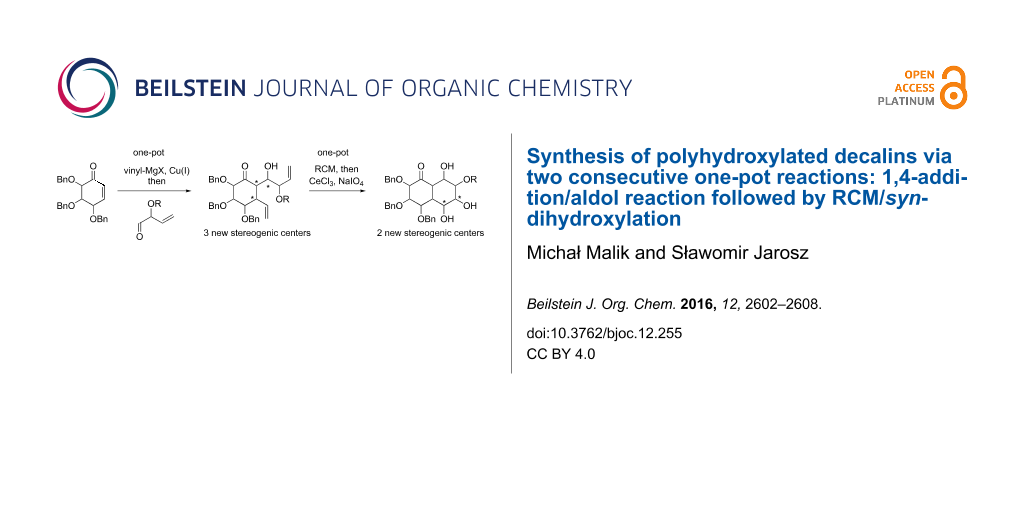
Abstract
Synthesis of novel polyhydroxylated derivatives of decalin is described. The presented methodology consists in a one-pot copper-catalyzed 1,4-addition of vinylmagnesium bromide to sugar-derived cyclohexenone, followed by an aldol reaction with a derivative of but-3-enal. The obtained diene is then subjected to an assisted tandem catalytic sequence: ring-closing metathesis with the subsequent reuse of the Ru-catalyst in the syn-dihydroxylation. The stereochemical outcome of these reactions is discussed.

Graphical Abstract
Introduction
Derivatives of carbohydrates, in which the endocyclic oxygen atom is replaced with a methylene group are known as carbasugars [1]. Due to their similarity to carbohydrates, these compounds often possess interesting biological properties, e.g., they may act as glycosidase inhibitors [2-4]. One of such compounds, acarbose [5-7], found an application as a drug and is marketed worldwide.
Five- and six-membered cyclitols are a well-known and recognizable group of carbasugars. The synthesis of these monocyclic compounds (such as 1 and 2, Figure 1) and evaluation of their biological properties remain a challenging and vital research area [8-11].
![[1860-5397-12-255-1]](/bjoc/content/figures/1860-5397-12-255-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: General structures of mono- and bicyclic carbasugars.
Figure 1: General structures of mono- and bicyclic carbasugars.
On the other hand, bicyclic analogs of cyclitols are not as well-documented as their monocylic counterparts. However, polyhydroxylated derivatives of decalin and hydrindane are interesting synthetic targets, since they can be regarded as conformationally locked carbasugars (3 and 4, Figure 1) [12-15]. Indeed, some of the bicyclic carbasugars were proven to possess strong and selective anti-glycosidase activity [16-18]. The polyoxygenated bicyclic motif is also found in some members of the nargenicin antibiotics family [19-21].
In our group, we have already proposed an approach to the synthesis of polyoxygenated bicyclic systems such as cis-decalins and trans-hydrindanes (Scheme 1) [22-25].
![[1860-5397-12-255-i1]](/bjoc/content/inline/1860-5397-12-255-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Approach to the synthesis of bicyclic carbasugars based on the use of sugar allyltins (previous works).
Scheme 1: Approach to the synthesis of bicyclic carbasugars based on the use of sugar allyltins (previous wor...
It relies on the use of sugar-derived allyltin (such as 5) followed by ZnCl2-induced fragmentation into dienoaldehyde 6, subsequent Horner–Wadsworth–Emmons olefination, and intramolecular Diels–Alder reaction.
In this paper, we explore the possibility of using a copper-catalyzed one-pot 1,4-addition of vinylmagnesium bromide to sugar-derived cyclic α,β-unsaturated ketones (such as 9), followed by an aldol reaction with an optically pure derivative of but-2-enal 10 to obtain a mixture of diastereomeric dienes, such as 11 (Scheme 2).
![[1860-5397-12-255-i2]](/bjoc/content/inline/1860-5397-12-255-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Approach to the synthesis of bicyclic decalins based on a 1,4-addition/aldol reaction followed by RCM with subsequent syn-dihydroxylation (this work).
Scheme 2: Approach to the synthesis of bicyclic decalins based on a 1,4-addition/aldol reaction followed by R...
