Abstract
The trans-2-deoxyribosylation of 4-thiouracil (4SUra) and 2-thiouracil (2SUra), as well as 6-azauracil, 6-azathymine and 6-aza-2-thiothymine was studied using dG and E. coli purine nucleoside phosphorylase (PNP) for the in situ generation of 2-deoxy-α-D-ribofuranose-1-phosphate (dRib-1P) followed by its coupling with the bases catalyzed by either E. coli thymidine (TP) or uridine (UP) phosphorylases. 4SUra revealed satisfactory substrate activity for UP and, unexpectedly, complete inertness for TP; no formation of 2’-deoxy-2-thiouridine (2SUd) was observed under analogous reaction conditions in the presence of UP and TP. On the contrary, 2SU, 2SUd, 4STd and 2STd are good substrates for both UP and TP; moreover, 2SU, 4STd and 2’-deoxy-5-azacytidine (Decitabine) are substrates for PNP and the phosphorolysis of the latter is reversible. Condensation of 2SUra and 5-azacytosine with dRib-1P (Ba salt) catalyzed by the accordant UP and PNP in Tris∙HCl buffer gave 2SUd and 2’-deoxy-5-azacytidine in 27% and 15% yields, respectively. 6-Azauracil and 6-azathymine showed good substrate properties for both TP and UP, whereas only TP recognizes 2-thio-6-azathymine as a substrate. 5-Phenyl and 5-tert-butyl derivatives of 6-azauracil and its 2-thioxo derivative were tested as substrates for UP and TP, and only 5-phenyl- and 5-tert-butyl-6-azauracils displayed very low substrate activity. The role of structural peculiarities and electronic properties in the substrate recognition by E. coli nucleoside phosphorylases is discussed.

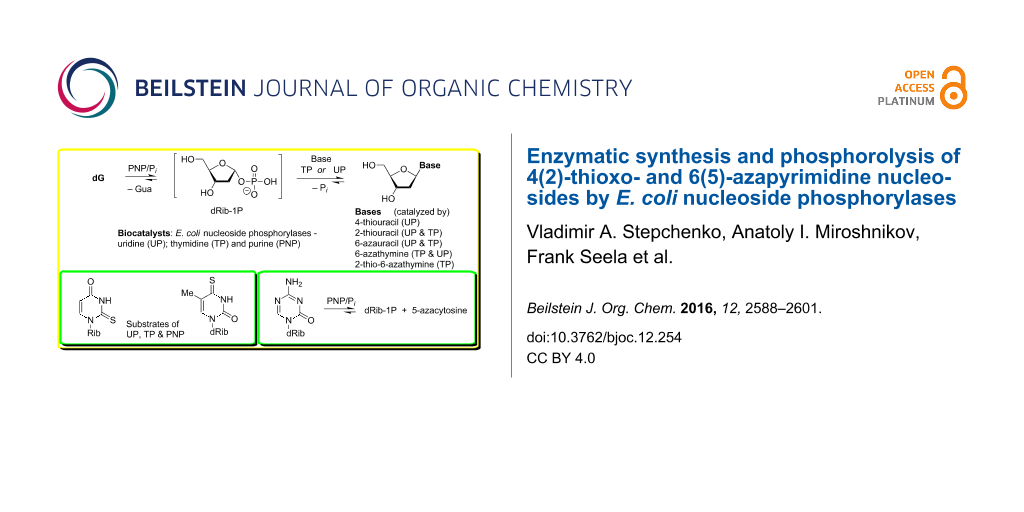
Graphical Abstract
Introduction
Nucleosides of 4- and 2-thioxopyrimidines and 6-azapyrimidines attract much attention from the time of pioneering works in the early 1950s on the chemical synthesis and investigation of their physicochemical and biological properties (early works are reviewed in [1,2]). Studies on thioxo- and azapyrimidine nucleosides are an inspiring subject of investigation due to their very special biochemical [3-7] and biophysical properties in comparison with the natural pyrimidine nucleosides in order to understand the impact of such modifications as monomers or constituents of oligonucleotides [8-14]. Thioxopyrimidine nucleosides as such, as well as building blocks of artificial oligonucleotides demonstrate promising antiviral activity in various experiments [15-22].
Regarding the chemical synthesis of this class of pyrimidine nucleosides various approaches were published (see, e.g., [8-11,14-16,23,24]; reviewed by Vorbrüggen and Ruh-Pohlenz [25]). On the contrary, only few publications are available on the enzymatic synthesis of these nucleosides. W. H. Prusoff reported on the first efficient transformation of 6-azathymine into its 2'-deoxy-D-riboside in phosphate buffer (50 mM, pH 8.0; 37 °C) in the presence of thymidine as a pentofuranose donor and washed cells or cell-free extract of Streptococcus faecalis as biocatalysts [26]. Later on, the conversion of 6-azapyrimidines into their ribonucleosides was observed during the cultivation of Streptococcus faecalis [27,28] and E. coli [29] cells, as well as an enzymatic glycosylation of 14C-labeled 6-azapyrimidines employing the preparation of trans-N-deoxyribosylase from Lactobacillus helveticus NCIB 6557 [30] had been described [31]. As for an enzymatic synthesis of thioxopyrimidine nucleosides, Kalckar [32] as well as Friedkin and co-workers [33,34] disclosed the formation of 2-thiouracil riboside and 2'-deoxyriboside using 2-thiouracil and the corresponding α-D-pentofuranose-1-phosphates (dicyclohexylammonium salts) as substrates of horse liver thymidine phosphorylases (hlTP) (reviewed in [1]).
Recently, Hatano et al. reported on the synthesis of 2-thiothymidine (2STd) and 1-(2-deoxy-β-D-ribofuranosyl)-2-thiouracil (2SUd) by the transglycosylation reaction of the corresponding thioxopyrimidines employing thymidine as a donor of the carbohydrate residue and E. coli TP as a biocatalyst [35]; 6-azauracil (3a) and 6-azathymine (4a) displayed no substrate activity in analogous reactions (cf. [26-29]). These studies prompted us to investigate the enzymatic transformations of 2(4)-thioxo- and 6(5)-azapyrimidines and their nucleosides in more detail and to outline (i) the scope and limitations of the enzymatic synthesis of 2'-deoxy-β-D-ribonucleosides catalyzed by the recombinant E. coli uridine (UP; EC 2.4.2.3) and thymidine (TP; EC 2.4.2.4) phosphorylases [36], and (ii) the role of structural features and electronic properties of the pyrimidine bases and nucleosides in the recognition by E. coli nucleoside phosphorylases.
Results and Discussion
Thioxo- and 6-aza-pyrimidines used in the transglycosylation reaction with recombinant E. coli nucleoside phosphorylases and 2'-deoxyguanosine as a donor of 2-deoxy-D-ribofuranose: The substrate properties of 4-thiouracil (1a; 4SUra), 2-thiouracil (2a; 2SUra), 6-azauracil (3a), 6-azathymine (4a) and 6-aza-2-thiothymine (5a) for the recombinant E. coli TP and UP in the transglycosylation reaction [37,38] using 2'-deoxyguanosine (dG) as a donor of the pentofuranose moiety in a combination with the recombinant E. coli purine nucleoside phosphorylase (PNP; product of deoD gene; EC 2.4.2.1) [36] were tested under standard reaction conditions and then individual nucleosides 1b, 3b–5b were prepared and their structure was proved by the integrity of spectral methods and comparison with the published spectral data. The most essential synthetic results are shown on Scheme 1, 1H and 13C NMR data are presented in Supporting Information File 1, Tables S1 and S2.
![[1860-5397-12-254-i1]](/bjoc/content/inline/1860-5397-12-254-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Enzymatic synthesis of 2-deoxy-β-D-ribofuranosides 1b–5b of the heterocyclic bases 1a–5a. Regents and conditions: dG/base ratio: 1.5:1.0 (mol), 10 mM K,Na-phosphate buffer; 40 °C, 48–72 h; HPLC analysis of the reaction mixtures see Experimental section and Supporting Information File 1.
Scheme 1: Enzymatic synthesis of 2-deoxy-β-D-ribofuranosides 1b–5b of the heterocyclic bases 1a–5a. Regents a...
Enzymatic synthesis and phosphorolysis pathways of 4(2)-thioxopyrimidine nucleosides: 4-Thiouracil (1a; 4SUra) revealed satisfactory substrate activity for E. coli UP giving rise to the formation of 4-thio-2'-deoxyuridine (1b; 4SUd) that was prepared in 39% yield (not optimized). Unexpectedly, 4SUra did show complete inertness for TP. Neither UP nor TP were able to catalyze the transformation of 2-thiouracil (2a) into 1-(2-deoxy-β-D-erythro-pentofuranosyl)-2-thiouracil (2SUd; 2b) under analogous reaction conditions (Scheme 1). The substrate properties of 4-thiouracil clearly point to essential differences in the modes of substrate binding and activation at the catalytic sites of UP and TP. Whereas the substrate recognition of 4-thiouracil by the latter enzyme depends strongly on the electronic structure and/or the van der Waals radius of the substituent at C-4, UP is less sensitive to these differences, in particular, to modification at the C-4 carbonyl group.
The results of the reverse reaction, the phosphorolysis of 4-thio-2'-deoxyuridine (1b; 4SUd) and 4-thiothymidine (11a, 4STd) by UP and TP, are in good agreement with the data on the synthesis (Scheme 2; Figure 1 and Figure 2). Indeed, the phosphorolysis of 4SUd and 4STd by UP proceeds very quickly and reaches equilibrium in the reaction 4SUra (4SThy) + dRib-1P ![[Graphic 2]](/bjoc/content/inline/1860-5397-12-254-i4.svg?max-width=637&scale=0.354546) 4SUd (4STd) + inorganic phosphate (Pi) [base–nucleoside ratio 65(70):35(30)] within several minutes. The phosphorolysis pattern for 4SUd is in satisfactory agreement with the net output (39%) of individual nucleoside obtained in the synthesis. Notably, phosphorolysis of (i) 2'-deoxyuridine (Ud) by UP proceeds at somewhat lower rate, but also reaches an equilibrium point at a similar ratio of uracil-Ud within one hour and then remains practically unchanged, and (ii) thymidine (Td) occurs at a lower rate than Ud and significantly slower than 4SUd, but comes to the similar ratio of the starting substrate and thymine formed after 10 h (Figure 1).
4SUd (4STd) + inorganic phosphate (Pi) [base–nucleoside ratio 65(70):35(30)] within several minutes. The phosphorolysis pattern for 4SUd is in satisfactory agreement with the net output (39%) of individual nucleoside obtained in the synthesis. Notably, phosphorolysis of (i) 2'-deoxyuridine (Ud) by UP proceeds at somewhat lower rate, but also reaches an equilibrium point at a similar ratio of uracil-Ud within one hour and then remains practically unchanged, and (ii) thymidine (Td) occurs at a lower rate than Ud and significantly slower than 4SUd, but comes to the similar ratio of the starting substrate and thymine formed after 10 h (Figure 1).
![[1860-5397-12-254-i2]](/bjoc/content/inline/1860-5397-12-254-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Phosphorolysis of nucleosides 1b–5b and related pyrimidine nucleosides (2’-deoxyuridine, thymidine, 2- and 4-thiothymidines) catalyzed by E. coli UP (Figure 1) and TP (Figure 2).
Scheme 2: Phosphorolysis of nucleosides 1b–5b and related pyrimidine nucleosides (2’-deoxyuridine, thymidine,...
![[1860-5397-12-254-1]](/bjoc/content/figures/1860-5397-12-254-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Phosphorolysis of a number of 2’-deoxy-β-D-ribofuranosides of uracil and thymine, and their 6-aza derivatives in comparison with the corresponding 4- and 2-thio derivatives catalyzed by E. coli UP. Reaction conditions: reaction mixture 1.0 mL; 25 mМ K,Na-phosphate buffer (рН 7.0), 20 °C; 2 mМ testing nucleoside. Enzyme: 0.016 units of E. coli UP (substrates drawn with solids lines) or 1.9 units of E. coli UP (substrates drawn with dotted lines); reaction progress was monitored by HPLC (see Supporting Information File 1); yields refer to the percentage of the resulting heterocyclic base.
Figure 1: Phosphorolysis of a number of 2’-deoxy-β-D-ribofuranosides of uracil and thymine, and their 6-aza d...
On the contrary, (i) the close similarity of substrate activity of Ud and Td towards TP was observed, and (ii) the TP catalyzed phosphorolysis of 4-thiothymidine (11a, 4STd) and 4-thio-2'-deoxyuridine (1b; 4SUd) proceeds very slowly suggesting the negative impact of the 4C=O → 4C=S replacement on the binding or/and activation of both pairs of substrates 4-thiothymine (4SThy) and 4-thiothymidine (4STd), and, to a greater extent, 4-thiouracil (4SUra) and its 2'-deoxyriboside 4SUd (Figure 2).
