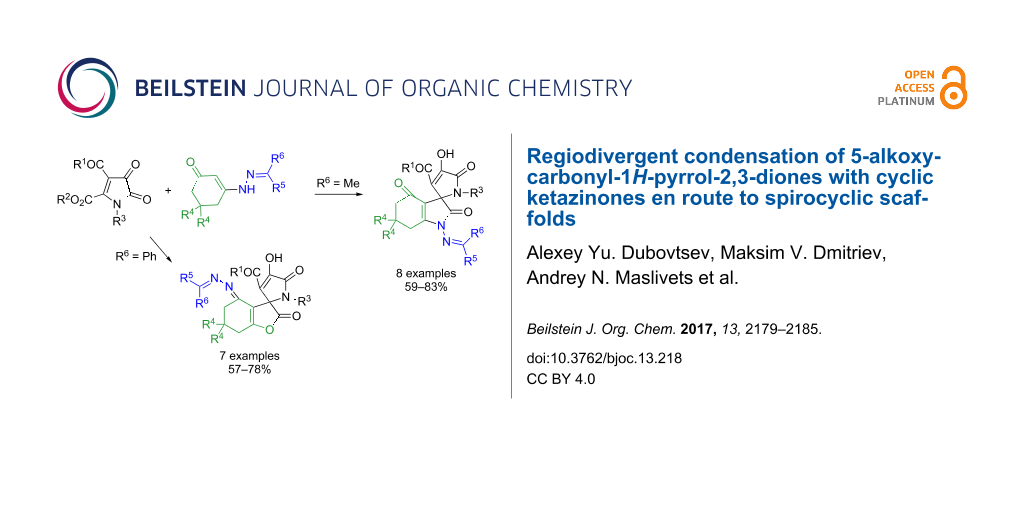
Abstract
The condensation of 5-alkoxycarbonyl-1H-pyrrolediones with cyclic ketazinones was systematically investigated. It was discovered that the regioselectivity of this reaction can be easily swapped between two alternative directions affording derivatives of partially hydrogenated indole or benzofurane. The control of this regioselectivity is efficiently governed by steric effects at the hydrazone moiety of the ketazinone reagent.

Graphical Abstract
Introduction
Molecular structures based on partially or exhaustively hydrogenated indole and benzofuran cores are omnipresent in nature. Both types of ring systems are found in a variety of important biologically active natural products [1-20], which continue to remain in the focus of attention for many research groups worldwide as targets for total synthesis and to serve as inspiration for exercises in drug design. Although many preparative methods of assembly of these structural units have been demonstrated, the development of new efficient and highly selective synthetic tools is always desired. From this prospective, we have become greatly interested in the chemistry of 1H-pyrrole-2,3-diones, polyfunctional building blocks that have great potential for the synthesis of heterocyclic structures. Indeed, these highly electrophilic cyclic vinylogous amides are known to undergo facile nucleophilic additions [21-25], sometimes accompanied with subsequent pericyclic rearrangements [26-33]. Not surprisingly, these versatile synthons have been successfully employed in the target-oriented synthesis of pyrrole-based natural alkaloids [34-38]. Herein we wish to report a new synthetic route towards spirocyclic scaffolds possessing partially hydrogenated indole or benzofuran cores. The featured approach is based on the highly efficient regiodivergent spirocondensation of 5-alkoxycarbonyl-1H-pyrrole-2,3-diones (serving as 1,2-bis-electrophiles) with cyclic ketazinones (serving as either 1,3-C,N- or 1,3-C,O-bis-nucleophiles).