As a result of this transformation, three new stereogenic centers are generated. A copper-catalyzed 1,4-addition of organometallic reagents to cyclic α,β-unsaturated ketones, followed by an aldol reaction has been already used in the synthesis of complex natural products [26-33]. Although this approach is challenging in terms of diastereoselectivity (up to eight possible diastereoisomers), it enables a rapid increase of molecular complexity. The major diastereoisomer of 11 can be then subjected to the assisted tandem catalytic sequence: RCM reaction, followed by the reuse of the ruthenium catalyst in the subsequent syn-dihydroxylation. As a result, the polyhydroxylated decalin derivative 12 was obtained. The syn-dihydroxylation of cyclic alkenes is among the most widely used methods for the introduction of hydroxy groups onto the ring. Such transformations are usually conducted with osmium-based catalysts [34,35], but due to their high cost and toxicity, other methodologies (in vast majority based on ruthenium) were also proposed during the last years [36-40]. An interesting approach, based on a ruthenium catalysis, was independently proposed by Blechert and Snapper [41,42]. Both groups described a methodology, in which the ring-closing metathesis (RCM) reaction is followed by the reuse of the Ru catalyst in the syn-dihydroxylation step. In our recent papers [43,44], we extended this concise and effective approach to the synthesis of bicyclic iminosugars.
To our knowledge, a sequence of a one-pot 1,4-addition/aldol reaction and RCM/syn-dihydroxylation has not been used yet (in such a consecutive manner) for the preparation of highly functionalized bicyclic structures.
Results and Discussion
The synthesis of polyhydroxylated cyclohexenones was initiated from known iododerivatives 13 and 14, prepared from suitably protected carbohydrates (Scheme 3) [45-47]. Each of these compounds was subjected to the zinc-mediated fragmentation, followed by the Jones oxidation.
![[1860-5397-12-255-i3]](/bjoc/content/inline/1860-5397-12-255-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: (a) i. Zn, MeOH/H2O, 60 °C, 2 h, ii. Jones reagent, acetone, rt, 1 h, iii. MeNHOMe∙HCl, py, Ph3P, CBr4, rt, 24 h, 67% (15, 3 steps) or 68% (16, 3 steps); (b) i. vinyl-MgBr, THF, 0 °C, 1 h, ii. Hoveyda–Grubbs II cat. (5 mol %), toluene, 50 °C, 24 h, 61% (17, 2 steps) or 75% (18, 2 steps).
Scheme 3: Reagents and conditions: (a) i. Zn, MeOH/H2O, 60 °C, 2 h, ii. Jones reagent, acetone, rt, 1 h, iii....
The resulting acid (not purified) was transformed into a Weinreb amide. As a result of this three-step sequence of reactions, derivatives 15 and 16 were formed, respectively. Addition of vinylmagnesium bromide to 15 and 16 afforded the corresponding dienes, which – without further purification – were subjected to the RCM reaction with the Hoveyda–Grubbs II generation catalyst. As a result, derivatives 17 and 18 were obtained, respectively.
The synthesis of aldehyde (S)-10 was initiated from D-mannitol. A known procedure led to compound 19 [48], which was easily transformed into diol 20, a convenient precursor of aldehyde (S)-10 (Scheme 4). Since such an aldehyde would be most likely unstable, we decided to deprotect 20 directly before the planned one-pot procedure and use it without purification.
![[1860-5397-12-255-i4]](/bjoc/content/inline/1860-5397-12-255-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: (a) i. BzCl, DCM, Et3N, DMAP, rt, 24 h, ii. HCl, MeOH/H2O, rt, 24 h, 55% (2 steps); (b) NaIO4, NaHCO3, DCM/H2O.
Scheme 4: Reagents and conditions: (a) i. BzCl, DCM, Et3N, DMAP, rt, 24 h, ii. HCl, MeOH/H2O, rt, 24 h, 55% (...
In order to obtain (R)-10, we decided to inverse the configuration at the free hydroxy group in allylic alcohol 19 by Mitsunobu reaction. Despite the fact that the use of allylic alcohols as substrates in Mitsunobu reactions can be a challenging task, some successful examples can be found in the literature [49,50]. However, the reaction with benzoic acid did not proceed at all, whereas application of p-nitrobenzoic acid yielded a complicated mixture of products. Therefore, an alternative approach had to be used to obtain (R)-10. A known procedure based on addition of vinylmagnesium bromide to isopropylidene-protected D-glyceraldehyde followed by silylation and separation of an equimolar mixture of diastereoisomers led to optically pure olefins 21 and 22 (Scheme 5) [51]. After desilylation followed by protection of the free hydroxy with a benzoyl group and deprotection of the isopropylidene moiety, diols 20 and 24 were obtained.
![[1860-5397-12-255-i5]](/bjoc/content/inline/1860-5397-12-255-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reagents and conditions: (a) TBAF∙3H2O, THF, rt, 24 h, 96% (19) or 94% (23); (b) i. BzCl, DCM, Et3N, DMAP, rt, 24 h, ii. HCl, MeOH/H2O, rt, 24 h, 58% (2 steps).
Scheme 5: Reagents and conditions: (a) TBAF∙3H2O, THF, rt, 24 h, 96% (19) or 94% (23); (b) i. BzCl, DCM, Et3N...
