Alder and Michael reaction, are catalyzed by guanidine 10 in THF (see Table 1), at 45% conversion the R isomer of 28 and the R,R isomer of 25 are the dominant species. However, due to the faster reaction of S,S-25 the numerical simulation shows that not the R,R but the S,S isomer is the preferred product of the Diels–Alder step with approximately 33% ee. This value comes close to the numbers obtained for the production of 26 (25% ee; 30% ee in THF) and 27 (28% ee) in CH2Cl2 when no secondary conversion occurs. In contrast, when compound 21 reacts with 24 ring opening is so fast that no Diels–Alder product accumulates. Here the ee value of the Michael product 31 (21%) must correspond to the stereoselectivity of the Diels–Alder step. Due to low stereoselectivities and fast retro-aldol reactions guanidine 10 is not optimal for the preparation of Diels–Alder adducts.
![[1860-5397-12-176-3]](/bjoc/content/figures/1860-5397-12-176-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: A) Chromatogram of rac-25 after incubation with 0.1 equiv of 10 in THF at −15 °C for 64 h. The faster running isomer shows increased conversion into 28. B) The slower running isomer (R) of 28 is formed preferentially. The faster running isomer of 25, therefore, must be S,S configurated.
Figure 3: A) Chromatogram of rac-25 after incubation with 0.1 equiv of 10 in THF at −15 °C for 64 h. The fast...
We therefore focussed our efforts on the Michael products: Anthrone 20 and maleimide 22 reacted with 0.1 equiv of 10 in THF at −15 °C for 64 h. Chromatographic separation then yielded 51% of 25 and 42% of Michael product 28 (83% ee). Recrystallization afforded 32% of 28 with 99% ee. Thus, 12.5 mg of catalyst 10 produced 51 mg of almost pure R-28. Analogous treatment of 20 and 23 yielded 21% of 29 (61% ee). After recrystallization 15% of 29 with 98% ee were obtained. Finally, Michael product 30, obtained from 20 and 24 (42%, 48% ee) yielded 26% of material with 95% ee.
Assignment of absolute configurations (Scheme 3): Well grown crystals of enantiopure Michael product 30 suitable for X-ray structural analysis could be obtained by a second recrystallization from CH2Cl2/cyclohexane. The 4-bromophenyl residue allowed us to assign the R configuration by anomalous dispersion (Supporting Information File 4). This isomer corresponds to the slower running isomer on a Chiralpak IA column. By catalytic hydrogenation with Pd on charcoal the bromo residue of enantiopure 30 was replaced with hydrogen thus converting R configurated 30 into R configurated Michael product 28 (slower running isomer on Chiralpak IA and preferred product under catalysis with 10). Both compounds must result from the S,S configurated Diels–Alder adducts. In the case of 25 this is the faster running isomer on Chiralpak IA. In an earlier study [28] with bisoxazoline catalyst 33, we have assigned the S,S configuration to the major isomer of product 32. By hydrogenolysis, we could now convert such a sample (40% ee) into 25. The product S,S-25 was formed as the dominant product (35% ee) thus confirming our previous assignment. Under mild basic conditions, S,S-32 finally could be opened into R configurated Michael product 30 [25,26] (31% ee) to generate a stereochemically consistent view.
![[1860-5397-12-176-i3]](/bjoc/content/inline/1860-5397-12-176-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Assignment of the absolute configurations by chemical correlation. The R configuration of compound 30 is derived from the crystal structure.
Scheme 3: Assignment of the absolute configurations by chemical correlation. The R configuration of compound ...
Conclusion
(S)-β-Phenylalanine (13), readily accessible by enantioselective hydrolysis of ester rac-12, is the starting material for an efficient synthesis of the chiral TBD analog 10. The cyclization steps in particular, which convert the triamine 19 into the final guanidine 10, give considerably higher yields compared to guanidines with five-membered rings such as compounds 6–9. Apart from the better synthetic accessibility the formal ring expansion to the six-membered structure of 10 does not increase the stereoselectivity when it is used as a catalyst. We could not observe the near-perfect selectivities of the Tan catalyst 8 in the base-induced reaction of anthrones and maleimides. Nevertheless ee values of up to 83% for the formal Michael products allowed us to isolate 3 compounds in almost enantiopure form after a single recrystallization. The Tan guanidine 8 [19], the bisoxazoline 33 [28], and a stereochemically related bisamidine [27] consistently favor the S,S isomer of Diels–Alder adduct 25. Although guanidine 10 has the opposite configuration, it also forms S,S-25 preferentially. Accordingly, transition-state structures in the reactions of guanidines 8 and 10 must differ from each other.