Results and Discussion
Previously, we demonstrated a convenient approach towards spiro[indole-3,2’-pyrroles] 3 based on catalyst-free cyclocondensation of six-membered cyclic enamines 1 (vinylogous secondary amides) with 5-methoxycarbonyl-1H-pyrrolediones 2 [39-41]. This transformation involved the Michael addition of an enamine to the α,β-unsaturated carbonyl fragment of a pyrroledione and subsequent 5-exo-trig intramolecular nucleophilic attack of the amine moiety on the ester substituent (Scheme 1). Interestingly, it seems that the substitution at the nitrogen atom in structure 1 is very crucial in governing the desired reactivity. Indeed, our previous attempts to expand the substrate scope to include “vinylogous primary amides” 1a resulted in the discovery of an alternative mechanistic pathway. Apparently, in this case the primary amine moiety in intermediate 4 preferred a nucleophilic attack on the keto function, affording bridged hemiaminal structures 5 as kinetic products (Scheme 1) [42]. Upon extended heating, however, recyclization into the thermodynamically more favorable “normal” spirocyclic products 3a occurs.
![[1860-5397-13-218-i1]](/bjoc/content/inline/1860-5397-13-218-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Spirocyclization of enamines with 5-methoxycarbonyl-1H-pyrrolediones.
Scheme 1: Spirocyclization of enamines with 5-methoxycarbonyl-1H-pyrrolediones.
It should be further noted that in contrast to reactions of enamines, which readily provide the corresponding adducts with pyrrolediones in the absence of catalysts, the similar transformation involving enols 6 (vinylogous carbonates and carbamates) normally requires more forcing conditions, but usually can be facilitated by addition of catalytic amounts of organic base (Scheme 2) [43].
![[1860-5397-13-218-i2]](/bjoc/content/inline/1860-5397-13-218-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Non-catalyzed spirocyclization of enoles (vinylogous carbonates and carbamates) with 5-methoxycarbonyl-1H-pyrrolediones.
Scheme 2: Non-catalyzed spirocyclization of enoles (vinylogous carbonates and carbamates) with 5-methoxycarbo...
Interestingly, we figured out that the presence of heteroatom X in the structure of enol 6 is important for the normal course of the spirocyclization reaction. Our multiple attempts to carry out this transformation with the participation of enolates generated from cyclohexane-1,3-diones 8 (vinylogous carboxylates) in the presence of bases were unsuccessful. This reaction did not proceed in the presence of weak bases (such as tertiary amines), while the use of stronger bases (hydroxides or alkoxides) caused decomposition of the base-sensitive 1H-pyrrole-2,3-dione moiety 9. An attempt to perform the reaction in the presence of catalytic amounts of Brønsted acid (TsOH) also did not provide the spirocyclic products. Instead, bridged 1,3-oxazepine products 12 were formed in marginal yields, resulting from an initial aldol reaction involving the carbonyl group at C-3 of pyrroledione 9 and a subsequent intramolecular 6-endo-trig O-nucleophilic attack of the enol species at a conjugate unsaturated ketone moiety in the five-membered ring of intermediate 11 (Scheme 3).
![[1860-5397-13-218-i3]](/bjoc/content/inline/1860-5397-13-218-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Acid-catalyzed spirocyclization of enoles (vinylogous carboxylates) with 5-alkoxycarbonyl-1H-pyrrolediones.
Scheme 3: Acid-catalyzed spirocyclization of enoles (vinylogous carboxylates) with 5-alkoxycarbonyl-1H-pyrrol...
Although the reasons for such divergence in the reactivity are not completely understood, one could argue that the lower nucleophilicity of enolates derived from 8 as compared to that of enamine 1a could be responsible for this effect. Indeed in this case, the product of 1,2-addition of the C-nucleophile to the most reactive keto function and subsequent nucleophilic attack by the O-enolate on the conjugate C=C bond activated by two electron acceptors could become more preferable as compared to the alternative “normal” pathway, leading to adducts 10 and involving Michael addition followed by intramolecular transesterification. Remarkably, the resulting bridged products 12 have reversed regiochemistry as compared to the earlier-described cycloadducts 5 (Scheme 1). It should also be pointed out that all of these products were formed as single 10-endo diastereomers (see Scheme 3 for atom numbering). This configuration was unambiguously confirmed by a single crystal X-ray crystallography of compound 12ab (CCDC 1546062) shown in Figure 1.