![[1860-5397-12-254-2]](/bjoc/content/figures/1860-5397-12-254-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Phosphorolysis of 2′-deoxyuridine and thymidine, their 4- and 2-thio derivatives and 6-aza-2-thiothymidine (5b) catalyzed by E. coli TP (for reaction conditions, see caption of Figure 1. Enzyme: 6.6 × 10−4 units of TP was used for all substrates, except for 6-aza-2-thiothymidine, for which 26.5 units of the enzyme was used. Phosphorolysis of 6-aza-2′-deoxyuridine and 6-azathymidine did not exceed a few percent even at high enzyme concentrations and is not presented on the plot.
Figure 2: Phosphorolysis of 2′-deoxyuridine and thymidine, their 4- and 2-thio derivatives and 6-aza-2-thioth...
The inertness of 2SUra towards glycosylation catalyzed by both UP and TP was surprising in the light of the works of Kalckar and Friedkin which studied TP from horse liver [1,32-34]. Moreover, Hatano et al. [35] briefly described a very efficient conversion of 2-thiouracil and 2-thiothymine into the corresponding 2'-deoxyribosides in 54 and 61% yields, respectively, employing thymidine as a donor of the pentofuranose moiety and TP (Sigma) as a biocatalyst; the TP quantity and the reaction conditions are not indicated. The discrepancy between our results and those by Hatano et al. remains unclear. To shed more light into the problem, we reexamined the phosphorolysis profiles of 2'-deoxy-2-thiouridine (2b; 2SUd) and 2-thiothymidine (11b; 2STd) by E. coli UP and TP and compared them with those for corresponding natural nucleosides. We observed that the phosphorolytic cleavage of the glycoside linkages (i) of 2-thioxo analogues and corresponding natural nucleosides by UP proceeds at the same rate at the initial stages of reactions, (ii) of Ud and 2SUd occurs somewhat faster vs that of Td and 2STd, and (iii) of Ud and Td stops at the level of ca. 55% conversion. Contrary to the natural substrates, the reactions of 2SUd and 2STd continued and reached equilibrium at a ratio of substrates ca. 95:5 after 5–7 h (Figure 1). As distinct from UP, phosphorolysis of 2SUd and 2STd catalyzed by TP proceeded much more quickly vs the corresponding natural nucleosides attaining 85–95% conversion in ca. 2 h (Figure 2). In general, replacement of the 2-oxo group by a thioxo function leads to a significant increase of the rate of phosphorolysis of 2SUd and 2STd by E. coli UP (conversion 80–90% in 5–6 h) and to a greater extent, by TP (conversion 90–95% in 2–2.5 h) compared with the respective natural substrates.
In addition, a number of experiments on the synthesis of 2SUd have been conducted and it was verified that (i) the use of thymidine as a 2-deoxy-D-ribofuranose donor (thymidine:2SUra ratio of 2.5:1 (mol); rt, 24 h) at decreasing molarity of phosphate buffer to 0.4 mM (pH 7.0) resulted in the formation of 2SUd in 3% and 25% yields (HPLC) in the reactions catalyzed by UP and TP, respectively; (ii) the reaction of 2SUra and 2-deoxy-α-D-ribofuranose-1-phosphate and E. coli UP in Tris∙HCl buffer resulted in an equilibrium of starting 2SUra and formed 2SUd of ca. 7:3 in the reaction mixture after 1 h, from which the desired nucleoside was isolated by column chromatography in 27% yield (see Experimental section).
The role of hydrogen bonding between Gln166 and the pyrimidine base in the interaction with E. coli UP: The E. coli UP catalyzes the reversible phosphorolysis of a number of the base and pentofuranose modified pyrimidine nucleosides to the corresponding bases and α-D-pentofuranose-1-phosphates (α-D-PF-1P) (reviewed in [1,2,37,38]). The crystal structure of E. coli UP in a complex with substrates was analyzed (see [39,40] and the works cited therein) and the role of some amino acid residues of the catalytic site was characterized by the single-site mutagenesis [41]. It was suggested that the uracil binding site includes Gln166, the carboxamide group of which forms two strong hydrogen bonds 3N(H)···(O=)C(R)-NH2···(O=)C-2. Moreover, the carbonyl group of the Glu166 side-chain forms an additional hydrogen bond with a water molecule, which, in turn, takes a part in hydrogen bonding to the 4C(=O) oxo function of uracil and to the adjacent guanidinium group of Arg223 [40]. In addition, the side-chain of Arg168 is directly hydrogen-bonded to the 4C(=O) of uracil and the hydrogen bonds network of the side-chains of Gln166, Arg223 and Arg168 are responsible for the strict specificity to uracil and to a lesser extent to thymine [5,6] recognizing the 2C(=O)-3N(H)-4C(=O) fragment as distinct from the 2C(=O)-3N=4C(NH2) part of cytosine. Notably, the eight-membered ring comprising two H-bonds formed by the side-chain of Gln166 plays an important role in the phosphorolytic cleavage of the glycoside bond and in all likelihood in the reversed reaction of the glycoside bond formation. It is conceivable that the Gln166/base hydrogen bonding provides the correct positioning of uracil and its closely related analogues, e.g., 5-fluorouracil, thymine and its 5-trifluoro analogue etc, within the catalytic site of E. coli UP, which is accompanied by the substrate activation ensuring the subsequent attack of the N-1 nitrogen atom on the electrophilic C-1 atom of α-D-PF-1P. In the case of E. coli TP, there are controversial suggestions regarding the amino acid residues participating in the nucleoside binding and activation at the catalytic site precluding the similar analysis based on the most important interaction (vide infra).
Suggested mechanistic differences of tautomeric structures of pyrimidines studied and their recognition by E. coli UP vs TP in the glycoside bond formation: Activation of uracil and thymine as well as their related analogues in the chemical synthesis of nucleosides consists in the trimethylsilylation giving rise to the formation of 2,4-di-O-TMS derivatives accompanied by the sp3 → sp2 transformation of the nitrogen atoms. The sp2 hybridized nitrogen atoms attack the electrophilic C-1 carbon atom of the appropriately protected sugar derivatives leading to the glycosylic bond formation (reviewed in [25]). A similar mechanism occurs during the attack of the sp2 hybridized N-1 atom of the pyrimidine base to the anomeric C-1 carbon atom of α-D-PF-1P in the nucleophilic substitution of phosphoric acid residue of the latter catalyzed by E. coli UP and TP.
It was unequivocally proven by the physicochemical and theoretical methods that natural pyrimidine bases exist in the gas phase and in water solution in the canonical 2,4-diketo form (e.g. [42,43]). The tautomeric structures of 4- and 2-thioanalogues of thymine and uracil have been also investigated by diverse methods and the predominant population of the oxo/thioxo tautomers was established [44-47]. We have analyzed the electronic structures of 2,4-diketo and regioisomeric oxo/thioxo tautomers of uracil, 4-thiouracil and 2-thiouracil, as well as their 4(2)-enol(mercapto) forms by the ab initio method (6-31G** level; basic set of parameters). We found that oxo/thioxo structures are thermodynamically more stable by 10–15 kcal/mol on all the occasions; similar analysis of 6-azapyrimidines showed even higher thermodynamic stability of the keto/thioketo tautomers by 20–30 kcal/mol (Supporting Information File 1, Table S3).
Furthermore, the monoanionic forms of 4-thiouracil and 2-thiouracil in aqueous medium were analyzed by the UV and IR spectroscopic methods and the structure 6 with charge delocalization for the former and two tautomeric monoanions 7a and 7b (ca. 1:1) with charge localization on the C-4 oxygen atom for the latter were suggested [48,49] (Figure 3).
![[1860-5397-12-254-3]](/bjoc/content/figures/1860-5397-12-254-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Supposed monoanionic forms of 4-thiouracil and 2-thiouracil in aqueous medium [48,49].
Figure 3: Supposed monoanionic forms of 4-thiouracil and 2-thiouracil in aqueous medium [48,49].
The aforementioned observations, as well as the very likely main contribution of the side-chain of Glu166 in the correct positioning of a pyrimidine substrate at the E. coli UP catalytic site suggests a possible mechanism of the activation of the substrate and we became interested in whether this interaction contributes to the sp3 → sp2 transformation of the N-1 nitrogen atoms. With this aim in view, we constructed and geometry optimized [Bio+(CHARMM27) force field] and then re-optimized using semi-empirical PM3 method (HyperChem 8.1) the eight-member cyclic structures comprising two hydrogen bonds between acetamide (AA) as a Gln166 side-chain mimic and tautomers of bases with sp3 (8a) vs sp2 (8b and 8c) hybridized N-1 nitrogen atoms (Table 1; for details, see Supporting Information File 1, Table S4).
Table 1: Geometry optimized supposed tautomeric structures of uracil and bases 1a–5a at E. coli UP catalytic site with sp3 and sp2 hybridized N-1 nitrogen atoms (thermodynamically stable tautomers are highlighted in boldface, and the more stable N-1 sp2 structures are in italics and underlined)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-254-i3.svg?max-width=637&scale=1.0)
|
|||||||
| Substrate |
ETOTAL kcal/mol
(ΔETOTAL)b |
Partial charge of the N-1 atom (e) |
ETOTAL kcal/mol
(ΔETOTAL)b |
Partial charge of the N-1 atom (e) |
ETOTAL kcal/mol
(ΔETOTAL)b |
Partial charge of the N-1 atom (e) | Remarks |
|---|---|---|---|---|---|---|---|
| uracil | –51,160.1 | 0.083 | −51,151.2 (+8.9) | −0.202 | −51,144.0 (+16.1) | −0.214 | UP and TP accept uracil as a substrate |
| 4-thiouracil (1a) | –48,654.5 (+6.5) | 0.114 | −48,661.0 | −0.166 | −48,640.4 (+20.6) | −0.167 | only UP recognizes 1a as a substrate |
| 2-thiouracil (2a) | –48,657.0 (+0.7) | 0.197 | −48,644.0 (+13.0) | −0.128 | −48,657.7 | −0.146 | TP recognizes 2a better vs UP |
| 6-azauracil (3a) | –51,792.7 | 0.050 | −51,783.7 (+10.0) | −0.136 | −51,779.9 (+12.8) | −0.191 | UP recognizes 3a better vs TP |
| 6-azathymine (4a) | –55,244,3 | 0.057 | −55,235.6 (+8.7) | −0.132 | −55,234.0 (+10.3) | −0.193 | TP recognizes 4a better vs UP |
|
6-aza-2-thio-
thymine (5a) |
–52,739.7 (+1.1) | 0.174 | −52,731.1 (+9.7) | −0.064 | −52,740.8 | −0.109 | only TP accapts 5a as a substrate |
aOnly total energy values (ETOTAL, kcal/mol) and partial charges (e) of the N-1 atoms are given (for detailed information, see Supporting Information File 1, Table S4); thermodynamically stable tautomers are highlighted in boldface, and the more stable N-1 sp2 structures are highlighted in italic and underlined. bThe ΔETOTAL values (are given in parenthesis; kcal/mol) mean the differences between the ETOTAL values of the thermodynamically most stable tautomer and the less stable tautomers.
The uracil/AA tautomer with the sp3 hybridized N-1 atom is thermodynamically more stable by 9 and 16 kcal/mol vs those of the N-1 sp2 structures implying that (i) the base/Gln166 hydrogen bonding alone is not apparently sufficient to realize the sp3 → sp2 transformation, and (ii) the concerted interaction of side-chains of the triad Gln166, Arg223 and Arg168 of the UP catalytic site makes it possible to overcome the barrier of 9 kcal/mol required to activate the substrate. At the same time, the corresponding sp2 structures 8b and 8c are characterized by the highest partial charges of the N-1 atoms (−0.202/−0.214 e) reflecting the nucleophilicity of these atoms that attack the electrophilic C-1 of α-D-PF-1P.
Among the bases analyzed, only the sp2 N-1 form of the 4-thiouracil/AA structure 8b is thermodynamically favorable over the N-1 sp3 tautomer 8a by −6 kcal/mol and the partial charge of the N-1 is calculated to be −0.166 e, which is compatible with its satisfactory substrate activity for E. coli UP. Under similar reaction conditions, 4-thiouracil revealed no substrate activity against E. coli TP suggesting the various types of the substrate binding and activation at E. coli UP and TP catalytic sites in spite of the some similarity of the interaction of the respective side-chains of Arg168 (UP) [40] and Arg171 (TP) [50] with a substituent at the C-4 atom. Notably, phosphorolysis of 4STd and 4SUd by TP (Figure 2 and Figure 4) proceeds essentially slower vs the one catalyzed by UP (Figure 1). It appears to be reasonable that TP manifests strict requirements to dimension and electronic structure of the C-4 substituent [4], as well as the TP catalyzed transformations depend in a greater extent on the experimental condition [51].