Supporting Information
| Supporting Information File 1: Synthetic procedures, characterization data, copies of chromatograms on chiral columns and of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.5 MB | Download |
| Supporting Information File 2: X-ray data of guanidine 10 as benzoate salt (CCDC-1482611). | ||
| Format: CIF | Size: 32.4 KB | Download |
| Supporting Information File 3: X-ray data of compound 29 (CCDC-1482612). | ||
| Format: CIF | Size: 1.2 MB | Download |
| Supporting Information File 4: X-ray data of compound 30 (CCDC-1482613). | ||
| Format: CIF | Size: 788.9 KB | Download |
References
-
Ma, Y.; De, S.; Chen, C. Tetrahedron 2015, 71, 1145–1173. doi:10.1016/j.tet.2014.11.056
Return to citation in text: [1] -
Ishikawa, T.; Kumamoto, T. Synthesis 2006, 737–752. doi:10.1055/s-2006-926325
Return to citation in text: [1] -
Selig, P. Synthesis 2013, 45, 703–718. doi:10.1055/s-0032-1318154
Return to citation in text: [1] [2] -
McKay, A. F.; Kreling, M.-E. Can. J. Chem. 1957, 35, 1438–1445. doi:10.1139/v57-190
Return to citation in text: [1] -
Schmidtchen, F. P. Chem. Ber. 1980, 113, 2175–2182. doi:10.1002/cber.19801130612
Return to citation in text: [1] [2] -
Echavarren, A.; Galán, A.; de Mendoza, J.; Salmerón, A.; Lehn, J.-M. Helv. Chim. Acta 1988, 71, 685–693. doi:10.1002/hlca.19880710402
Return to citation in text: [1] [2] -
Echavarren, A.; Galán, A.; Lehn, J.-M.; de Mendoza, J. J. Am. Chem. Soc. 1989, 111, 4994–4995. doi:10.1021/ja00195a071
Return to citation in text: [1] -
Kurzmeier, H.; Schmidtchen, F. P. J. Org. Chem. 1990, 55, 3749–3755. doi:10.1021/jo00299a013
Return to citation in text: [1] [2] -
Jadhav, V. D.; Schmidtchen, F. P. J. Org. Chem. 2008, 73, 1077–1087. doi:10.1021/jo7021026
Return to citation in text: [1] [2] -
Turočkin, A.; Honeker, R.; Raven, W.; Selig, P. J. Org. Chem. 2016, 81, 4516–4529. doi:10.1021/acs.joc.6b00283
Return to citation in text: [1] [2] -
Davis, A. P.; Dempsey, K. J. Tetrahedron: Asymmetry 1995, 6, 2829–2840. doi:10.1016/0957-4166(95)00374-X
Return to citation in text: [1] -
Kita, T.; Georgieva, A.; Hashimoto, Y.; Nakata, T.; Nagasawa, K. Angew. Chem., Int. Ed. 2002, 41, 2832–2834. doi:10.1002/1521-3773(20020802)41:15<2832::AID-ANIE2832>3.0.CO;2-Q
Return to citation in text: [1] -
Kita, T.; Shin, B.; Hashimoto, Y.; Nagasawa, K. Heterocycles 2007, 73, 241–247. doi:10.3987/COM-07-S(U)49
Return to citation in text: [1] -
Khuong-Huu, F.; Le Forestier, J.-P.; Goutarel, R. Tetrahedron 1972, 28, 5207–5220. doi:10.1016/S0040-4020(01)88940-7
Return to citation in text: [1] -
Büchi, G.; Rodriguez, A. D.; Yakushijin, K. J. Org. Chem. 1989, 54, 4494–4496. doi:10.1021/jo00280a009
Return to citation in text: [1] -
Corey, E. J.; Ohtani, M. Tetrahedron Lett. 1989, 30, 5227–5230. doi:10.1016/S0040-4039(01)93748-7
Return to citation in text: [1] [2] -
Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157–160. doi:10.1021/ol990623l
Return to citation in text: [1] [2] [3] -
Ye, W.; Leow, D.; Goh, S. L. M.; Tan, C.-T.; Chian, C.-H.; Tan, C.-H. Tetrahedron Lett. 2006, 47, 1007–1010. doi:10.1016/j.tetlet.2005.11.133
Return to citation in text: [1] [2] -
Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Rodionow, W. M.; Postovskaja, E. A. J. Am. Chem. Soc. 1929, 51, 841–847. doi:10.1021/ja01378a027