![[1860-5397-13-218-1]](/bjoc/content/figures/1860-5397-13-218-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP drawing of compound 12ab (CCDC 1546062) showing 50% probability amplitude displacement ellipsoids.
Figure 1: ORTEP drawing of compound 12ab (CCDC 1546062) showing 50% probability amplitude displacement ellips...
Puzzled by this unexpected reactivity, we reasoned that the enolate moiety can be activated towards the desired spirocyclization via conversion of 1,3-diones 8 into mono-hydrazones. Indeed, while mono-imines of these ketones strongly prefer keto-enamine tautomeric form 13 over enol-imine form 14 (Scheme 4) [44,45], the corresponding hydrazones have been reported to favor enol-hydrazone tautomer 16 (Scheme 4) [46-48].
![[1860-5397-13-218-i4]](/bjoc/content/inline/1860-5397-13-218-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Formation of mono-imines and mono-hydrazones of 1,3-cyclohexanediones and tautomeric equilibrium between enol-imine and keto-enamine forms.
Scheme 4: Formation of mono-imines and mono-hydrazones of 1,3-cyclohexanediones and tautomeric equilibrium be...
Keeping this in mind we decided to test the reactivity of ketazinones 17 that were obtained via condensation of cyclohexanediones 8 with hydrazone of acetophenone. We anticipated the formation of spirolactones 20 in this process, resulting from intramolecular transesterification involving the enol moiety in tautomeric form 18 (Scheme 5). Surprisingly, an alternative direction of spirocyclization involving the reaction of tautomeric form 19 and affording lactam rings proceeded exclusively. The corresponding spiro[indole-3,2’-pyrroles] 21 were obtained exclusively in good yields (Scheme 5).
![[1860-5397-13-218-i5]](/bjoc/content/inline/1860-5397-13-218-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Spirocyclizations involving non-bulky ketazinones 17 and 5-alkoxycarbonyl-1H-pyrrolediones 9.
Scheme 5: Spirocyclizations involving non-bulky ketazinones 17 and 5-alkoxycarbonyl-1H-pyrrolediones 9.
The formation of the indoline ring was unambiguously confirmed by the crystal structure of compound 21ab (CCDC 1546063, Figure 2). It seems that the nucleophilicity of the hydrazine moiety prevailed, and the formation of the thermodynamically more favorable amide bond governed the overall direction of this transformation. It should be also taken into account that, unlike the aforementioned hydrazone structures 15 and 16, ketazinones appear to be more stable in keto-enamine form 17.
![[1860-5397-13-218-2]](/bjoc/content/figures/1860-5397-13-218-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP drawing of compound 21ab (CCDC 1546063) showing 50% probability amplitude displacement ellipsoids.
Figure 2: ORTEP drawing of compound 21ab (CCDC 1546063) showing 50% probability amplitude displacement ellips...
We reasoned that the nucleophilicity of the ketazinone moiety can be substantially reduced via incorporation of excessive steric bulk, which ultimately could help us to redirect the course of the reaction towards formation of spirolactones of type 20. To evaluate this idea, we prepared ketazinones 22 (crystal structure of ketazinone 22a was confirmed by X-ray crystallography (CCDC 1546065, Figure 3)), derived from benzophenone and tested their reactivity with pyrrolediones 9 (Scheme 6). Gratifyingly, this reasoning was correct, as we obtained the corresponding lactones 23 as the sole products in reasonable yields (Scheme 6).
![[1860-5397-13-218-3]](/bjoc/content/figures/1860-5397-13-218-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP drawing of compound 22a (CCDC 1546065) showing 50% probability amplitude displacement ellipsoids.
Figure 3: ORTEP drawing of compound 22a (CCDC 1546065) showing 50% probability amplitude displacement ellipso...