The formation of the productive complex matching the structure 8b at the catalytic site of E. coli UP [40] corresponds well to the spectroscopic data for the mono-anionic form of the tautomer 6 of 4-thiouracil [48,49] (Figure 3). Decrease in the rate of the transglycosylation reaction of 4-thiouracil vs uracil can be explained by the decrease of the hydrogen binding capacity of C-4 sulfur atom compared with the oxygen (cf. the corresponding data for uracil and 4-thiouracil, Supporting Information File 1, Tables S3 and S4) and to some extent by a distortion of architecture of the productive complex due to differences of the van der Waals radii of the substituents at the C-4 atom.
In the case of 2-thiouracil, two tautomeric structures 8a and 8c are characterized by rather similar thermodynamic parameters, which implies the possibility of the binding and activation of 2-thiouracil due only to the base/Gln166 hydrogen bonding at the E. coli UP catalytic site. Unfortunately, there exist different assumptions concerning the amino acid residues of the E. coli TP catalytic site that are directly involved in the hydrogen bonding with pyrimidines and types of these interactions [3,52-56] (vide infra). Indeed, except for the side-chain Arg171/C-4 carbonyl interaction, several variants of direct hydrogen binding between the side-chains of Ser186 and Lys190 and the respective 3N(H) and 2C(=O) functions of substrate (open form) and its activation (catalytically competent closed form) are proposed, wherein there is no prevailing factor determining the substrate recognition (as opposed to E. coli UP), giving an impetus to the sp3 → sp2 transformation of the N-1 nitrogen atom. Moreover, Gago et al. had analyzed the possibility of changing the direct 2C(=O)/Lys190 (side-chain) interaction of thymidine in the open form on the mixed type of the direct hydrogen bonding of C-2 carbonyl with the side-chains of Lys190 and His85 in a transitional state and then to the direct 2C(=O)/His85 hydrogen bonding of thymine (product of reaction) in a closed form [3] (cf. [52,55]). This uncertainty does not allow identifying the most important type of hydrogen bond and analyzing its impact on the N-1 sp3 → sp2 transformation of the substrate.
The enzymatic synthesis of 6-azapyrimidine nucleosides: the C-2 thioxo motif in the reactions catalyzed by E. coli nucleoside phosphorylases: Like uracil, complexes of 6-azauracil (3a; aUra) and 6-azathymine (4a; aThy) with Gln166 with sp3-hybridized bases of the type 8a are thermodynamically more stable (Table 1); each of the bases is a substrate for both enzymes, UP and TP, and the glycosylation efficiency depends, as might be expected, on the C-5 substituent of the heterocycle, viz., glycosylation of 6-azauracil occurs more efficiently under UP catalysis, whereas the 6-azathymine synthesis proceeds more efficiently in the presence of TP and these reactions strongly displaced to the nucleoside formation (Scheme 1).
As distinct from 2-thiouracil, 6-aza-2-thiothymine (5a) was found to be a satisfactory substrate for E. coli TP and 6-aza-1-(2-deoxy-β-D-erythro-pentofuranosyl)-2-thiothymine (5b) was prepared in 50% yield (not optimized). Analysis of 6-aza-2-thiothymine/Gln166 complexes revealed that, like 2-thiouracil, more stable is the structure 8c; however, unlike 2-thiouracil and 6-azathymine, only TP catalyzed reversible glycosylation of 5a (see phosphorolysis of 5b, Figure 2 and Figure 4) that proceeds satisfactorily. On the contrary, UP is not able to catalyze the nucleophilic attack of the structure 8c of 2SaThy owing probably to the lower nucleophilicity of the N-1 nitrogen atom (−0.109 e) compared to 2SUra for which the partial charge of −0.146 e of the N-1 atom was calculated (Table 1). With respect to E. coli TP, the glycosylation of 6-azathymine (4a) appears to proceed virtually irreversibly (data not shown) with somewhat higher efficacy compared to 6-aza-2-thiothymine (5a) and, as a consequence, results in the high yield of the desired 2'-deoxyriboside 4b. Replacement of the C-2 oxygen atom of 6-azathymine (4a) by a sulfur atom changes the pattern of the TP catalyzed glycosylation of 6-aza-2-thiothymine (5a), imparting the reaction reversible character (Figure 4).
![[1860-5397-12-254-4]](/bjoc/content/figures/1860-5397-12-254-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Phosphorolysis of 6-aza-2-thiothymidine (5b), 4-thiothymidine (11a) and 4-thio-2′-deoxyuridine (1b) by E. coli TP for extended time period (for details, see caption of Figure 2).
Figure 4: Phosphorolysis of 6-aza-2-thiothymidine (5b), 4-thiothymidine (11a) and 4-thio-2′-deoxyuridine (1b)...
UP was practically unable to catalyze the synthesis of the nucleoside 5b (Figure 1). Again, unlike the base/Gln166 H-bonding mode, another kind of the interaction of the C-2 thioketo or mercapto function with the side-chain(s) of amino acid residue(s) of the TP active center plays a crucial role in the establishment of the observed 5a + dRib-1P 5b + Pi equilibrium.
As mentioned above, we did not observe the formation of 1-(2-deoxy-β-D-erythro-pentofuranosyl)-2-thiouracil (2SUd; 2b) under standard reaction conditions of the trans-2'-deoxyribosylation (Scheme 1), contrary to the previously published data [1,33-35] and speculated that in our experiment, the reaction equilibrium is almost completely displaced to the starting substrates, 2SUra and dRib-1P (vide supra). Indeed, we have earlier proved that replacement of the C-2 carbonyl group of 1-(2,3-dideoxy-3-fluoro-β-D-erythro-pentofuranosyl)thymine (FLT), which is strongly resistant towards E. coli TP [16,57], with the thiocarbonyl group gives 2-thio-FLT derivative that is phosphorolyzed by E. coli TP [16]. Later on, Gago et al. found that E. coli TP showed lower affinity (Km) for 2-thiothymidine vs that of thymidine (1.587 vs 0.8 mM), and vice versa maximal initial velocity value (Vmax) for 2STd is higher (65.4 vs 24.2 nanoM × mL−1·min−1) pointing to a higher substrate activity of 2STd vs that of thymidine in the phosphorolysis. In the present paper, we have shown that in the reactions catalyzed by UP, conversion of natural substrates, Ud and Td, to products stops at ca. 55%, while the phosphorolysis of the corresponding C-2 thio analogues reaches more than 90% (Figures 1, 2 and 4). In this regard, the absence of substrate activity of 2-thiouridine towards UP and TP observed recently [5,58] appears to be completely unexpected since it implies a dramatic change of the substrate specificity of the enzyme.
Substrate properties of related nucleosides and bases for E. coli UP, TP and PNP: The aforementioned contradictory data prompted us to test the substrate properties of 2-thiouridine (9), 2-thio-5-methoxyuridine (10), 4-thiothymidine (11a), 2-thiothymidine (11b), 6-methyl-2-thiouridine (12), 5-azacytidine (13; aC) and 5-aza-2′-deoxycytidine (14; aCd; anticancer drug Decitabine) (Figure 5) [25,59] for E. coli UP, TP and PNP (for reaction conditions, see Experimental section).
![[1860-5397-12-254-5]](/bjoc/content/figures/1860-5397-12-254-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Structures of 2-thiopyrimidine(9–12) and 5-azacytidine (13 and 14) nucleosides.
Figure 5: Structures of 2-thiopyrimidine(9–12) and 5-azacytidine (13 and 14) nucleosides.
We found that compounds 9–11a,b are good substrates for both UP and TP (see also Figures 1, 2 and 4); 6-methyl-2-thiouridine (12) showed no substrate activity for both nucleoside phosphorylases. This latter observation points to the importance of the syn/anti base orientation around the glycosyl bond in the definition of the substrate properties of pyrimidine nucleosides towards UP and TP. It should be noted that (i) 6-methyl-2'-deoxyuridine (6MeUd) was shown to undergo an irreversible phosphorolysis in the E. coli living cells or in the presence of non-dialyzed cell-free extract from E. coli B and the 15T-mutant [60] and (ii) 6-methyluridine is very week substrate for UP [42,61]. Analysis of the NMR spectra of 6MeUd in D2O pointed to the predominant base population in the syn-conformation [62]. The stereochemistry of 6-methyluridine was also investigated by physicochemical methods [63-65] and in crystal [66] and the dominating syn base conformation around the glycosyl bond was established.
We have earlier shown that 2'-deoxy-β-D-ribosides of cytosine, uracil and thymine are substrates for E. coli PNP (Cd >>> Ud >> Td), whereas the corresponding ribosides devoid of substrate activity [67]. The substrate activity of 5-aza-2'-deoxycytidine (14) (but not 5-azadeoxycytidine (13)) and 4-thiothymidine (11a) for PNP (aCd >>> 4STd) correlate well with that of the corresponding natural 2-deoxyribosides. Unexpectedly, 2-thiouridine (9) showed weak activity for PNP whereas 2-thiothymidine (11b) entirely was lacking such an activity. It is conceivable that in the case of such unusual substrates of PNP as pyrimidine nucleosides their structural features define the substrate properties, viz., 2-thiouridine (very weak substrate) vs uridine (non-substrate), thymidine (very weak substrate) vs 2-thiothymidine (non-substrate).
Shugar and co-workers studied the substrate properties of N-3-regioisomers of adenosine and inosine, N3-(β-D-ribofuranosyl)-adenine (N3-Ado) and -hypoxanthine (N3-Ino), respectively, towards the calf-spleen and E. coli PNPs and disclosed their satisfactory substrate activity [68]. The authors discussed the results in terms of similar spatial organization of natural nucleosides and their N-3-isomers admitting the binding and activation of the latter at the catalytic sites of the PNPs. Noteworthy that phosphorolysis of both N-3-regioisomers proceeds irreversibly. In the present work, we compared the energy minimized structures of N3-Ado and 5-aza-2'-deoxycytidine (14) and found a rather similar stereochemistry of both structures, in particular, a quite similar orientation of pyrimidine and pentofuranose rings (Figure 6). Moreover, the phosphorolysis of the latter was reversible and the reaction of 5-azacytosine with 2-deoxy-α-D-ribofuranose-1-phosphate (Ba salt) (1:1 molar ratio; 2 mM) in the presence of E. coli PNP (10 units) in 50 mM Tris∙HCl buffer (pH 7.0) at 20 °C for 30 min resulted in the formation of nucleoside 14 in 15% yield (HPLC; System A, tR, min: 5-azacytosine – 1.6, 5-aza-2'-deoxycytidine – 5.3).
![[1860-5397-12-254-6]](/bjoc/content/figures/1860-5397-12-254-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Energy minimized structures of N3-(β-D-ribofuranosyl)adenine (left) and 5-aza-2′-deoxycytidine (right) and in the mid both structures are overlapped by the glycosyl bonds (calculations by ab initio, 3-21G level; Polak–Ribiere (conjugate gradient); basis set of parameters; HyperChem 8.1).
Figure 6: Energy minimized structures of N3-(β-D-ribofuranosyl)adenine (left) and 5-aza-2′-deoxycytidine (rig...
An analogous comparative analysis of stereochemistry of N3-Ino and 2-thiouridine (9) gave very similar results (data not shown). It is obvious that in the case of such non-conventional substrates of E. coli PNP, introduction of any additional factors giving rise to spatial distortion (e.g. 2-thioxo function) will lead to the loss of substrate activity.
Substrate properties of C-5-phenyl and C-5-t-butyl substituted 6-azauracil and its C-2-thioxo derivatives: In addition, we evaluated the tolerance of E. coli UP and TP with respect to the bulkiness of the substituents at the C-5 carbon atom of pyrimidines and for this purpose the substrate properties of a number of derivatives of 6-azapyrimidines 15–18 were tested (Figure 7).
![[1860-5397-12-254-7]](/bjoc/content/figures/1860-5397-12-254-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Structures of 6-azapyrimidines 15–18 tested for E. coli UP and TP.
Figure 7: Structures of 6-azapyrimidines 15–18 tested for E. coli UP and TP.
Beginning with the pioneering works of Friedkin and co-workers (testing of 5-amino- and halogeno derivatives of uracil for TP from horse liver) and Heidelberger and co-workers (practical synthesis of 5-fluoro- and 5-trifluoromethyl-2'-deoxyurines) substrate properties of 5-substituted uracil derivatives were studied in a number of publications (reviewed in [1,2,37]) due to the great potential of these bases and nucleosides for the treatment of tumours and viral diseases. 5-Ethyl- and (E)-5-(2-bromovinyl)uracil and C-5 halogenated derivatives of uracil revealed good substrate activity for TP and/or UP employed within the whole E. coli cells and the β-D-2'-deoxyribosides of aforementioned bases have been synthesized in 55–65% yields [1,37,69]. Recently, the comparative effectiveness of immobilized pyrimidine nucleoside phosphorylase from Bacillus subtilis (BsPyNP) and E. coli TP in (i) the phosphorolysis of thymidine, uridine and its deoxy- (2'- and 5'-monodeoxy and 2',3'-dideoxy) and arabino derivatives, and (ii) the conversion of 5-fluoro (Br, I, CF3 and –CH=CHBr)-uracil into respective 2'-deoxyribosides (2'-deoxyuridine as a donor of the sugar moiety) was studied in detail by Ubiali et al. [70].