Return to citation in text: [1] -
Rodionow, W. M. J. Am. Chem. Soc. 1929, 51, 847–852. doi:10.1021/ja01378a028
Return to citation in text: [1] -
Faulconbridge, S. J.; Holt, K. E.; Garcia Sevillano, L.; Lock, C. J.; Tiffin, P. D.; Tremayne, N.; Winter, S. Tetrahedron Lett. 2000, 41, 2679–2681. doi:10.1016/S0040-4039(00)00222-7
Return to citation in text: [1] [2] -
Tasnádi, G.; Forró, E.; Fülöp, F. Tetrahedron: Asymmetry 2008, 19, 2072–2077. doi:10.1016/j.tetasy.2008.08.009
Return to citation in text: [1] [2] -
Grayson, J. I.; Roos, J.; Osswald, S. Org. Process Res. Dev. 2011, 15, 1201–1206. doi:10.1021/op200084g
Return to citation in text: [1] [2] [3] -
Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024
Return to citation in text: [1] [2] [3] [4] [5] -
Riant, O.; Kagan, H. B.; Ricard, L. Tetrahedron 1994, 50, 4543–4554. doi:10.1016/S0040-4020(01)89385-6
Return to citation in text: [1] [2] [3] [4] -
Akalay, D.; Dürner, G.; Bats, J. W.; Göbel, M. W. Beilstein J. Org. Chem. 2008, 4, No. 28. doi:10.3762/bjoc.4.28
Return to citation in text: [1] [2] -
Akalay, D.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 2365–2368. doi:10.1002/ejoc.200800179
Return to citation in text: [1] [2] [3] [4] -
Uemae, K.; Masuda, S.; Yamamoto, Y. J. Chem. Soc., Perkin Trans. 1 2001, 1002–1006. doi:10.1039/b100961n
Return to citation in text: [1] -
Hau, C.-K.; He, H.; Lee, A. W. M.; Chik, D. T. W.; Cai, Z.; Wong, H. N. C. Tetrahedron 2010, 66, 9860–9874. doi:10.1016/j.tet.2010.10.064
Return to citation in text: [1] -
Zea, A.; Valerao, G.; Alba, A.-N. R.; Moyano, A.; Rios, R. Adv. Synth. Catal. 2010, 352, 1102–1106. doi:10.1002/adsc.201000031
Return to citation in text: [1] -
Zea, A.; Alba, A.-N. R.; Bravo, N.; Moyano, A.; Rios, R. Tetrahedron 2011, 67, 2513–2529. doi:10.1016/j.tet.2011.02.032
Return to citation in text: [1] -
Bai, J.-F.; Guo, Y.-L.; Peng, L.; Jia, L.-N.; Xu, X.-Y.; Wang, L.-X. Tetrahedron 2013, 69, 1229–1233. doi:10.1016/j.tet.2012.11.011
Return to citation in text: [1] -
Kumagai, J.; Otsuki, T.; Reddy, U. V. S.; Kohari, Y.; Seki, C.; Uwai, K.; Okuyama, Y.; Kwon, E.; Tokiwa, M.; Takeshita, M.; Nakano, H. Tetrahedron: Asymmetry 2015, 26, 1423–1429. doi:10.1016/j.tetasy.2015.10.022
Return to citation in text: [1] -
Tawde, T. S.; Wagh, S. J.; Sapre, J. V.; Khose, V. N.; Badani, P. M.; Karnik, A. V. Tetrahedron: Asymmetry 2016, 27, 130–135. doi:10.1016/j.tetasy.2016.01.002
Return to citation in text: [1]
| 29. | Uemae, K.; Masuda, S.; Yamamoto, Y. J. Chem. Soc., Perkin Trans. 1 2001, 1002–1006. doi:10.1039/b100961n |
| 30. | Hau, C.-K.; He, H.; Lee, A. W. M.; Chik, D. T. W.; Cai, Z.; Wong, H. N. C. Tetrahedron 2010, 66, 9860–9874. doi:10.1016/j.tet.2010.10.064 |
| 31. | Zea, A.; Valerao, G.; Alba, A.-N. R.; Moyano, A.; Rios, R. Adv. Synth. Catal. 2010, 352, 1102–1106. doi:10.1002/adsc.201000031 |
| 32. | Zea, A.; Alba, A.-N. R.; Bravo, N.; Moyano, A.; Rios, R. Tetrahedron 2011, 67, 2513–2529. doi:10.1016/j.tet.2011.02.032 |
| 33. | Bai, J.-F.; Guo, Y.-L.; Peng, L.; Jia, L.-N.; Xu, X.-Y.; Wang, L.-X. Tetrahedron 2013, 69, 1229–1233. doi:10.1016/j.tet.2012.11.011 |