![[1860-5397-13-218-i6]](/bjoc/content/inline/1860-5397-13-218-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Spirocyclizations involving bulky ketazinones 22 and 5-alkoxycarbonyl-1H-pyrrolediones 9.
Scheme 6: Spirocyclizations involving bulky ketazinones 22 and 5-alkoxycarbonyl-1H-pyrrolediones 9.
The crystal structure of compound 23aa (CCDC 1546064) depicted in Figure 4 confirmed the formation of this elusive scaffold.
![[1860-5397-13-218-4]](/bjoc/content/figures/1860-5397-13-218-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP drawing of compound 23aa (CCDC 1546064) showing 50% probability amplitude displacement ellipsoids.
Figure 4: ORTEP drawing of compound 23aa (CCDC 1546064) showing 50% probability amplitude displacement ellips...
Conclusion
In conclusion, we discovered a new mode of cyclocondensations with “inverted” regiochemistry of addition, which involved the acid-catalyzed reaction of 5-alkoxycarbonyl-4-aroyl-1H-pyrrole-2,3-diones with cyclohexane-1,3-diones and lead to the formation of bridged 2,5-methanobenzo[f][1,3]oxazepines. We also found efficient regiodivergent spirocondensation of the same pyrrolediones with cyclic ketazinones affording the formation of spirocyclic scaffolds with either hydroindoles or hydrobenzofuran moieties. Remarkably, the direction of this condensation can be efficiently switched towards the formation of either of the products by tweaking steric parameters of the employed ketazinones.
Acknowledgements
This study was supported by the Russian Foundation for Basic Research (project #16-43-590613) and by the Ministry of Education and Science of the Russian Federation (project #4.6774.2017). Financial support in the frame of the Grant from the President of the Russian Federation (project #МК-1657.2017.3) is also gratefully acknowledged (MVD).
References
-
Raghavan, S.; Ravi, A. Org. Biomol. Chem. 2016, 14, 10222–10229. doi:10.1039/C6OB01966H
Return to citation in text: [1] -
Zhong, S.; Sauter, P. F.; Nieger, M.; Bräse, S. Chem. – Eur. J. 2015, 21, 11219–11225. doi:10.1002/chem.201501199
Return to citation in text: [1] -
Zhang, W.; Ma, L.; Li, S.; Liu, Z.; Chen, Y.; Zhang, H.; Zhang, G.; Zhang, Q.; Tian, X.; Yuan, C.; Zhang, S.; Zhang, W.; Zhang, C. J. Nat. Prod. 2014, 77, 1887–1892. doi:10.1021/np500362p
Return to citation in text: [1] -
Diethelm, S.; Schindler, C. S.; Carreira, E. M. Chem. – Eur. J. 2014, 20, 6071–6080. doi:10.1002/chem.201400046
Return to citation in text: [1] -
Hostetler, G.; Dunn, D.; McKenna, B. A.; Kopec, K.; Chatterjee, S. Chem. Biol. Drug Des. 2014, 83, 149–153. doi:10.1111/cbdd.12240
Return to citation in text: [1] -
Zhang, F.-M.; Tu, Y.-Q.; Liu, J.-D.; Fan, X.-H.; Shi, L.; Hu, X.-D.; Wang, S.-H.; Zhang, Y.-Q. Tetrahedron 2006, 62, 9446–9455. doi:10.1016/j.tet.2006.07.027
Return to citation in text: [1] -
Wipf, P.; Rector, S. R.; Takahashi, H. J. Am. Chem. Soc. 2002, 124, 14848–14849. doi:10.1021/ja028603t