Tested in this paper 6-azapyrimidines 15–18 turned out to be extremely poor substrates for both E. coli TP and UP and the formation only 2'-deoxyribosides of 6-aza-5-tert-butyluracil 15 (ca. 1%) and 6-aza-5-phenyluracil 16 (ca. 2%) was detected by the HPLC/MS analysis of the respective reaction mixtures (data not shown). Thus, both enzymes demonstrated severe restrictions on the bulkiness and three-dimensional structure of the substituent at the C-5 atom of the pyrimidines. The spatial organization of the very poor substrates 5-tert-butyluracil (15) and 5-phenyluracil (16) vs good substrates 5-ethyluracil and (E)-5-(2-bromovinyl)uracil was evaluated by a geometry optimization employing the ab initio method and data are presented in Figure 8. These data suggest that the structure of the catalytically competent substrate-enzyme complex cannot accommodate spatially dispersed bases 15 and 16 in a rather tight place of the catalytic site of the enzymes.
![[1860-5397-12-254-8]](/bjoc/content/figures/1860-5397-12-254-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Geometry optimized structures (PM3 method) of 5-tert-butyl-6-azauracil (15) and 5-phenyl-6-azauracil (16) (upper structures) vs those of 5-ethyluracil and (E)-5-(2-bromovinyl)uracil (lower structures).
Figure 8: Geometry optimized structures (PM3 method) of 5-tert-butyl-6-azauracil (15) and 5-phenyl-6-azauraci...
Conclusion
The substrate properties of 4(2)-thioxo- and 6-azapyrimidines as well as their nucleosides for E. coli UP and TP have been investigated leading to a number of observations that are of importance for the biotechnology of pyrimidine nucleosides. Only E. coli UP can effectively catalyze the N1-2'-deoxy-D-ribosylation of 4-thiouracil; 2'-deoxy-4-thiouridine (4SUd) and 4-thiothymidine (4STd) are excellent substrates for the enzyme, whereas phosphorolysis of Ud and Td proceeded with a somewhat lesser efficiency. 4SUd and 4STd revealed moderate substrate activity for E. coli TP. Thus, in the case of TP, the 4C(=O) → 4C(=S) replacement resulted in the loss of substrate activity of the pyrimidine base and in an decrease of substrate activity of 4STd and to a greater extent of 4SUd (the absence of the 5C-methyl group!) vs the relevant natural nucleosides, Td and Ud. Completely opposite effects were observed in the case of UP, viz., the substrate activity of 4SUra vs that of uracil is marginally diminished (data not shown), whereas the substrate activity of 4SUd and 4STd is essentially enhanced (without discriminating 5C-H/Me) compared with the respective parent Ud and Td, where UP showed slight substrate preference for the former.
Replacement of the 2C(=O) carbonyl of Ud and Td with thiocarbonyl function resulted in a dramatic enhancement of the substrate activities of 2SUd and 2STd vs those of the parent nucleosides [5,58]. The reactions 2SUra (2SThy) + dRib-1P 2SUd (2STd) + Pi catalyzed by UP and to a greater extent by TP are displaced to the initial bases; as a consequence, 2SUd was synthesized in 27% yield by the reaction of 2SUra and dRib-1P in Tris∙HCl buffer in the presence of UP.
An enzymatic trans-2-deoxyribosylation of 6-azauracil (3a) and 6-azathymine (4a) catalyzed by UP and TP proceeds practically irreversibly [26-29,35]. In contrast, only TP accepts 6-aza-2-thiothymine (5a) as a substrate yielding its 2'-deoxyriboside 5b as a result of the reversible glycosylation pointing again to the surprising ability of the 2C(=S) sulfur atom to enhance the substrate activity of the relevant nucleosides.
2-Thiouridine (9) (but not uridine [67] and 2-thiothymidine (11b)) and 4-thiothymidine (11a) (like thymidine [67]) are weak substrates for E. coli PNP. The most unexpected finding consists in that the phosphorolysis of 5-aza-2'-deoxycytidine (14; anticancer drug Decitabine) catalyzed by E. coli PNP is reversible and condensation of 5-azacytosine with 2-deoxy-α-D-ribofuranose-1-phosphate resulted in the formation of nucleoside 14.
The C-5 tert-butyl and phenyl derivatives of 6-azapyrimidines 15 and 16 as well as their C-2 thio-counterparts 17 and 18 showed negligible substrate activity for E. coli UP and TP pointing to an excess of the allowed bulkiness of C-5 substituent compared with ethyl and bromovinyl groups, respectively.
Experimental
General methods
All chemicals and solvents were of laboratory grade as obtained from commercial suppliers and were used without further purification. The NMR and UV–vis spectra were recorded on Brucker Avance 500-DRX (Bruker, Germany) and Carry 100 spectrometers (Varian, USA), respectively. TLC was performed on TLC aluminium sheets covered with silica gel 60 F254. Low resolution mass spectra were measured on a LCQ Fleet ion trap mass spectrometer (Thermo Electron, USA) in 80% aq acetonitrile.
HPLC system: HPLC COMPACT Pump 2050 with Lambda 1010 UV detector (BISCHOFF Chromatography, Germany); for chromatographic conditions and retention times (tR) see Supporting Information File 1. The 1H and 13C NMR spectra of nucleosides synthesized are given in Supporting Information File 1.
Flash column chromatography was carried out on silica gel 60, 35–70 µm (Merck, USA).
The following recombinant E. coli enzymes [16] were used in the present study: UP, a solution in 5 mM potassium phosphate buffer (pH 7.5) with activity of 18.7 IU/mL, and TP, a solution in 5 mM potassium phosphate buffer (pH 7.0) with activity of 265 IU/mL; PNP (the product of the deoD gene; EC 2.4.2.1) specific activity 54 IU per mg, 17 mg per mL. All the reactions were conducted at room temperature and pH 7.0 if it is not stated otherwise.
2-Deoxy-α-D-ribofuranose-1-phosphate (barium salt) was synthesized as described previously [71].
Initial test for substrate activity in the transglycosylation reaction: Total volume of reaction mixture was 4 mL: 5 mM K,Na-phosphate buffer, 1 mM 2'-deoxyguanosine, 0.8 mM tested base, 40 units of PNP, 3 units of UP or TP; 40 °C. Reaction progress was monitored by HPLC (System A; see Supporting Information File 1).
Phosphorolysis conditions (Figures 1,2 and 4): Total volume of reaction mixture was 1 mL: 25 mM K,Na-phosphate buffer, 2 mM tested nucleoside: 0.016 units of UP or 6.6 × 10−4 units of TP (substrates drawn with solid lines); 1.9 units of UP or 26.5 units of TP (substrate drawn with dotted lines); room temperature. Reaction progress was monitored by HPLC (System B or C; see Supporting Information File 1).
Substrate properties of pyrimidine nucleosides 9–14 for E. coli UP, TP and PNP: Total volume of reaction mixture was 2 mL; nucleoside 1 mg/mL, 50 mM K,Na-phosphate buffer (pH 7.0), 150 IU of the corresponding enzyme, room temperature, 1 h; phosphorolysis (HPLC) of: 9 – 62% (UP), 97% (TP), 13% (PNP); 10 – 92% (UP), 96% (TP); 11a – 34% (UP), 87% (TP), 22% (PNP); 14 – 58% (PNP); no phosphorolysis was observed in the case of UP and TP.
4-Thio-2'-deoxyuridine (1b): 4-Thiouracil (100 mg, 0.780 mmol) and 2′-deoxyguanosine (313 mg, 1.17 mmol) were suspended in 10 mM K,Na-phosphate buffer (40 mL), PNP (1200 units) and UP (238 units) were added and reaction mixture was stirred at 40 °C for 48 h, and the formation of the nucleoside 1b was monitored by HPLC (system A). After 20 h precipitated guanine was filtered off; silica gel (2 mL) was added to the filtrate and the solvent was removed in vacuo. The residue was co-evaporated with ethanol (10 mL) and put on the top of silica gel column (2 × 10 cm) that was eluted with chloroform/methanol, 30:1 (v/v). 4-Thio-2'-deoxyuridine (1b) was obtained as yellow powder (74 mg, 0.303 mmol, 39%), that was recrystallized from methanol; mp 164–165 °C; Lit. data [72]: 154–155 °C (from ethanol); UV (H2O) λmax, nm (ε, M−1∙cm−1): 331 (27,700) and 245 (4,800) at pH 7.0; 316 (24,600) and ca. 230 (shoulder; ca. 7,500) at pH 10.0; 331 (26,800) and 245 (4,640) at pH 4.0; λmin: 276 (1,600) and 225 (3,200) at pH 7.0; 258 (3,400) at pH 10.0, 276 (1,600) and 225 (3,000) at pH 4.0 [72,73]; ESIMS (positive ion mode): 267 [M + Na]+; ESIMS (negative ion mode): 243 [M – H]−
![[1860-5397-12-254-9]](/bjoc/content/figures/1860-5397-12-254-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: The UV spectra of 4-thio-2′-deoxyuridine (1b).
Figure 9: The UV spectra of 4-thio-2′-deoxyuridine (1b).
2-Thio-2'-deoxyuridine (2b): 2-Thiouracil (40 mg, 0.31 mmol), 2-deoxy-α-D-ribofuranosyl phosphate barium salt (105 mg, 0.30 mmol) and barium acetate (40 mg, 0.16 mmol) were dissolved in 20 mM Tris∙HCl buffer (pH 7.3). UP (10 units) was added, reaction mixture was gently stirred at room temperature and the formation of the nucleoside 2b was monitored by HPLC (System C). After 1 h, equilibrium between the starting base and its nucleoside was established, the reaction mixture was filtrated, silica gel (2 mL) was added to the filtrate, and the solvent was removed in vacuo. The residue was co-evaporated with ethanol (10 mL) and put on the top of silica gel column (2 × 10 cm) that was eluted with chloroform/methanol, (10:1, v/v). 2-Thio-2'-deoxyuridine (2b) was obtained as white powder (20 mg, 0.097 mmol, 27%) of 98.7% purity; lit. data [74]: 133–134 °C (from abs. MeOH and technical hexane); UV (H2O) λmax, nm (ε, M−1∙cm−1): 217 (15,000) and 274 (12,950); (1 N NaOH) 238 (19,600) and 269 (19,600); UV (H2O) λmax, nm (ε, M−1∙cm−1): 272 (13,100) and 218 (14,700) at pH 7.0; 263 (13,700) and ≈240 (sh, ≈10,000) at pH 10; 275 (13,700) and 217 (16,400) at pH 4; λmin: 240 (8,100) at pH 7.0; 216 (9,200) at pH 10.0; 240 (8,300) at pH 4.0.; ESIMS (positive ion mode): 245 [M + H]+; ESIMS (negative ion mode): 195 [M − •C5'H2OH − •O3'H – H]−.
6-Aza-2'-deoxyuridine (3b): 6-Azauracil (106 mg, 0.937 mmol) and 2'-deoxyguanosine (376 mg, 1.406 mmol) were suspended in 10 mM K,Na-phosphate buffer (10 mL), UP (238 units) and PNP (11 units) were added and reaction mixture was stirred at 40 °C for 60 h. After that time, the HPLC (Sytem A) indicated almost complete transformation of starting base into nucleoside 3b. Precipitated guanine was filtered off; silica gel (3 mL) was added and the mixture was evaporated to dryness. The residue was twice co-evaporated with ethanol (10 mL) and put on the top of silica gel column (2 × 15 cm) that was eluted with chloroform/methanol, 15:1 (v/v). 6-Aza-2′-deoxyuridine (3b, 193 mg, 0.842 mmol, 90%) was obtained as colorless oil [30,75]. UV (H2O) λmax, nm (ε, M−1∙cm−1): 262 (4,000) at pH 7.0, 255 (4,300) at pH 10.0, 263 (4,000) at pH 4.0; λmin: 231 (1,900) at pH 7.0, 224 (2,300) at pH 10.0, 232 (1,900) at pH 4.0. Lit. data [9]: UV (phosphate buffer): λmax, nm (ε, M−1∙cm−1): 262 (6,900) at pH 5.0, 258 (6,900) at pH 7.0, 255 (7,200) at pH 9.0; ESIMS (positive ion mode): 252 [M + Na]+, 273 [M + 2Na − 2H]+; ESIMS (negative ion mode): 228 [M – H]+.