| 34. | Kumagai, J.; Otsuki, T.; Reddy, U. V. S.; Kohari, Y.; Seki, C.; Uwai, K.; Okuyama, Y.; Kwon, E.; Tokiwa, M.; Takeshita, M.; Nakano, H. Tetrahedron: Asymmetry 2015, 26, 1423–1429. doi:10.1016/j.tetasy.2015.10.022 |
| 35. | Tawde, T. S.; Wagh, S. J.; Sapre, J. V.; Khose, V. N.; Badani, P. M.; Karnik, A. V. Tetrahedron: Asymmetry 2016, 27, 130–135. doi:10.1016/j.tetasy.2016.01.002 |
| 28. | Akalay, D.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 2365–2368. doi:10.1002/ejoc.200800179 |
| 25. | Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024 |
| 26. | Riant, O.; Kagan, H. B.; Ricard, L. Tetrahedron 1994, 50, 4543–4554. doi:10.1016/S0040-4020(01)89385-6 |
| 1. | Ma, Y.; De, S.; Chen, C. Tetrahedron 2015, 71, 1145–1173. doi:10.1016/j.tet.2014.11.056 |
| 5. | Schmidtchen, F. P. Chem. Ber. 1980, 113, 2175–2182. doi:10.1002/cber.19801130612 |
| 20. | Rodionow, W. M.; Postovskaja, E. A. J. Am. Chem. Soc. 1929, 51, 841–847. doi:10.1021/ja01378a027 |
| 21. | Rodionow, W. M. J. Am. Chem. Soc. 1929, 51, 847–852. doi:10.1021/ja01378a028 |
| 22. | Faulconbridge, S. J.; Holt, K. E.; Garcia Sevillano, L.; Lock, C. J.; Tiffin, P. D.; Tremayne, N.; Winter, S. Tetrahedron Lett. 2000, 41, 2679–2681. doi:10.1016/S0040-4039(00)00222-7 |
| 23. | Tasnádi, G.; Forró, E.; Fülöp, F. Tetrahedron: Asymmetry 2008, 19, 2072–2077. doi:10.1016/j.tetasy.2008.08.009 |
| 4. | McKay, A. F.; Kreling, M.-E. Can. J. Chem. 1957, 35, 1438–1445. doi:10.1139/v57-190 |
| 6. | Echavarren, A.; Galán, A.; de Mendoza, J.; Salmerón, A.; Lehn, J.-M. Helv. Chim. Acta 1988, 71, 685–693. doi:10.1002/hlca.19880710402 |
| 8. | Kurzmeier, H.; Schmidtchen, F. P. J. Org. Chem. 1990, 55, 3749–3755. doi:10.1021/jo00299a013 |
| 10. | Turočkin, A.; Honeker, R.; Raven, W.; Selig, P. J. Org. Chem. 2016, 81, 4516–4529. doi:10.1021/acs.joc.6b00283 |
| 16. | Corey, E. J.; Ohtani, M. Tetrahedron Lett. 1989, 30, 5227–5230. doi:10.1016/S0040-4039(01)93748-7 |
| 17. | Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157–160. doi:10.1021/ol990623l |
| 2. | Ishikawa, T.; Kumamoto, T. Synthesis 2006, 737–752. doi:10.1055/s-2006-926325 |
| 3. | Selig, P. Synthesis 2013, 45, 703–718. doi:10.1055/s-0032-1318154 |
| 9. | Jadhav, V. D.; Schmidtchen, F. P. J. Org. Chem. 2008, 73, 1077–1087. doi:10.1021/jo7021026 |
| 18. | Ye, W.; Leow, D.; Goh, S. L. M.; Tan, C.-T.; Chian, C.-H.; Tan, C.-H. Tetrahedron Lett. 2006, 47, 1007–1010. doi:10.1016/j.tetlet.2005.11.133 |
| 14. | Khuong-Huu, F.; Le Forestier, J.-P.; Goutarel, R. Tetrahedron 1972, 28, 5207–5220. doi:10.1016/S0040-4020(01)88940-7 |
| 15. | Büchi, G.; Rodriguez, A. D.; Yakushijin, K. J. Org. Chem. 1989, 54, 4494–4496. doi:10.1021/jo00280a009 |
| 17. | Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157–160. doi:10.1021/ol990623l |
| 27. | Akalay, D.; Dürner, G.; Bats, J. W.; Göbel, M. W. Beilstein J. Org. Chem. 2008, 4, No. 28. doi:10.3762/bjoc.4.28 |
| 12. | Kita, T.; Georgieva, A.; Hashimoto, Y.; Nakata, T.; Nagasawa, K. Angew. Chem., Int. Ed. 2002, 41, 2832–2834. doi:10.1002/1521-3773(20020802)41:15<2832::AID-ANIE2832>3.0.CO;2-Q |
| 13. | Kita, T.; Shin, B.; Hashimoto, Y.; Nagasawa, K. Heterocycles 2007, 73, 241–247. doi:10.3987/COM-07-S(U)49 |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 11. | Davis, A. P.; Dempsey, K. J. Tetrahedron: Asymmetry 1995, 6, 2829–2840. doi:10.1016/0957-4166(95)00374-X |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 5. | Schmidtchen, F. P. Chem. Ber. 1980, 113, 2175–2182. doi:10.1002/cber.19801130612 |