With α,β-unsaturated ketones 17 and 18 and two enantiomeric, optically pure aldehydes (R)- and (S)-10 in hand, we started to study the 1,4-addition of vinyl-MgBr followed by an aldol reaction (Scheme 6).
![[1860-5397-12-255-i6]](/bjoc/content/inline/1860-5397-12-255-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reagents and conditions: (a) vinyl-MgBr, CuBr∙Me2S, THF, −45 °C, 15 min, then (S)- or (R)-10, −45 °C to −20 °C, 15 min, 54% (25) or 48% (26).
Scheme 6: Reagents and conditions: (a) vinyl-MgBr, CuBr∙Me2S, THF, −45 °C, 15 min, then (S)- or (R)-10, −45 °...
The addition of a Grignard reagent to 17 was performed in THF at −45 °C in the presence of CuBr∙Me2S (0.5 equiv). After 15 min, a solution of aldehyde (either (R)- or (S)-10) in THF was added; the reaction mixture was warmed to −20 °C, and quenched with sat. NH4Cl. In the case of (S)-10, diolefin 25 was separated as major product (54%) out of 8 possible diastereoisomers. Moreover, small amounts of other diastereoisomers (ca. 8%) were also isolated as a complicated mixture. In the case of (R)-10, diolefin 26 was formed as major isomer (48%, contaminated with small amounts of other diastereoisomers of unknown stereochemistry). Small amounts of other isomers (ca. 5%) were also isolated as a complicated mixture. In both reactions, small amounts (ca. 10%) of intermediary product of 1,4-addition was also isolated. The stereochemistry of the obtained products was determined at the later stage (Figure 2). It turned out, that the transformation proceeds similarly in case of both enantiomers of 10, regardless of the stereochemistry at the α-position. First, the addition of the vinyl-MgBr to 17 proceeded anti in relation to the neighbouring benzyloxy group. Then, the resulting enolate was approached by an aldehyde from the opposite side to the newly attached vinyl group. A similar stereochemical outcome of such transformation has been already reported [26-33]. Moreover, the addition of aldehyde gave rise to the anti aldol product (both in 25 and 26). This is in accordance with the general observation, that E-enolates (enolates in six-membered rings are always E) give predominantly the anti-products in aldol reactions (Zimmerman–Traxler model) [52-54].
The application of the above-described procedure to cyclohexenone 18 was not successful, since it yielded a complicated, inseparable mixture of diastereoisomers instead of one major product.
Both diolefins 25 and 26 were subjected to the ring-closing metathesis (Scheme 7) with the Hoveyda–Grubbs II generation catalyst (5 mol %), followed by the reuse of the Ru catalyst in the subsequent syn-dihydroxylation with NaIO4 as a stoichiometric oxidant in the presence of catalytic amounts of CeCl3 (15 mol %).
![[1860-5397-12-255-i7]](/bjoc/content/inline/1860-5397-12-255-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reagents and conditions: (a) Hoveyda–Grubbs II cat. (5 mol %), toluene, 50 °C, 2 h, then evaporation, then NaIO4, CeCl3∙7H2O (15 mol %), MeCN/AcOEt, 0 °C, 20 min, 56% (27 + 28, dr = 8:1) or 53% (29, dr >99:1).
Scheme 7: Reagents and conditions: (a) Hoveyda–Grubbs II cat. (5 mol %), toluene, 50 °C, 2 h, then evaporatio...
As a result, in the case of diolefin 25 as substrate, a separable mixture of diols (dr = 8:1; 27 being the major isomer) was obtained in fair yield (56%). The same procedure applied to compound 26 resulted in the formation of virtually one diastereoisomer 29 (53%). The structure of all compounds was assigned by 2D-NOESY experiments (Figure 2). In the case of both olefins, syn-dihydroxylation proceeded from the less congested face of the double bond (anti in the relation to the neighbouring benzoyl group).
![[1860-5397-12-255-2]](/bjoc/content/figures/1860-5397-12-255-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Possible course of the syn-dihydroxylation leading to 27, 28, and 29.
Figure 2: Possible course of the syn-dihydroxylation leading to 27, 28, and 29.
Compounds 27 and 29 were subjected to Evans–Saksena reduction with NaBH(OAc)3 in MeCN/THF in the presence of AcOH (Scheme 8). Under these conditions, derivative 30 was obtained from 27 in good yield (67%) and with high diastereoselectivity (dr = 10:1; inseparable mixture). The same procedure applied to 29 yielded a separable mixture of diastereoisomers (dr = 4:1; 31 being the major isomer) in good yield (74%). The structure of all these compounds was confirmed by 2D-NOESY experiments (Scheme 8). In case of both substrates (27 and 29), the reduction proceeded anti in the relation to the β-hydroxy group [55,56]. These observations were confirmed by the NMR data in which proton H-4 (doublet of doublets) in 30 has one large coupling constant (9.5 Hz with H-3) and one small with H-5 (2.8 Hz), which indicates the syn relation between H-4 and H-5. Similar analysis in decalin 31 also points at the syn relation of H-4 and H-5. On the other hand, proton H-4 in 32 has two large coupling constants (~9.5 Hz), which indicates the anti relation between H-4 and H-5.