6-Azathymidine (4b): 6-Azathymine (100 mg, 0.787 mmol) and 2'-deoxyguanosine (315 mg, 1.18 mmol) were suspended in 10 mM K,Na-phosphate buffer (20 mL), TP (390 units) and PNP (400 units) were added and reaction mixture was stirred at 40 °C for 72 h. After that time HPLC (System A) indicated almost complete conversion of starting base into its nucleoside. Precipitated guanine was filtered off and filtrate was evaporated to dryness. The residue was dissolved in methanol, filtrated and to the filtrate silica gel (2 mL) was added. The mixture was evaporated to dryness and the residue was put on top of a silica gel column (2 × 10 cm) that was eluted with chloroform/methanol, 15:1 (v/v). 6-Azathymidine (153 mg, 0.630 mmol, 80%) was obtained as colorless oil [30,75]. UV (H2O) λmax, nm (ε, M−1∙cm−1): 263 (5,300) at pH 4.0, 263(5,300) at pH 7.0, 252 (6,000) at pH 10.0; λmin: 234 (2,400) at pH 4.0, 234 (2,300) at pH 7.0, 223 (3,800) at pH 10.0. Lit. data [26]: UV λmax, nm (ε, M−1∙cm−1): 250 (0.1 N HCl) and 265 (0.1 N NaOH); λmin, nm: 225 and 235. ESIMS (positive ion mode): 266 [M + Na]+, 282 [M + K]+; ESIMS (negative ion mode): 242 (M – H]+.
6-Aza-2-thiothymidine (5b). 2-Thio-6-azathymine (28 mg, 0.193 mmol) and 2′-deoxyguanosine (77 mg, 0.288 mmol) were suspended in 10 mM K,Na-phosphate buffer (15 mL), PNP (274 units) and TP (145 units) were added and the reaction mixture was stirred at 40 °C and the formation of the products was monitored by HPLC (System A). After 72 h the reaction rate significantly decreased. Precipitated guanine was filtered off; silica gel (2 mL) was added to the filtrate and solvent removed in vacuum. The residue was co-evaporated with ethanol (10 mL) and put on top of silica gel column (2 × 10 cm) that was eluted with chloroform/methanol, 30:1 (v/v). 2-Thio-6-azathymidine (5b) was obtained as white powder (25 mg, 0.097 mmol, 50%), that was recrystallized from acetonitrile; mp 174–176 °C.
UV spectra (H2O): λmax, nm (ε, M−1∙cm−1): 272 (18,100) and 220 (11,900) at pH 7.0; 263 (21,800) and ca. 238 (shoulder; ca. 14,000) at pH 10.0, 272 (18,000) and 219 (12,100) at pH 4.0; λmin: 240 (6,400) at pH 7.0, 217 (9,300) at pH 10.0, 240 (5,600) at pH 4.0.
![[1860-5397-12-254-10]](/bjoc/content/figures/1860-5397-12-254-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: The UV spectra of 6-aza-2-thiothymidine (5b).
Figure 10: The UV spectra of 6-aza-2-thiothymidine (5b).
Vorbrüggen and Strelke [59] quoted the following UV data for 2-thio-6-azauridine: λmax, nm: (H2O) 269 (8,200) and 218 (12,600); (0.01 N NaOH) 267 (18,850) and 236 (12,600). ESIMS (positive ion mode): 282 [M + Na]+, 298 [M + K]+; ESIMS (negative ion mode): 258 [M – H]−.
Supporting Information
| Supporting Information File 1: Analytical and computational data. | ||
| Format: PDF | Size: 872.8 KB | Download |
Acknowledgments
The authors are indebted to Prof. Dr. Helmut Vorbrüggen (Institut für Chemie und Biochemie - Organische Chemie; Freie Universität Berlin, Germany) for providing the nucleosides 9–14 for this study. Financial support by the International Science and Technology Centre (http://www.istc.ru; project #B-1640) and the Byelorussian Republican Foundation for Fundamental Research (http://www.fond.bas-net.by; project #X13MC-027) is gratefully acknowledged. The authors are indebted to Dr. L.A. Alexandrova (Engelhardt Institute of Molecular Biology RAS, Vavilova 32, 119991 Moscow, Russia) for supplying us with heterocyclic bases 15–18, and to Dr. A.V. Baranovsky (Institute of Bioorganic Chemistry, Minsk) for assistance in the analysis of the NMR spectra of synthesized nucleosides and Dr. R.S. Esipov (Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, RAS, Moscow) for enzyme preparations used in the present work. I.A. Mikhailopulo is deeply thankful to the Alexander von Humboldt-Stiftung (Bonn – Bad-Godesberg, Germany) for kind support of my work.
References
-
Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Mikhailopulo, I. A.; Miroshnikov, A. I. ActaNaturae 2010, 2, 36–58.
Return to citation in text: [1] [2] [3] -
Mendieta, J.; Martín-Santamaría, S.; Priego, E.-M.; Balzarini, J.; Camarasa, M.-J.; Pérez-Pérez, M.-J.; Gago, F. Biochemistry 2004, 43, 405–414. doi:10.1021/bi034793o
Return to citation in text: [1] [2] [3] -
Panova, N. G.; Shcheveleva, E. V.; Alexeev, K. S.; Mukhortov, V. G.; Zuev, A. N.; Mikhailov, S. N.; Esipov, R. S.; Chuvikovskii, D. V.; Miroshnikov, A. I. Mol. Biol. 2004, 38, 770–776. doi:10.1023/b:mbil.0000043946.44742.c8
Return to citation in text: [1] [2] -
Panova, N. G.; Alexeev, C. S.; Kuzmichov, A. S.; Shcheveleva, E. V.; Gavryushov, S. A.; Polyakov, K. M.; Kritzyn, A. M.; Mikhailov, S. N.; Esipov, R. S.; Miroshnikov, A. I. Biochemistry (Moscow) 2007, 72, 21–28. doi:10.1134/S0006297907010026
Return to citation in text: [1] [2] [3] [4] -
Panova, N. G.; Alexeev, C. S.; Polyakov, K. M.; Gavryushov, S. A.; Kritzyn, A. M.; Mikhailov, S. N. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 1211–1214. doi:10.1080/15257770802257895
Return to citation in text: [1] [2] -
Miazga, A.; Hamy, F.; Louvel, S.; Klimkait, T.; Pietrusiewicz, Z.; Kurzyńska-Kokorniak, A.; Figlerowicz, M.; Wińska, P.; Kulikowski, T. Antiviral Res. 2011, 92, 57–63. doi:10.1016/j.antiviral.2011.05.012
And references cited therein.
Return to citation in text: [1] -
Carlucci, M.; Kierzek, E.; Olejnik, A.; Turner, D. H.; Kierzek, R. Biochemistry 2009, 48, 10882–10893. doi:10.1021/bi901506f
Return to citation in text: [1] [2] -
Seela, F.; Chittepu, P. J. Org. Chem. 2007, 72, 4358–4366. doi:10.1021/jo0702903
Return to citation in text: [1] [2] [3] -
Kumar, R. K.; Davis, D. R. Nucleic Acids Res. 1997, 25, 1272–1280. doi:10.1093/nar/25.6.1272
Return to citation in text: [1] [2] -
Sanghvi, Y. S.; Hoke, G. D.; Freier, S. M.; Zounes, M. C.; Gonzalez, C.; Cummins, L.; Sasmor, H.; Cook, P. D. Nucleic Acids Res. 1993, 21, 3197–3203. doi:10.1093/nar/21.14.3197
Return to citation in text: [1] [2] -
Sintim, H. O.; Kool, E. T. J. Am. Chem. Soc. 2006, 128, 396–397. doi:10.1021/ja0562447
Return to citation in text: [1] -
Bretner, M.; Felczak, K.; Dzik, J. M.; Golos, B.; Rode, W.; Drabikowska, A.; Poznanski, J.; Krawiec, K.; Piasek, A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1997, 16, 1295–1299. doi:10.1080/07328319708006174
Return to citation in text: [1] -
Rajeev, K. G.; Prakash, T. P.; Manoharan, M. Org. Lett. 2003, 5, 3005–3008. doi:10.1021/ol0348607
Return to citation in text: [1] [2] -
Zhang, Q.; Mi, Z.; Huang, Y.; Ma, L.; Ding, J.; Wang, J.; Zhang, Y.; Chen, Y.; Zhou, J.; Guo, F.; Li, X.; Cen, S. Retrovirology 2016, 13, 13. doi:10.1186/s12977-016-0247-z
Return to citation in text: [1] [2] -
Poopeiko, N. E.; Poznanski, J.; Drabikowska, A.; Balzarini, J.; De Clercq, E.; Mikhailopulo, I. A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1995, 14, 435–437. doi:10.1080/15257779508012402
Return to citation in text: [1] [2] [3] [4] [5] -
Reelfs, O.; Macpherson, P.; Ren, X.; Xu, Y.-Z.; Karran, P.; Young, A. Nucleic Acids Res. 2011, 39, 9620–9632. doi:10.1093/nar/gkr674
Return to citation in text: [1] -
Pridgeon, S. W.; Heer, R.; Taylor, G. A.; Newell, D. R.; O’Toole, K.; Robinson, M.; Xu, Y.-Z.; Karran, P.; Boddy, A. V. Br. J. Cancer 2011, 104, 1869–1876. doi:10.1038/bjc.2011.180
Return to citation in text: [1] -
Zhang, X.; Jeffs, G.; Ren, X.; O’Donovan, P.; Montaner, B.; Perrett, C. M.; Karran, P.; Xu, Y.-Z. DNA Repair 2007, 6, 344–354. doi:10.1016/j.dnarep.2006.11.003
Return to citation in text: [1] -
Horváth, A.; Beck, Z.; Bardos, T. J.; Dunnd, J. A.; Aradi, J. Bioorg. Med. Chem. Lett. 2006, 16, 5321–5323. doi:10.1016/j.bmcl.2006.07.082
Return to citation in text: [1] -
Morrey, J. D.; Smee, D. F.; Sidwell, R. W.; Tseng, C. Antiviral Res. 2002, 55, 107–116. doi:10.1016/S0166-3542(02)00013-X
Return to citation in text: [1] -
Shigeta, S.; Mori, S.; Watanabe, F.; Takahashi, K.; Nagata, T.; Koike, N.; Wakayama, T.; Saneyoshi, M. Antiviral Chem. Chemother. 2002, 13, 67–82. doi:10.1177/095632020201300201
Return to citation in text: [1] -
Kulikowski, T.; Shugar, D. J. Med. Chem. 1974, 17, 269–273. doi:10.1021/jm00249a003
Return to citation in text: [1] -
Xu, Y.-Z.; Zheng, Q. G.; Swann, P. F. Tetrahedron Lett. 1991, 32, 2817–2820. doi:10.1016/0040-4039(91)85095-M
Return to citation in text: [1] -
Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis Of Nucleosides. Organic Reactions; John Wiley & Sons, Inc., 2004; Vol. 55, pp 1–630. doi:10.1002/0471264180.or055.01
Return to citation in text: [1] [2] [3] -
Prusoff, W. H. J. Biol. Chem. 1955, 215, 809–821.
Return to citation in text: [1] [2] [3] [4] -
Handschumacher, R. E. Biochim. Biophys. Acta 1957, 23, 428–430. doi:10.1016/0006-3002(57)90348-7
Return to citation in text: [1] [2] [3] -
Handschumacher, R. E. J. Biol. Chem. 1960, 235, 764–768.
Return to citation in text: [1] [2] [3] -
Škoda, J.; Hess, V. F.; Šorm, F. Experientia 1957, 13, 150–151. doi:10.1007/bf02158141
Return to citation in text: [1] [2] [3] -
Kara, J.; Šorm, F. Collect. Czech. Chem. Commun. 1963, 28, 1441–1448. doi:10.1135/cccc19631441
Return to citation in text: [1] [2] [3] -
Stout, M. G.; Hoard, D. E.; Holman, M. J.; Wu, E. S.; Siegel, J. M. Methods Carbohydr. Chem. 1976, 7, 19–24.
Return to citation in text: [1] -
Kalckar, H. M. Biochim. Biophys. Acta 1953, 12, 250–264. doi:10.1016/0006-3002(53)90144-9
Return to citation in text: [1] [2] -
Friedkin, M.; Roberts, D. J. Biol. Chem. 1954, 207, 257–266.
Return to citation in text: [1] [2] [3] -
Strominger, D. B.; Friedkin, M. J. Biol. Chem. 1954, 208, 663–668.