| 6. | Echavarren, A.; Galán, A.; de Mendoza, J.; Salmerón, A.; Lehn, J.-M. Helv. Chim. Acta 1988, 71, 685–693. doi:10.1002/hlca.19880710402 |
| 7. | Echavarren, A.; Galán, A.; Lehn, J.-M.; de Mendoza, J. J. Am. Chem. Soc. 1989, 111, 4994–4995. doi:10.1021/ja00195a071 |
| 8. | Kurzmeier, H.; Schmidtchen, F. P. J. Org. Chem. 1990, 55, 3749–3755. doi:10.1021/jo00299a013 |
| 9. | Jadhav, V. D.; Schmidtchen, F. P. J. Org. Chem. 2008, 73, 1077–1087. doi:10.1021/jo7021026 |
| 10. | Turočkin, A.; Honeker, R.; Raven, W.; Selig, P. J. Org. Chem. 2016, 81, 4516–4529. doi:10.1021/acs.joc.6b00283 |
| 16. | Corey, E. J.; Ohtani, M. Tetrahedron Lett. 1989, 30, 5227–5230. doi:10.1016/S0040-4039(01)93748-7 |
| 17. | Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157–160. doi:10.1021/ol990623l |
| 18. | Ye, W.; Leow, D.; Goh, S. L. M.; Tan, C.-T.; Chian, C.-H.; Tan, C.-H. Tetrahedron Lett. 2006, 47, 1007–1010. doi:10.1016/j.tetlet.2005.11.133 |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 28. | Akalay, D.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 2365–2368. doi:10.1002/ejoc.200800179 |
| 22. | Faulconbridge, S. J.; Holt, K. E.; Garcia Sevillano, L.; Lock, C. J.; Tiffin, P. D.; Tremayne, N.; Winter, S. Tetrahedron Lett. 2000, 41, 2679–2681. doi:10.1016/S0040-4039(00)00222-7 |
| 23. | Tasnádi, G.; Forró, E.; Fülöp, F. Tetrahedron: Asymmetry 2008, 19, 2072–2077. doi:10.1016/j.tetasy.2008.08.009 |
| 24. | Grayson, J. I.; Roos, J.; Osswald, S. Org. Process Res. Dev. 2011, 15, 1201–1206. doi:10.1021/op200084g |
| 24. | Grayson, J. I.; Roos, J.; Osswald, S. Org. Process Res. Dev. 2011, 15, 1201–1206. doi:10.1021/op200084g |
| 24. | Grayson, J. I.; Roos, J.; Osswald, S. Org. Process Res. Dev. 2011, 15, 1201–1206. doi:10.1021/op200084g |
| 26. | Riant, O.; Kagan, H. B.; Ricard, L. Tetrahedron 1994, 50, 4543–4554. doi:10.1016/S0040-4020(01)89385-6 |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 25. | Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024 |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 25. | Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024 |
| 26. | Riant, O.; Kagan, H. B.; Ricard, L. Tetrahedron 1994, 50, 4543–4554. doi:10.1016/S0040-4020(01)89385-6 |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 25. | Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024 |
| 19. | Shen, J.; Nguyen, T. T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. J. Am. Chem. Soc. 2006, 128, 13692–13693. doi:10.1021/ja064636n |
| 25. | Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024 |
| 26. | Riant, O.; Kagan, H. B.; Ricard, L. Tetrahedron 1994, 50, 4543–4554. doi:10.1016/S0040-4020(01)89385-6 |
| 27. | Akalay, D.; Dürner, G.; Bats, J. W.; Göbel, M. W. Beilstein J. Org. Chem. 2008, 4, No. 28. doi:10.3762/bjoc.4.28 |
| 28. | Akalay, D.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 2365–2368. doi:10.1002/ejoc.200800179 |
| 28. | Akalay, D.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 2365–2368. doi:10.1002/ejoc.200800179 |
© 2016 Goldberg et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)