![[1860-5397-12-255-i8]](/bjoc/content/inline/1860-5397-12-255-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reagents and conditions: (a) NaBH(OAc)3, MeCN/THF/AcOH, rt, 24 h, 67% (30, dr >99:1) or 74% (31 + 32, dr = 4:1).
Scheme 8: Reagents and conditions: (a) NaBH(OAc)3, MeCN/THF/AcOH, rt, 24 h, 67% (30, dr >99:1) or 74% (31 + 32...
Conclusion
We have demonstrated a convenient and simple route to optically pure polyhydroxylated trans-decalins. The methodology is based on two consecutive one-pot procedures. The first one consists in Cu(I)-catalyzed 1,4-addition of vinylmagnesium bromide followed by an aldol reaction, which is then followed by an assisted tandem catalytic sequence: Ru-catalyzed ring-closing metathesis with subsequent syn-dihydroxylation with the re-use of Ru catalyst. These transformations applied to optically pure 4,5,6-tribenzyloxycyclohexenone 17 derived from D-glucose gave the desired bicyclic structures in good yields and very good diastereoselectivities. To sum up, the proposed methodology is an efficient way for the preparation of highly oxygenated trans-decalins. It offers a possibility to significantly increase molecular complexity in just two steps.
Supporting Information
| Supporting Information File 1: Experimental procedures, spectral data, and copies of the 1H and 13C spectra for all new compounds. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Arjona, O.; Gómez, A. M.; López, J. C.; Plumet, J. Chem. Rev. 2007, 107, 1919–2036. doi:10.1021/cr0203701
Return to citation in text: [1] -
Ganem, B. Glycomimetics that Inhibit Carbohydrate Metabolites. In Carbohydrate Mimics. Concepts and Methods; Chapleur, Y., Ed.; Wiley: Weinheim, 1998; pp 239–258.
Return to citation in text: [1] -
Tatsuta, K. Total Synthesis and Chemical Design of Useful Glycosidase Inhibitors. In Carbohydrate Mimics. Concepts and Methods; Chapleur, Y., Ed.; Wiley: Weinheim, 1998; pp 283–306.
Return to citation in text: [1] -
Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k
Return to citation in text: [1] -
Junge, B.; Heiker, F.-R.; Kurz, J.; Müller, L.; Schmidt, D. D.; Wünsche, C. Carbohydr. Res. 1984, 128, 235–268. doi:10.1016/0008-6215(84)85333-1
Return to citation in text: [1] -
Riccardi, G.; Giacco, R.; Parillo, M.; Turco, S.; Rivellese, A. A.; Ventura, M. R.; Contadini, S.; Marra, G.; Monteduro, M.; Santeusanio, F.; Brunetti, P.; Librenti, M. C.; Pontiroli, A. E.; Vedani, P.; Pozza, G.; Bergamini, L.; Bianchi, C. Diabetic Med. 1999, 16, 228–232. doi:10.1046/j.1464-5491.1999.00047.x
Return to citation in text: [1] -
Chaisson, J.-L.; Josse, R.-G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Lancet 2002, 359, 2072–2077. doi:10.1016/S0140-6736(02)08905-5
Return to citation in text: [1] -
Duchek, J.; Adams, D. R.; Hudlicky, T. Chem. Rev. 2011, 111, 4223–4258. doi:10.1021/cr1004138
Return to citation in text: [1] -
Baran, A.; Çambul, S.; Nebioglu, M.; Balci, M. J. Org. Chem. 2012, 77, 5086–5097. doi:10.1021/jo300655p
Return to citation in text: [1] -
Baran, A.; Aydin, G.; Savran, T.; Sahin, E.; Balci, M. Org. Lett. 2013, 15, 4350–4353. doi:10.1021/ol401823m
Return to citation in text: [1] -
El Bkassiny, S.; N’Go, I.; Sevrain, C. M.; Tikad, A.; Vincent, S. P. Org. Lett. 2014, 16, 2462–2465. doi:10.1021/ol500848q
Return to citation in text: [1] -
Nowogródzki, M.; Jarosz, S. Curr. Org. Chem. 2010, 14, 533–545. doi:10.2174/138527210790820230
Return to citation in text: [1] -
Nowogródzki, M.; Jarosz, S. Curr. Org. Chem. 2010, 14, 601–613. doi:10.2174/138527210790820267
Return to citation in text: [1] -
Jarosz, S.; Nowogródzki, M.; Magdycz, M.; Potopnyk, M.; Rauter, A.; Lindhorst, T. Carbobicyclic Sugar Mimics. In Carbohydrate Chemistry: Chemical and Biological Approaches; Rauter, A. P.; Lindhorst, T., Eds.; RSC Publishing, 2011; Vol. 37, pp 303–325.