Return to citation in text: [1] [2] [3] -
Hatano, A.; Harano, A.; Kirihara, M. Chem. Lett. 2006, 35, 232–233. doi:10.1246/cl.2006.232
Return to citation in text: [1] [2] [3] [4] -
Esipov, R. S.; Gurevich, A. I.; Chuvikovsky, D. V.; Chupova, L. A.; Muravyova, T. I.; Miroshnikov, A. I. Protein Expression Purif. 2002, 24, 56–60. doi:10.1006/prep.2001.1524
Return to citation in text: [1] [2] -
Mikhailopulo, I. A. Curr. Org. Chem. 2007, 11, 317–335. doi:10.2174/138527207780059330
Return to citation in text: [1] [2] [3] [4] -
Mikhailopulo, I. A.; Miroshnikov, A. I. Mendeleev Commun. 2011, 21, 57–68. doi:10.1016/j.mencom.2011.03.001
Return to citation in text: [1] [2] -
Caradoc-Davies, T. T.; Cutfield, S. M.; Lamont, I. L.; Cutfield, J. F. J. Mol. Biol. 2004, 337, 337–354. doi:10.1016/j.jmb.2004.01.039
Return to citation in text: [1] -
Tran, T. H.; Christoffersen, S.; Allan, P. W.; Parker, W. B.; Piškur, J.; Serra, I.; Terreni, M.; Ealick, S. E. Biochemistry 2011, 50, 6549–6558. doi:10.1021/bi200707z
Return to citation in text: [1] [2] [3] [4] -
Oliva, I.; Zuffi, G.; Barile, D.; Orsini, G.; Tonon, G.; De Gioia, L.; Ghisotti, D. J. Biochem. 2004, 135, 495–499. doi:10.1093/jb/mvh057
Return to citation in text: [1] -
Rejnek, J.; Hanus, M.; Kabeláč, M.; Ryjáček, F.; Hobza, P. Phys. Chem. Chem. Phys. 2005, 7, 2006–2017. doi:10.1039/B501499A
Return to citation in text: [1] [2] -
Kryachko, E. N.; Nguyen, M. T. Adv. Quantum Chem. 2001, 40, 79–102. doi:10.1016/S0065-3276(01)40010-4
Return to citation in text: [1] -
Shukla, M. K.; Leszczynski, J. J. Mol. Struct.: THEOCHEM 2006, 771, 149–155. doi:10.1016/j.theochem.2006.03.031
Return to citation in text: [1] -
Sarzyńska, J.; Kuliński, T. Comput. Methods Sci. Technol. 2005, 11, 49–55. doi:10.12921/cmst.2005.11.01.49-55
Return to citation in text: [1] -
Palafox, M. A.; Rastogi, V. K.; Tanwar, R. P.; Mittal, L. Spectrochim. Acta, Part A 2003, 59, 2473–2486. doi:10.1016/S1386-1425(02)00409-2
Return to citation in text: [1] -
Yekeler, H. J. Comput.-Aided Mol. Des. 2000, 14, 243–250. doi:10.1023/A:1008132202838
Return to citation in text: [1] -
Psoda, A.; Kazimierczuk, Z.; Shugar, D. J. Am. Chem. Soc. 1974, 96, 6832–6839. doi:10.1021/ja00829a003
Return to citation in text: [1] [2] [3] -
Psoda, A.; Shugar, D. Acta Biochim. Pol. 1979, 26, 55–72.
Return to citation in text: [1] [2] [3] -
Rick, S. W.; Abashkin, Y. G.; Hilderbrandt, R. L.; Burt, S. K. Proteins: Struct., Funct., Genet. 1999, 37, 242–252. doi:10.1002/(SICI)1097-0134(19991101)37:2<242::AID-PROT9>3.0.CO;2-5.
Return to citation in text: [1] -
Pugmire, M. J.; Ealick, S. E. Biochem. J. 2002, 361, 1–25. doi:10.1042/bj3610001
And references cited therein.
Return to citation in text: [1] -
Walter, M. R.; Cook, W. J.; Cole, L. B.; Short, S. A.; Koszalka, G. W.; Krenitsky, T. A.; Ealick, S. E. J. Biol. Chem. 1990, 265, 14016–14022.
Return to citation in text: [1] [2] -
Pugmire, M. J.; Cook, W. J.; Jasanoff, A.; Walter, M. R.; Ealick, S. E. J. Mol. Biol. 1998, 281, 285–299. doi:10.1006/jmbi.1998.1941
Return to citation in text: [1] -
Edwards, P. N. J. Enzyme Inhib. Med. Chem. 2006, 21, 483–518. doi:10.1080/14756360600721075
Return to citation in text: [1] -
Timofeev, V. I.; Abramchik, Y. A.; Fateev, I. V.; Zhukhlistova, N. E.; Murav’eva, T. I.; Kuranova, I. P.; Esipov, R. S. Crystallogr. Rep. 2013, 58, 842–853. doi:10.1134/S1063774513060230
Return to citation in text: [1] [2] -
Schwartz, M. Methods Enzymol. 1978, 51, 442–445. doi:10.1016/S0076-6879(78)51061-6
Return to citation in text: [1] -
Schinazi, R. F.; Peck, A.; Sommadossi, J.-P. Biochem. Pharmacol. 1992, 44, 199–204. doi:10.1016/0006-2952(92)90001-Y
Return to citation in text: [1] -
Alexeev, C. S.; Panova, N. G.; Polyakov, K. M.; Mikhailov, S. N. Substrate specificity of E. coli uridine phosphorylase. In Abstracts of XIX International Round Table on Nucleosides, Nucleotides and Nucleic Acids, IRT 2010, Lyon, France, Aug 29–Sept 3, 2010; PA011, 2010; pp 94–95.
Return to citation in text: [1] [2] -
Vorbrüggen, H.; Strehlke, P. Chem. Ber. 1973, 106, 3039–3061. doi:10.1002/cber.19731060936
Return to citation in text: [1] [2] -
Votruba, I.; Holy, A.; Pichat, L. Nucleic Acids Res. 1974, 1, 689–698. doi:10.1093/nar/1.5.689
Return to citation in text: [1] -
Felczak, K.; Drabikowska, A. K.; Vilpo, J. A.; Kulikowski, T.; Shugar, D. J. Med. Chem. 1996, 39, 1720–1728. doi:10.1021/jm950675q
Return to citation in text: [1] -
Cadet, J.; Ducolomb, R.; Taieb, C. Tetrahedron Lett. 1975, 16, 3455–3458. doi:10.1016/S0040-4039(00)91381-9
Return to citation in text: [1] -
Miles, D. W.; Inskeep, D. H.; Robins, M. J.; Winkley, M. W.; Robins, R. K.; Eyring, H. J. Am. Chem. Soc. 1970, 92, 3872–3881. doi:10.1021/ja00716a007
Return to citation in text: [1] -
Rabczenko, A.; Jankowski, K.; Zakrzewska, K. Biochim. Biophys. Acta 1974, 353, 1–15. doi:10.1016/0005-2787(74)90092-6
Return to citation in text: [1] -
Cleve, G.; Hoyer, G.-A.; Schulz, G.; Vorbrüggen, H. Chem. Ber. 1973, 106, 3062–3072. doi:10.1002/cber.19731060937
Return to citation in text: [1] -
Suck, D.; Saenger, W. J. Am. Chem. Soc. 1972, 94, 6520–6526. doi:10.1021/ja00773a041
Return to citation in text: [1] -
Stepchenko, V. A.; Seela, F.; Esipov, R. S.; Miroshnikov, A. I.; Sokolov, Y. A.; Mikhailopulo, I. A. Synlett 2012, 1541–1545. doi:10.1055/s-0031-1290679
Return to citation in text: [1] [2] [3] -
Bzowska, A.; Kulikowska, E.; Poopeiko, N. E.; Shugar, D. Eur. J. Biochem. 1996, 239, 229–234. doi:10.1111/j.1432-1033.1996.0229u.x
And references cited therein.
Return to citation in text: [1] -
Kalinichenko, E. N.; Barai, V. N.; Bokut, S. B.; Romanova, V. V.; Zinchenko, A. I.; Herrmann, G.; Mikhailopulo, I. A. Biotechnol. Lett. 1989, 11, 621–626. doi:10.1007/BF01025269
Return to citation in text: [1] -
Serra, I.; Bavaro, T.; Cecchini, D. A.; Daly, S.; Albertini, A. M.; Terreni, M.; Ubiali, D. J. Mol. Catal. B: Enzym. 2013, 95, 16–22. doi:10.1016/j.molcatb.2013.05.007
Return to citation in text: [1] -
Fateev, I. V.; Kharitonova, M. I.; Antonov, K. V.; Konstantinova, I. D.; Stepanenko, V. N.; Esipov, R. S.; Seela, F.; Temburnikar, K. W.; Seley-Radtke, K. L.; Stepchenko, V. A.; Sokolov, Y. A.; Miroshnikov, A. I.; Mikhailopulo, I. A. Chem. – Eur. J. 2015, 21, 13401–13419. doi:10.1002/chem.201501334
Return to citation in text: [1] -
Elion, G. B.; Hitchings, G. H.; Samuel, B., (Burroughs Wellcome Co.). 4-thio-2'-deoxyuridine. U.S. Patent 3,163,639 A, Dec 29, 1964.
Return to citation in text: [1] [2] -
Zhang, X.; Xu, Y.-Z. Molecules 2011, 16, 5655–5664. doi:10.3390/molecules16075655
Return to citation in text: [1] -
Hunter, J. H.; Skulnick, H. I. Process for preparing 2-thiouracil nucleosides. (The Upjohn Company). U.S. Patent 3,975,374 A, Aug 17, 1976.