Return to citation in text: [1] -
Ansari, A. A.; Rajasekaran, P.; Khan, M. M.; Vankar, Y. D. J. Org. Chem. 2014, 79, 1690–1699. doi:10.1021/jo402574h
Return to citation in text: [1] -
Mehta, G.; Ramesh, S. S. Chem. Commun. 2000, 2429–2430. doi:10.1039/b007224i
Return to citation in text: [1] -
Mehta, G.; Ramesh, S. S. Tetrahedron Lett. 2001, 42, 1987–1990. doi:10.1016/S0040-4039(01)00050-8
Return to citation in text: [1] -
Mehta, G.; Ramesh, S. S. Can. J. Chem. 2005, 83, 581–594. doi:10.1139/v05-032
Return to citation in text: [1] -
Enev, V. S.; Drescher, M.; Mulzer, J. Tetrahedron 2007, 63, 5930–5939. doi:10.1016/j.tet.2007.02.126
Return to citation in text: [1] -
Marchart, S.; Mulzer, J.; Enev, V. S. Org. Lett. 2007, 9, 813–816. doi:10.1021/ol0630189
Return to citation in text: [1] -
Enev, V. S.; Felzmann, W.; Gromov, A.; Marchart, S.; Mulzer, J. Chem. – Eur. J. 2012, 18, 9651–9668. doi:10.1002/chem.201200257
Return to citation in text: [1] -
Jarosz, S.; Nowogródzki, M.; Kołaczek, M. Tetrahedron: Asymmetry 2007, 18, 2674–2679. doi:10.1016/j.tetasy.2007.10.016
Return to citation in text: [1] -
Błońska, A.; Cmoch, P.; Jarosz, S. Tetrahedron: Asymmetry 2009, 20, 2728–2732. doi:10.1016/j.tetasy.2009.11.008
Return to citation in text: [1] -
Nowogródzki, M.; Malik, M.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1501–1511. doi:10.1016/j.tetasy.2012.09.015
Return to citation in text: [1] -
Magdycz, M.; Jarosz, S. Tetrahedron: Asymmetry 2013, 24, 1402–1411. doi:10.1016/j.tetasy.2013.09.018
Return to citation in text: [1] -
Imuta, S.; Ochiai, S.; Kuribayashi, M.; Chida, N. Tetrahedron Lett. 2003, 44, 5047–5051. doi:10.1016/S0040-4039(03)01186-9
Return to citation in text: [1] [2] -
Imuta, S.; Tanimoto, H.; Momose, M. K.; Chida, N. Tetrahedron 2006, 62, 6926–6944. doi:10.1016/j.tet.2006.04.079
Return to citation in text: [1] [2] -
Aldegunde, M. J.; Castedo, L.; Granja, J. R. Org. Lett. 2008, 10, 3789–3792. doi:10.1021/ol801469h
Return to citation in text: [1] [2] -
Dowling, M. S.; Vanderwal, C. D. J. Am. Chem. Soc. 2009, 131, 15090–15091. doi:10.1021/ja906241w
Return to citation in text: [1] [2] -
Matsuda, Y.; Endo, Y.; Saikawa, Y.; Nakata, M. J. Org. Chem. 2011, 76, 6258–6263. doi:10.1021/jo2010186
Return to citation in text: [1] [2] -
Li, H.; Wang, X.; Lei, X. Angew. Chem., Int. Ed. 2012, 51, 491–495. doi:10.1002/anie.201106753
Return to citation in text: [1] [2] -
Arisetti, N.; Reiser, O. Org. Lett. 2015, 17, 94–97. doi:10.1021/ol5032975
Return to citation in text: [1] [2] -
Hong, B.; Li, H.; Wu, J.; Zhang, J.; Lei, X. Angew. Chem., Int. Ed. 2015, 54, 1011–1015. doi:10.1002/anie.201409503
Return to citation in text: [1] [2] -
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009
Return to citation in text: [1] -
Donohoe, T. J.; Bataille, C. J. R.; Innocenti, P. Hydrogen-Bonding-Mediated Directed Osmium Dihydroxylation; Organic Reactions; John Wiley and Sons Ltd., 2012; pp 76 ff.