Return to citation in text: [1] -
Kögler, M.; Busson, R.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Louat, T.; Munier-Lehmann, H.; Herdewijn, P. Chem. Biodiversity 2012, 9, 536–556. doi:10.1002/cbdv.201100285
Return to citation in text: [1] [2]
| 42. | Rejnek, J.; Hanus, M.; Kabeláč, M.; Ryjáček, F.; Hobza, P. Phys. Chem. Chem. Phys. 2005, 7, 2006–2017. doi:10.1039/B501499A |
| 43. | Kryachko, E. N.; Nguyen, M. T. Adv. Quantum Chem. 2001, 40, 79–102. doi:10.1016/S0065-3276(01)40010-4 |
| 44. | Shukla, M. K.; Leszczynski, J. J. Mol. Struct.: THEOCHEM 2006, 771, 149–155. doi:10.1016/j.theochem.2006.03.031 |
| 45. | Sarzyńska, J.; Kuliński, T. Comput. Methods Sci. Technol. 2005, 11, 49–55. doi:10.12921/cmst.2005.11.01.49-55 |
| 46. | Palafox, M. A.; Rastogi, V. K.; Tanwar, R. P.; Mittal, L. Spectrochim. Acta, Part A 2003, 59, 2473–2486. doi:10.1016/S1386-1425(02)00409-2 |
| 47. | Yekeler, H. J. Comput.-Aided Mol. Des. 2000, 14, 243–250. doi:10.1023/A:1008132202838 |
| 48. | Psoda, A.; Kazimierczuk, Z.; Shugar, D. J. Am. Chem. Soc. 1974, 96, 6832–6839. doi:10.1021/ja00829a003 |
| 49. | Psoda, A.; Shugar, D. Acta Biochim. Pol. 1979, 26, 55–72. |
| 48. | Psoda, A.; Kazimierczuk, Z.; Shugar, D. J. Am. Chem. Soc. 1974, 96, 6832–6839. doi:10.1021/ja00829a003 |
| 49. | Psoda, A.; Shugar, D. Acta Biochim. Pol. 1979, 26, 55–72. |
| 3. | Mendieta, J.; Martín-Santamaría, S.; Priego, E.-M.; Balzarini, J.; Camarasa, M.-J.; Pérez-Pérez, M.-J.; Gago, F. Biochemistry 2004, 43, 405–414. doi:10.1021/bi034793o |
| 52. | Walter, M. R.; Cook, W. J.; Cole, L. B.; Short, S. A.; Koszalka, G. W.; Krenitsky, T. A.; Ealick, S. E. J. Biol. Chem. 1990, 265, 14016–14022. |
| 53. | Pugmire, M. J.; Cook, W. J.; Jasanoff, A.; Walter, M. R.; Ealick, S. E. J. Mol. Biol. 1998, 281, 285–299. doi:10.1006/jmbi.1998.1941 |
| 54. | Edwards, P. N. J. Enzyme Inhib. Med. Chem. 2006, 21, 483–518. doi:10.1080/14756360600721075 |
| 55. | Timofeev, V. I.; Abramchik, Y. A.; Fateev, I. V.; Zhukhlistova, N. E.; Murav’eva, T. I.; Kuranova, I. P.; Esipov, R. S. Crystallogr. Rep. 2013, 58, 842–853. doi:10.1134/S1063774513060230 |
| 56. | Schwartz, M. Methods Enzymol. 1978, 51, 442–445. doi:10.1016/S0076-6879(78)51061-6 |
| 51. |
Pugmire, M. J.; Ealick, S. E. Biochem. J. 2002, 361, 1–25. doi:10.1042/bj3610001
And references cited therein. |
| 40. | Tran, T. H.; Christoffersen, S.; Allan, P. W.; Parker, W. B.; Piškur, J.; Serra, I.; Terreni, M.; Ealick, S. E. Biochemistry 2011, 50, 6549–6558. doi:10.1021/bi200707z |
| 50. | Rick, S. W.; Abashkin, Y. G.; Hilderbrandt, R. L.; Burt, S. K. Proteins: Struct., Funct., Genet. 1999, 37, 242–252. doi:10.1002/(SICI)1097-0134(19991101)37:2<242::AID-PROT9>3.0.CO;2-5. |
| 4. | Panova, N. G.; Shcheveleva, E. V.; Alexeev, K. S.; Mukhortov, V. G.; Zuev, A. N.; Mikhailov, S. N.; Esipov, R. S.; Chuvikovskii, D. V.; Miroshnikov, A. I. Mol. Biol. 2004, 38, 770–776. doi:10.1023/b:mbil.0000043946.44742.c8 |
| 48. | Psoda, A.; Kazimierczuk, Z.; Shugar, D. J. Am. Chem. Soc. 1974, 96, 6832–6839. doi:10.1021/ja00829a003 |
| 49. | Psoda, A.; Shugar, D. Acta Biochim. Pol. 1979, 26, 55–72. |
| 40. | Tran, T. H.; Christoffersen, S.; Allan, P. W.; Parker, W. B.; Piškur, J.; Serra, I.; Terreni, M.; Ealick, S. E. Biochemistry 2011, 50, 6549–6558. doi:10.1021/bi200707z |
| 3. | Mendieta, J.; Martín-Santamaría, S.; Priego, E.-M.; Balzarini, J.; Camarasa, M.-J.; Pérez-Pérez, M.-J.; Gago, F. Biochemistry 2004, 43, 405–414. doi:10.1021/bi034793o |
| 52. | Walter, M. R.; Cook, W. J.; Cole, L. B.; Short, S. A.; Koszalka, G. W.; Krenitsky, T. A.; Ealick, S. E. J. Biol. Chem. 1990, 265, 14016–14022. |
| 55. | Timofeev, V. I.; Abramchik, Y. A.; Fateev, I. V.; Zhukhlistova, N. E.; Murav’eva, T. I.; Kuranova, I. P.; Esipov, R. S. Crystallogr. Rep. 2013, 58, 842–853. doi:10.1134/S1063774513060230 |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 33. | Friedkin, M.; Roberts, D. J. Biol. Chem. 1954, 207, 257–266. |
| 34. | Strominger, D. B.; Friedkin, M. J. Biol. Chem. 1954, 208, 663–668. |
| 35. | Hatano, A.; Harano, A.; Kirihara, M. Chem. Lett. 2006, 35, 232–233. doi:10.1246/cl.2006.232 |
| 62. | Cadet, J.; Ducolomb, R.; Taieb, C. Tetrahedron Lett. 1975, 16, 3455–3458. doi:10.1016/S0040-4039(00)91381-9 |
| 63. | Miles, D. W.; Inskeep, D. H.; Robins, M. J.; Winkley, M. W.; Robins, R. K.; Eyring, H. J. Am. Chem. Soc. 1970, 92, 3872–3881. doi:10.1021/ja00716a007 |
| 64. | Rabczenko, A.; Jankowski, K.; Zakrzewska, K. Biochim. Biophys. Acta 1974, 353, 1–15. doi:10.1016/0005-2787(74)90092-6 |
| 65. | Cleve, G.; Hoyer, G.-A.; Schulz, G.; Vorbrüggen, H. Chem. Ber. 1973, 106, 3062–3072. doi:10.1002/cber.19731060937 |
| 60. | Votruba, I.; Holy, A.; Pichat, L. Nucleic Acids Res. 1974, 1, 689–698. doi:10.1093/nar/1.5.689 |
| 42. | Rejnek, J.; Hanus, M.; Kabeláč, M.; Ryjáček, F.; Hobza, P. Phys. Chem. Chem. Phys. 2005, 7, 2006–2017. doi:10.1039/B501499A |
| 61. | Felczak, K.; Drabikowska, A. K.; Vilpo, J. A.; Kulikowski, T.; Shugar, D. J. Med. Chem. 1996, 39, 1720–1728. doi:10.1021/jm950675q |
| 5. | Panova, N. G.; Alexeev, C. S.; Kuzmichov, A. S.; Shcheveleva, E. V.; Gavryushov, S. A.; Polyakov, K. M.; Kritzyn, A. M.; Mikhailov, S. N.; Esipov, R. S.; Miroshnikov, A. I. Biochemistry (Moscow) 2007, 72, 21–28. doi:10.1134/S0006297907010026 |
| 58. | Alexeev, C. S.; Panova, N. G.; Polyakov, K. M.; Mikhailov, S. N. Substrate specificity of E. coli uridine phosphorylase. In Abstracts of XIX International Round Table on Nucleosides, Nucleotides and Nucleic Acids, IRT 2010, Lyon, France, Aug 29–Sept 3, 2010; PA011, 2010; pp 94–95. |
| 25. | Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis Of Nucleosides. Organic Reactions; John Wiley & Sons, Inc., 2004; Vol. 55, pp 1–630. doi:10.1002/0471264180.or055.01 |
| 59. | Vorbrüggen, H.; Strehlke, P. Chem. Ber. 1973, 106, 3039–3061. doi:10.1002/cber.19731060936 |
| 16. | Poopeiko, N. E.; Poznanski, J.; Drabikowska, A.; Balzarini, J.; De Clercq, E.; Mikhailopulo, I. A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1995, 14, 435–437. doi:10.1080/15257779508012402 |
| 57. | Schinazi, R. F.; Peck, A.; Sommadossi, J.-P. Biochem. Pharmacol. 1992, 44, 199–204. doi:10.1016/0006-2952(92)90001-Y |
| 16. | Poopeiko, N. E.; Poznanski, J.; Drabikowska, A.; Balzarini, J.; De Clercq, E.; Mikhailopulo, I. A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1995, 14, 435–437. doi:10.1080/15257779508012402 |
| 67. | Stepchenko, V. A.; Seela, F.; Esipov, R. S.; Miroshnikov, A. I.; Sokolov, Y. A.; Mikhailopulo, I. A. Synlett 2012, 1541–1545. doi:10.1055/s-0031-1290679 |
| 68. |
Bzowska, A.; Kulikowska, E.; Poopeiko, N. E.; Shugar, D. Eur. J. Biochem. 1996, 239, 229–234. doi:10.1111/j.1432-1033.1996.0229u.x
And references cited therein. |
| 66. | Suck, D.; Saenger, W. J. Am. Chem. Soc. 1972, 94, 6520–6526. doi:10.1021/ja00773a041 |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 2. | Mikhailopulo, I. A.; Miroshnikov, A. I. ActaNaturae 2010, 2, 36–58. |
| 8. | Carlucci, M.; Kierzek, E.; Olejnik, A.; Turner, D. H.; Kierzek, R. Biochemistry 2009, 48, 10882–10893. doi:10.1021/bi901506f |
| 9. | Seela, F.; Chittepu, P. J. Org. Chem. 2007, 72, 4358–4366. doi:10.1021/jo0702903 |
| 10. | Kumar, R. K.; Davis, D. R. Nucleic Acids Res. 1997, 25, 1272–1280. doi:10.1093/nar/25.6.1272 |
| 11. | Sanghvi, Y. S.; Hoke, G. D.; Freier, S. M.; Zounes, M. C.; Gonzalez, C.; Cummins, L.; Sasmor, H.; Cook, P. D. Nucleic Acids Res. 1993, 21, 3197–3203. doi:10.1093/nar/21.14.3197 |
| 14. | Rajeev, K. G.; Prakash, T. P.; Manoharan, M. Org. Lett. 2003, 5, 3005–3008. doi:10.1021/ol0348607 |
| 15. | Zhang, Q.; Mi, Z.; Huang, Y.; Ma, L.; Ding, J.; Wang, J.; Zhang, Y.; Chen, Y.; Zhou, J.; Guo, F.; Li, X.; Cen, S. Retrovirology 2016, 13, 13. doi:10.1186/s12977-016-0247-z |
| 16. | Poopeiko, N. E.; Poznanski, J.; Drabikowska, A.; Balzarini, J.; De Clercq, E.; Mikhailopulo, I. A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1995, 14, 435–437. doi:10.1080/15257779508012402 |
| 23. | Kulikowski, T.; Shugar, D. J. Med. Chem. 1974, 17, 269–273. doi:10.1021/jm00249a003 |
| 24. | Xu, Y.-Z.; Zheng, Q. G.; Swann, P. F. Tetrahedron Lett. 1991, 32, 2817–2820. doi:10.1016/0040-4039(91)85095-M |
| 35. | Hatano, A.; Harano, A.; Kirihara, M. Chem. Lett. 2006, 35, 232–233. doi:10.1246/cl.2006.232 |
| 67. | Stepchenko, V. A.; Seela, F.; Esipov, R. S.; Miroshnikov, A. I.; Sokolov, Y. A.; Mikhailopulo, I. A. Synlett 2012, 1541–1545. doi:10.1055/s-0031-1290679 |
| 15. | Zhang, Q.; Mi, Z.; Huang, Y.; Ma, L.; Ding, J.; Wang, J.; Zhang, Y.; Chen, Y.; Zhou, J.; Guo, F.; Li, X.; Cen, S. Retrovirology 2016, 13, 13. doi:10.1186/s12977-016-0247-z |
| 16. | Poopeiko, N. E.; Poznanski, J.; Drabikowska, A.; Balzarini, J.; De Clercq, E.; Mikhailopulo, I. A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1995, 14, 435–437. doi:10.1080/15257779508012402 |
| 17. | Reelfs, O.; Macpherson, P.; Ren, X.; Xu, Y.-Z.; Karran, P.; Young, A. Nucleic Acids Res. 2011, 39, 9620–9632. doi:10.1093/nar/gkr674 |
| 18. | Pridgeon, S. W.; Heer, R.; Taylor, G. A.; Newell, D. R.; O’Toole, K.; Robinson, M.; Xu, Y.-Z.; Karran, P.; Boddy, A. V. Br. J. Cancer 2011, 104, 1869–1876. doi:10.1038/bjc.2011.180 |
| 19. | Zhang, X.; Jeffs, G.; Ren, X.; O’Donovan, P.; Montaner, B.; Perrett, C. M.; Karran, P.; Xu, Y.-Z. DNA Repair 2007, 6, 344–354. doi:10.1016/j.dnarep.2006.11.003 |
| 20. | Horváth, A.; Beck, Z.; Bardos, T. J.; Dunnd, J. A.; Aradi, J. Bioorg. Med. Chem. Lett. 2006, 16, 5321–5323. doi:10.1016/j.bmcl.2006.07.082 |
| 21. | Morrey, J. D.; Smee, D. F.; Sidwell, R. W.; Tseng, C. Antiviral Res. 2002, 55, 107–116. doi:10.1016/S0166-3542(02)00013-X |
| 22. | Shigeta, S.; Mori, S.; Watanabe, F.; Takahashi, K.; Nagata, T.; Koike, N.; Wakayama, T.; Saneyoshi, M. Antiviral Chem. Chemother. 2002, 13, 67–82. doi:10.1177/095632020201300201 |
| 26. | Prusoff, W. H. J. Biol. Chem. 1955, 215, 809–821. |
| 27. | Handschumacher, R. E. Biochim. Biophys. Acta 1957, 23, 428–430. doi:10.1016/0006-3002(57)90348-7 |
| 28. | Handschumacher, R. E. J. Biol. Chem. 1960, 235, 764–768. |
| 29. | Škoda, J.; Hess, V. F.; Šorm, F. Experientia 1957, 13, 150–151. doi:10.1007/bf02158141 |
| 8. | Carlucci, M.; Kierzek, E.; Olejnik, A.; Turner, D. H.; Kierzek, R. Biochemistry 2009, 48, 10882–10893. doi:10.1021/bi901506f |
| 9. | Seela, F.; Chittepu, P. J. Org. Chem. 2007, 72, 4358–4366. doi:10.1021/jo0702903 |
| 10. | Kumar, R. K.; Davis, D. R. Nucleic Acids Res. 1997, 25, 1272–1280. doi:10.1093/nar/25.6.1272 |
| 11. | Sanghvi, Y. S.; Hoke, G. D.; Freier, S. M.; Zounes, M. C.; Gonzalez, C.; Cummins, L.; Sasmor, H.; Cook, P. D. Nucleic Acids Res. 1993, 21, 3197–3203. doi:10.1093/nar/21.14.3197 |
| 12. | Sintim, H. O.; Kool, E. T. J. Am. Chem. Soc. 2006, 128, 396–397. doi:10.1021/ja0562447 |
| 13. | Bretner, M.; Felczak, K.; Dzik, J. M.; Golos, B.; Rode, W.; Drabikowska, A.; Poznanski, J.; Krawiec, K.; Piasek, A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1997, 16, 1295–1299. doi:10.1080/07328319708006174 |
| 14. | Rajeev, K. G.; Prakash, T. P.; Manoharan, M. Org. Lett. 2003, 5, 3005–3008. doi:10.1021/ol0348607 |
| 33. | Friedkin, M.; Roberts, D. J. Biol. Chem. 1954, 207, 257–266. |
| 34. | Strominger, D. B.; Friedkin, M. J. Biol. Chem. 1954, 208, 663–668. |
| 26. | Prusoff, W. H. J. Biol. Chem. 1955, 215, 809–821. |
| 27. | Handschumacher, R. E. Biochim. Biophys. Acta 1957, 23, 428–430. doi:10.1016/0006-3002(57)90348-7 |
| 28. | Handschumacher, R. E. J. Biol. Chem. 1960, 235, 764–768. |
| 29. | Škoda, J.; Hess, V. F.; Šorm, F. Experientia 1957, 13, 150–151. doi:10.1007/bf02158141 |
| 35. | Hatano, A.; Harano, A.; Kirihara, M. Chem. Lett. 2006, 35, 232–233. doi:10.1246/cl.2006.232 |
| 3. | Mendieta, J.; Martín-Santamaría, S.; Priego, E.-M.; Balzarini, J.; Camarasa, M.-J.; Pérez-Pérez, M.-J.; Gago, F. Biochemistry 2004, 43, 405–414. doi:10.1021/bi034793o |
| 4. | Panova, N. G.; Shcheveleva, E. V.; Alexeev, K. S.; Mukhortov, V. G.; Zuev, A. N.; Mikhailov, S. N.; Esipov, R. S.; Chuvikovskii, D. V.; Miroshnikov, A. I. Mol. Biol. 2004, 38, 770–776. doi:10.1023/b:mbil.0000043946.44742.c8 |
| 5. | Panova, N. G.; Alexeev, C. S.; Kuzmichov, A. S.; Shcheveleva, E. V.; Gavryushov, S. A.; Polyakov, K. M.; Kritzyn, A. M.; Mikhailov, S. N.; Esipov, R. S.; Miroshnikov, A. I. Biochemistry (Moscow) 2007, 72, 21–28. doi:10.1134/S0006297907010026 |
| 6. | Panova, N. G.; Alexeev, C. S.; Polyakov, K. M.; Gavryushov, S. A.; Kritzyn, A. M.; Mikhailov, S. N. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 1211–1214. doi:10.1080/15257770802257895 |
| 7. |
Miazga, A.; Hamy, F.; Louvel, S.; Klimkait, T.; Pietrusiewicz, Z.; Kurzyńska-Kokorniak, A.; Figlerowicz, M.; Wińska, P.; Kulikowski, T. Antiviral Res. 2011, 92, 57–63. doi:10.1016/j.antiviral.2011.05.012
And references cited therein. |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 67. | Stepchenko, V. A.; Seela, F.; Esipov, R. S.; Miroshnikov, A. I.; Sokolov, Y. A.; Mikhailopulo, I. A. Synlett 2012, 1541–1545. doi:10.1055/s-0031-1290679 |
| 29. | Škoda, J.; Hess, V. F.; Šorm, F. Experientia 1957, 13, 150–151. doi:10.1007/bf02158141 |
| 31. | Stout, M. G.; Hoard, D. E.; Holman, M. J.; Wu, E. S.; Siegel, J. M. Methods Carbohydr. Chem. 1976, 7, 19–24. |
| 70. | Serra, I.; Bavaro, T.; Cecchini, D. A.; Daly, S.; Albertini, A. M.; Terreni, M.; Ubiali, D. J. Mol. Catal. B: Enzym. 2013, 95, 16–22. doi:10.1016/j.molcatb.2013.05.007 |
| 27. | Handschumacher, R. E. Biochim. Biophys. Acta 1957, 23, 428–430. doi:10.1016/0006-3002(57)90348-7 |
| 28. | Handschumacher, R. E. J. Biol. Chem. 1960, 235, 764–768. |
| 32. | Kalckar, H. M. Biochim. Biophys. Acta 1953, 12, 250–264. doi:10.1016/0006-3002(53)90144-9 |
| 5. | Panova, N. G.; Alexeev, C. S.; Kuzmichov, A. S.; Shcheveleva, E. V.; Gavryushov, S. A.; Polyakov, K. M.; Kritzyn, A. M.; Mikhailov, S. N.; Esipov, R. S.; Miroshnikov, A. I. Biochemistry (Moscow) 2007, 72, 21–28. doi:10.1134/S0006297907010026 |
| 58. | Alexeev, C. S.; Panova, N. G.; Polyakov, K. M.; Mikhailov, S. N. Substrate specificity of E. coli uridine phosphorylase. In Abstracts of XIX International Round Table on Nucleosides, Nucleotides and Nucleic Acids, IRT 2010, Lyon, France, Aug 29–Sept 3, 2010; PA011, 2010; pp 94–95. |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 2. | Mikhailopulo, I. A.; Miroshnikov, A. I. ActaNaturae 2010, 2, 36–58. |
| 37. | Mikhailopulo, I. A. Curr. Org. Chem. 2007, 11, 317–335. doi:10.2174/138527207780059330 |
| 25. | Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis Of Nucleosides. Organic Reactions; John Wiley & Sons, Inc., 2004; Vol. 55, pp 1–630. doi:10.1002/0471264180.or055.01 |
| 30. | Kara, J.; Šorm, F. Collect. Czech. Chem. Commun. 1963, 28, 1441–1448. doi:10.1135/cccc19631441 |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 37. | Mikhailopulo, I. A. Curr. Org. Chem. 2007, 11, 317–335. doi:10.2174/138527207780059330 |
| 69. | Kalinichenko, E. N.; Barai, V. N.; Bokut, S. B.; Romanova, V. V.; Zinchenko, A. I.; Herrmann, G.; Mikhailopulo, I. A. Biotechnol. Lett. 1989, 11, 621–626. doi:10.1007/BF01025269 |
| 36. | Esipov, R. S.; Gurevich, A. I.; Chuvikovsky, D. V.; Chupova, L. A.; Muravyova, T. I.; Miroshnikov, A. I. Protein Expression Purif. 2002, 24, 56–60. doi:10.1006/prep.2001.1524 |
| 36. | Esipov, R. S.; Gurevich, A. I.; Chuvikovsky, D. V.; Chupova, L. A.; Muravyova, T. I.; Miroshnikov, A. I. Protein Expression Purif. 2002, 24, 56–60. doi:10.1006/prep.2001.1524 |
| 37. | Mikhailopulo, I. A. Curr. Org. Chem. 2007, 11, 317–335. doi:10.2174/138527207780059330 |
| 38. | Mikhailopulo, I. A.; Miroshnikov, A. I. Mendeleev Commun. 2011, 21, 57–68. doi:10.1016/j.mencom.2011.03.001 |
| 72. | Elion, G. B.; Hitchings, G. H.; Samuel, B., (Burroughs Wellcome Co.). 4-thio-2'-deoxyuridine. U.S. Patent 3,163,639 A, Dec 29, 1964. |
| 72. | Elion, G. B.; Hitchings, G. H.; Samuel, B., (Burroughs Wellcome Co.). 4-thio-2'-deoxyuridine. U.S. Patent 3,163,639 A, Dec 29, 1964. |
| 73. | Zhang, X.; Xu, Y.-Z. Molecules 2011, 16, 5655–5664. doi:10.3390/molecules16075655 |
| 16. | Poopeiko, N. E.; Poznanski, J.; Drabikowska, A.; Balzarini, J.; De Clercq, E.; Mikhailopulo, I. A.; Shugar, D.; Kulikowski, T. Nucleosides Nucleotides 1995, 14, 435–437. doi:10.1080/15257779508012402 |
| 71. | Fateev, I. V.; Kharitonova, M. I.; Antonov, K. V.; Konstantinova, I. D.; Stepanenko, V. N.; Esipov, R. S.; Seela, F.; Temburnikar, K. W.; Seley-Radtke, K. L.; Stepchenko, V. A.; Sokolov, Y. A.; Miroshnikov, A. I.; Mikhailopulo, I. A. Chem. – Eur. J. 2015, 21, 13401–13419. doi:10.1002/chem.201501334 |
| 5. | Panova, N. G.; Alexeev, C. S.; Kuzmichov, A. S.; Shcheveleva, E. V.; Gavryushov, S. A.; Polyakov, K. M.; Kritzyn, A. M.; Mikhailov, S. N.; Esipov, R. S.; Miroshnikov, A. I. Biochemistry (Moscow) 2007, 72, 21–28. doi:10.1134/S0006297907010026 |
| 6. | Panova, N. G.; Alexeev, C. S.; Polyakov, K. M.; Gavryushov, S. A.; Kritzyn, A. M.; Mikhailov, S. N. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 1211–1214. doi:10.1080/15257770802257895 |
| 25. | Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis Of Nucleosides. Organic Reactions; John Wiley & Sons, Inc., 2004; Vol. 55, pp 1–630. doi:10.1002/0471264180.or055.01 |
| 41. | Oliva, I.; Zuffi, G.; Barile, D.; Orsini, G.; Tonon, G.; De Gioia, L.; Ghisotti, D. J. Biochem. 2004, 135, 495–499. doi:10.1093/jb/mvh057 |
| 40. | Tran, T. H.; Christoffersen, S.; Allan, P. W.; Parker, W. B.; Piškur, J.; Serra, I.; Terreni, M.; Ealick, S. E. Biochemistry 2011, 50, 6549–6558. doi:10.1021/bi200707z |
| 59. | Vorbrüggen, H.; Strehlke, P. Chem. Ber. 1973, 106, 3039–3061. doi:10.1002/cber.19731060936 |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 2. | Mikhailopulo, I. A.; Miroshnikov, A. I. ActaNaturae 2010, 2, 36–58. |
| 37. | Mikhailopulo, I. A. Curr. Org. Chem. 2007, 11, 317–335. doi:10.2174/138527207780059330 |
| 38. | Mikhailopulo, I. A.; Miroshnikov, A. I. Mendeleev Commun. 2011, 21, 57–68. doi:10.1016/j.mencom.2011.03.001 |
| 9. | Seela, F.; Chittepu, P. J. Org. Chem. 2007, 72, 4358–4366. doi:10.1021/jo0702903 |
| 39. | Caradoc-Davies, T. T.; Cutfield, S. M.; Lamont, I. L.; Cutfield, J. F. J. Mol. Biol. 2004, 337, 337–354. doi:10.1016/j.jmb.2004.01.039 |
| 40. | Tran, T. H.; Christoffersen, S.; Allan, P. W.; Parker, W. B.; Piškur, J.; Serra, I.; Terreni, M.; Ealick, S. E. Biochemistry 2011, 50, 6549–6558. doi:10.1021/bi200707z |
| 30. | Kara, J.; Šorm, F. Collect. Czech. Chem. Commun. 1963, 28, 1441–1448. doi:10.1135/cccc19631441 |
| 75. | Kögler, M.; Busson, R.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Louat, T.; Munier-Lehmann, H.; Herdewijn, P. Chem. Biodiversity 2012, 9, 536–556. doi:10.1002/cbdv.201100285 |
| 1. | Friedkin, M.; Kalckar, H. M. Nucleoside phosphorylases. In The Enzymes, 2nd ed.; Boyer, P. D.; Lardy, H.; Myrbäck, K., Eds.; Academic Press: New York, USA, 1961; Vol. 5, pp 237–255. |
| 32. | Kalckar, H. M. Biochim. Biophys. Acta 1953, 12, 250–264. doi:10.1016/0006-3002(53)90144-9 |
| 33. | Friedkin, M.; Roberts, D. J. Biol. Chem. 1954, 207, 257–266. |
| 34. | Strominger, D. B.; Friedkin, M. J. Biol. Chem. 1954, 208, 663–668. |
| 74. | Hunter, J. H.; Skulnick, H. I. Process for preparing 2-thiouracil nucleosides. (The Upjohn Company). U.S. Patent 3,975,374 A, Aug 17, 1976. |
| 35. | Hatano, A.; Harano, A.; Kirihara, M. Chem. Lett. 2006, 35, 232–233. doi:10.1246/cl.2006.232 |
| 30. | Kara, J.; Šorm, F. Collect. Czech. Chem. Commun. 1963, 28, 1441–1448. doi:10.1135/cccc19631441 |
| 75. | Kögler, M.; Busson, R.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Louat, T.; Munier-Lehmann, H.; Herdewijn, P. Chem. Biodiversity 2012, 9, 536–556. doi:10.1002/cbdv.201100285 |
© 2016 Stepchenko et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)