Return to citation in text: [1] -
Plietker, B.; Niggemann, M. Org. Lett. 2003, 5, 3353–3356. doi:10.1021/ol035335a
Return to citation in text: [1] -
Yip, W.-P.; Yu, W.-Y.; Zhu, N.; Che, C.-M. J. Am. Chem. Soc. 2005, 127, 14239–14249. doi:10.1021/ja0528230
Return to citation in text: [1] -
Plietker, B.; Niggemann, M. J. Org. Chem. 2005, 70, 2402–2405. doi:10.1021/jo048020x
Return to citation in text: [1] -
Bataille, C. J. R.; Donohoe, T. J. Chem. Soc. Rev. 2011, 40, 114–128. doi:10.1039/B923880H
Return to citation in text: [1] -
Piccialli, V. Molecules 2014, 19, 6534–6582. doi:10.3390/molecules19056534
Return to citation in text: [1] -
Beligny, S.; Eibauer, S.; Maechling, S.; Blechert, S. Angew. Chem., Int. Ed. 2006, 45, 1900–1903. doi:10.1002/anie.200503552
Return to citation in text: [1] -
Scholte, A. A.; An, M. H.; Snapper, M. L. Org. Lett. 2006, 8, 4759–4762. doi:10.1021/ol061837n
Return to citation in text: [1] -
Malik, M.; Witkowski, G.; Ceborska, M.; Jarosz, S. Org. Lett. 2013, 15, 6214–6217. doi:10.1021/ol403063v
Return to citation in text: [1] -
Malik, M.; Ceborska, M.; Witkowski, G.; Jarosz, S. Tetrahedron: Asymmetry 2015, 26, 29–34. doi:10.1016/j.tetasy.2014.11.013
Return to citation in text: [1] -
Hosokawa, S.; Isobe, M. J. Org. Chem. 1999, 64, 37–48. doi:10.1021/jo980088n
Return to citation in text: [1] -
Kleban, M.; Kautz, U.; Greul, J.; Hilgers, P.; Kugler, R.; Dong, H.-Q.; Jäger, V. Synthesis 2000, 1027–1033. doi:10.1055/s-2000-6305
Return to citation in text: [1] -
Skaanderup, P. R.; Poulsen, C. S.; Hyldtoft, L.; Jørgensen, M. R.; Madsen, R. Synthesis 2002, 1721–1727. doi:10.1055/s-2002-33641
Return to citation in text: [1] -
Schneider, C.; Kazmaier, U. Synthesis 1998, 1314–1320. doi:10.1055/s-1998-6103
Return to citation in text: [1] -
Swamy, K. C. K.; Kumar, N. N. B.; Balaraman, E.; Kumar, K. V. P. P. Chem. Rev. 2009, 109, 2551–2651. doi:10.1021/cr800278z
Return to citation in text: [1] -
Shull, B. K.; Sakai, T.; Nichols, J. B.; Koreeda, M. J. Org. Chem. 1997, 62, 8294–8303. doi:10.1021/jo9615155
Return to citation in text: [1] -
Bravo, F.; Castillón, S. Eur. J. Org. Chem. 2001, 507–516. doi:10.1002/1099-0690(200102)2001:3<507::AID-EJOC507>3.0.CO;2-R
Return to citation in text: [1] -
Zimmermann, H. E.; Traxler, M. D. J. Am. Chem. Soc. 1957, 79, 1920–1923. doi:10.1021/ja01565a041
Return to citation in text: [1] -
Kleschick, W. A.; Buse, C. T.; Heathcock, C. H. J. Am. Chem. Soc. 1977, 99, 247–248. doi:10.1021/ja00443a046
Return to citation in text: [1] -
Heathcock, C. H.; Buse, C. T.; Kleschick, W. A.; Pirrung, M. C.; Sohn, J. E.; Lampe, J. J. Org. Chem. 1980, 45, 1066–1081. doi:10.1021/jo01294a030
Return to citation in text: [1] -
Saksena, A. K.; Mangiaracina, P. Tetrahedron Lett. 1983, 24, 273–276. doi:10.1016/S0040-4039(00)81383-0
Return to citation in text: [1] -
Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035
Return to citation in text: [1]
| 52. | Zimmermann, H. E.; Traxler, M. D. J. Am. Chem. Soc. 1957, 79, 1920–1923. doi:10.1021/ja01565a041 |
| 53. | Kleschick, W. A.; Buse, C. T.; Heathcock, C. H. J. Am. Chem. Soc. 1977, 99, 247–248. doi:10.1021/ja00443a046 |
| 54. | Heathcock, C. H.; Buse, C. T.; Kleschick, W. A.; Pirrung, M. C.; Sohn, J. E.; Lampe, J. J. Org. Chem. 1980, 45, 1066–1081. doi:10.1021/jo01294a030 |
| 51. | Bravo, F.; Castillón, S. Eur. J. Org. Chem. 2001, 507–516. doi:10.1002/1099-0690(200102)2001:3<507::AID-EJOC507>3.0.CO;2-R |
| 26. | Imuta, S.; Ochiai, S.; Kuribayashi, M.; Chida, N. Tetrahedron Lett. 2003, 44, 5047–5051. doi:10.1016/S0040-4039(03)01186-9 |
| 27. | Imuta, S.; Tanimoto, H.; Momose, M. K.; Chida, N. Tetrahedron 2006, 62, 6926–6944. doi:10.1016/j.tet.2006.04.079 |
| 28. | Aldegunde, M. J.; Castedo, L.; Granja, J. R. Org. Lett. 2008, 10, 3789–3792. doi:10.1021/ol801469h |
| 29. | Dowling, M. S.; Vanderwal, C. D. J. Am. Chem. Soc. 2009, 131, 15090–15091. doi:10.1021/ja906241w |
| 30. | Matsuda, Y.; Endo, Y.; Saikawa, Y.; Nakata, M. J. Org. Chem. 2011, 76, 6258–6263. doi:10.1021/jo2010186 |
| 31. | Li, H.; Wang, X.; Lei, X. Angew. Chem., Int. Ed. 2012, 51, 491–495. doi:10.1002/anie.201106753 |
| 32. | Arisetti, N.; Reiser, O. Org. Lett. 2015, 17, 94–97. doi:10.1021/ol5032975 |
| 33. | Hong, B.; Li, H.; Wu, J.; Zhang, J.; Lei, X. Angew. Chem., Int. Ed. 2015, 54, 1011–1015. doi:10.1002/anie.201409503 |
| 1. | Arjona, O.; Gómez, A. M.; López, J. C.; Plumet, J. Chem. Rev. 2007, 107, 1919–2036. doi:10.1021/cr0203701 |
| 12. | Nowogródzki, M.; Jarosz, S. Curr. Org. Chem. 2010, 14, 533–545. doi:10.2174/138527210790820230 |
| 13. | Nowogródzki, M.; Jarosz, S. Curr. Org. Chem. 2010, 14, 601–613. doi:10.2174/138527210790820267 |
| 14. | Jarosz, S.; Nowogródzki, M.; Magdycz, M.; Potopnyk, M.; Rauter, A.; Lindhorst, T. Carbobicyclic Sugar Mimics. In Carbohydrate Chemistry: Chemical and Biological Approaches; Rauter, A. P.; Lindhorst, T., Eds.; RSC Publishing, 2011; Vol. 37, pp 303–325. |
| 15. | Ansari, A. A.; Rajasekaran, P.; Khan, M. M.; Vankar, Y. D. J. Org. Chem. 2014, 79, 1690–1699. doi:10.1021/jo402574h |
| 48. | Schneider, C.; Kazmaier, U. Synthesis 1998, 1314–1320. doi:10.1055/s-1998-6103 |
| 8. | Duchek, J.; Adams, D. R.; Hudlicky, T. Chem. Rev. 2011, 111, 4223–4258. doi:10.1021/cr1004138 |
| 9. | Baran, A.; Çambul, S.; Nebioglu, M.; Balci, M. J. Org. Chem. 2012, 77, 5086–5097. doi:10.1021/jo300655p |
| 10. | Baran, A.; Aydin, G.; Savran, T.; Sahin, E.; Balci, M. Org. Lett. 2013, 15, 4350–4353. doi:10.1021/ol401823m |
| 11. | El Bkassiny, S.; N’Go, I.; Sevrain, C. M.; Tikad, A.; Vincent, S. P. Org. Lett. 2014, 16, 2462–2465. doi:10.1021/ol500848q |
| 49. | Swamy, K. C. K.; Kumar, N. N. B.; Balaraman, E.; Kumar, K. V. P. P. Chem. Rev. 2009, 109, 2551–2651. doi:10.1021/cr800278z |
| 50. | Shull, B. K.; Sakai, T.; Nichols, J. B.; Koreeda, M. J. Org. Chem. 1997, 62, 8294–8303. doi:10.1021/jo9615155 |
| 5. | Junge, B.; Heiker, F.-R.; Kurz, J.; Müller, L.; Schmidt, D. D.; Wünsche, C. Carbohydr. Res. 1984, 128, 235–268. doi:10.1016/0008-6215(84)85333-1 |
| 6. | Riccardi, G.; Giacco, R.; Parillo, M.; Turco, S.; Rivellese, A. A.; Ventura, M. R.; Contadini, S.; Marra, G.; Monteduro, M.; Santeusanio, F.; Brunetti, P.; Librenti, M. C.; Pontiroli, A. E.; Vedani, P.; Pozza, G.; Bergamini, L.; Bianchi, C. Diabetic Med. 1999, 16, 228–232. doi:10.1046/j.1464-5491.1999.00047.x |
| 7. | Chaisson, J.-L.; Josse, R.-G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Lancet 2002, 359, 2072–2077. doi:10.1016/S0140-6736(02)08905-5 |
| 43. | Malik, M.; Witkowski, G.; Ceborska, M.; Jarosz, S. Org. Lett. 2013, 15, 6214–6217. doi:10.1021/ol403063v |
| 44. | Malik, M.; Ceborska, M.; Witkowski, G.; Jarosz, S. Tetrahedron: Asymmetry 2015, 26, 29–34. doi:10.1016/j.tetasy.2014.11.013 |
| 2. | Ganem, B. Glycomimetics that Inhibit Carbohydrate Metabolites. In Carbohydrate Mimics. Concepts and Methods; Chapleur, Y., Ed.; Wiley: Weinheim, 1998; pp 239–258. |
| 3. | Tatsuta, K. Total Synthesis and Chemical Design of Useful Glycosidase Inhibitors. In Carbohydrate Mimics. Concepts and Methods; Chapleur, Y., Ed.; Wiley: Weinheim, 1998; pp 283–306. |
| 4. | Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k |
| 45. | Hosokawa, S.; Isobe, M. J. Org. Chem. 1999, 64, 37–48. doi:10.1021/jo980088n |
| 46. | Kleban, M.; Kautz, U.; Greul, J.; Hilgers, P.; Kugler, R.; Dong, H.-Q.; Jäger, V. Synthesis 2000, 1027–1033. doi:10.1055/s-2000-6305 |
| 47. | Skaanderup, P. R.; Poulsen, C. S.; Hyldtoft, L.; Jørgensen, M. R.; Madsen, R. Synthesis 2002, 1721–1727. doi:10.1055/s-2002-33641 |
| 26. | Imuta, S.; Ochiai, S.; Kuribayashi, M.; Chida, N. Tetrahedron Lett. 2003, 44, 5047–5051. doi:10.1016/S0040-4039(03)01186-9 |
| 27. | Imuta, S.; Tanimoto, H.; Momose, M. K.; Chida, N. Tetrahedron 2006, 62, 6926–6944. doi:10.1016/j.tet.2006.04.079 |
| 28. | Aldegunde, M. J.; Castedo, L.; Granja, J. R. Org. Lett. 2008, 10, 3789–3792. doi:10.1021/ol801469h |
| 29. | Dowling, M. S.; Vanderwal, C. D. J. Am. Chem. Soc. 2009, 131, 15090–15091. doi:10.1021/ja906241w |
| 30. | Matsuda, Y.; Endo, Y.; Saikawa, Y.; Nakata, M. J. Org. Chem. 2011, 76, 6258–6263. doi:10.1021/jo2010186 |
| 31. | Li, H.; Wang, X.; Lei, X. Angew. Chem., Int. Ed. 2012, 51, 491–495. doi:10.1002/anie.201106753 |
| 32. | Arisetti, N.; Reiser, O. Org. Lett. 2015, 17, 94–97. doi:10.1021/ol5032975 |
| 33. | Hong, B.; Li, H.; Wu, J.; Zhang, J.; Lei, X. Angew. Chem., Int. Ed. 2015, 54, 1011–1015. doi:10.1002/anie.201409503 |
| 36. | Plietker, B.; Niggemann, M. Org. Lett. 2003, 5, 3353–3356. doi:10.1021/ol035335a |
| 37. | Yip, W.-P.; Yu, W.-Y.; Zhu, N.; Che, C.-M. J. Am. Chem. Soc. 2005, 127, 14239–14249. doi:10.1021/ja0528230 |
| 38. | Plietker, B.; Niggemann, M. J. Org. Chem. 2005, 70, 2402–2405. doi:10.1021/jo048020x |
| 39. | Bataille, C. J. R.; Donohoe, T. J. Chem. Soc. Rev. 2011, 40, 114–128. doi:10.1039/B923880H |
| 40. | Piccialli, V. Molecules 2014, 19, 6534–6582. doi:10.3390/molecules19056534 |
| 22. | Jarosz, S.; Nowogródzki, M.; Kołaczek, M. Tetrahedron: Asymmetry 2007, 18, 2674–2679. doi:10.1016/j.tetasy.2007.10.016 |
| 23. | Błońska, A.; Cmoch, P.; Jarosz, S. Tetrahedron: Asymmetry 2009, 20, 2728–2732. doi:10.1016/j.tetasy.2009.11.008 |
| 24. | Nowogródzki, M.; Malik, M.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1501–1511. doi:10.1016/j.tetasy.2012.09.015 |
| 25. | Magdycz, M.; Jarosz, S. Tetrahedron: Asymmetry 2013, 24, 1402–1411. doi:10.1016/j.tetasy.2013.09.018 |
| 41. | Beligny, S.; Eibauer, S.; Maechling, S.; Blechert, S. Angew. Chem., Int. Ed. 2006, 45, 1900–1903. doi:10.1002/anie.200503552 |
| 42. | Scholte, A. A.; An, M. H.; Snapper, M. L. Org. Lett. 2006, 8, 4759–4762. doi:10.1021/ol061837n |
| 19. | Enev, V. S.; Drescher, M.; Mulzer, J. Tetrahedron 2007, 63, 5930–5939. doi:10.1016/j.tet.2007.02.126 |
| 20. | Marchart, S.; Mulzer, J.; Enev, V. S. Org. Lett. 2007, 9, 813–816. doi:10.1021/ol0630189 |
| 21. | Enev, V. S.; Felzmann, W.; Gromov, A.; Marchart, S.; Mulzer, J. Chem. – Eur. J. 2012, 18, 9651–9668. doi:10.1002/chem.201200257 |
| 55. | Saksena, A. K.; Mangiaracina, P. Tetrahedron Lett. 1983, 24, 273–276. doi:10.1016/S0040-4039(00)81383-0 |
| 56. | Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035 |
| 16. | Mehta, G.; Ramesh, S. S. Chem. Commun. 2000, 2429–2430. doi:10.1039/b007224i |
| 17. | Mehta, G.; Ramesh, S. S. Tetrahedron Lett. 2001, 42, 1987–1990. doi:10.1016/S0040-4039(01)00050-8 |
| 18. | Mehta, G.; Ramesh, S. S. Can. J. Chem. 2005, 83, 581–594. doi:10.1139/v05-032 |
| 34. | Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009 |
| 35. | Donohoe, T. J.; Bataille, C. J. R.; Innocenti, P. Hydrogen-Bonding-Mediated Directed Osmium Dihydroxylation; Organic Reactions; John Wiley and Sons Ltd., 2012; pp 76 ff. |
© 2016 Malik and Jarosz; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)