Return to citation in text: [1] -
Padwa, A.; Brodney, M. A.; Dimitroff, M.; Liu, B.; Wu, T. J. Org. Chem. 2001, 66, 3119–3128. doi:10.1021/jo010020z
Return to citation in text: [1] -
Rigby, J. H.; Cavezza, A.; Heeg, M. J. J. Am. Chem. Soc. 1998, 120, 3664–3670. doi:10.1021/ja974317j
Return to citation in text: [1] -
Martin, S. F.; Davidsen, S. K. J. Am. Chem. Soc. 1984, 106, 6431–6433. doi:10.1021/ja00333a061
Return to citation in text: [1] -
Antúnez, D.-J. B.; Greenhalgh, M. D.; Fallan, C.; Slawin, A. M. Z.; Smith, A. D. Org. Biomol. Chem. 2016, 14, 7268–7274. doi:10.1039/C6OB01326K
Return to citation in text: [1] -
He, Z.-T.; Tian, B.; Fukui, Y.; Tong, X.; Tian, P.; Lin, G.-Q. Angew. Chem., Int. Ed. 2013, 52, 5314–5318. doi:10.1002/anie.201300137
Return to citation in text: [1] -
Takeda, N.; Ueda, M.; Kagehira, S.; Komei, H.; Tohnai, N.; Miyata, M.; Naito, T.; Miyata, O. Org. Lett. 2013, 15, 4382–4385. doi:10.1021/ol401897u
Return to citation in text: [1] -
Saliu, F.; Tolppa, E.-L.; Zoia, L.; Orlandi, M. Tetrahedron Lett. 2011, 52, 3856–3860. doi:10.1016/j.tetlet.2011.05.072
Return to citation in text: [1] -
Bertolini, F.; Pineschi, M. Org. Prep. Proced. Int. 2009, 41, 385–418. doi:10.1080/00304940903240836
Return to citation in text: [1] -
Barradas, S.; Carreño, M. C.; González-Lopez, M.; Latorre, A.; Urbano, A. Org. Lett. 2007, 9, 5019–5022. doi:10.1021/ol702236e
Return to citation in text: [1] -
Liu, Q.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 2552–2553. doi:10.1021/ja058337u
Return to citation in text: [1] -
Crimmins, M. T.; Brown, B. H.; Plake, H. R. J. Am. Chem. Soc. 2006, 128, 1371–1378. doi:10.1021/ja056334b
Return to citation in text: [1] -
Ogino, T.; Kurihara, C.; Baba, Y.; Kanematsu, K. J. Chem. Soc., Chem. Commun. 1994, 1979–1980. doi:10.1039/c39940001979
Return to citation in text: [1] -
Zambias, R. A.; Caldwell, C. G.; Kopka, I. E.; Hammond, M. L. J. Org. Chem. 1988, 53, 4135–4137. doi:10.1021/jo00252a056
Return to citation in text: [1] -
Li, J.-L.; Li, Q.; Yang, K.-C.; Li, Y.; Zhou, L.; Han, B.; Peng, C.; Gou, X.-J. RSC Adv. 2016, 6, 38875–38879. doi:10.1039/C6RA06441H
Return to citation in text: [1] -
Zhang, M.-L.; Yue, D.-F.; Wang, Z.-H.; Luo, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Beilstein J. Org. Chem. 2016, 12, 295–300. doi:10.3762/bjoc.12.31
Return to citation in text: [1] -
Bhanushali, M.; Zhao, C.-G. Tetrahedron Lett. 2012, 53, 359–362. doi:10.1016/j.tetlet.2011.11.056
Return to citation in text: [1] -
Abd El-Nabi, H. A. Tetrahedron 2002, 58, 135–141. doi:10.1016/S0040-4020(01)01029-8
Return to citation in text: [1] -
Fabian, W. M. F. J. Org. Chem. 2002, 67, 7475–7482. doi:10.1021/jo026142o
Return to citation in text: [1] -
Wang, C.; Jia, H.; Zhang, C.; Gao, Z.; Zhou, L.; Yuan, C.; Xiao, Y.; Guo, H. J. Org. Chem. 2017, 82, 633–641. doi:10.1021/acs.joc.6b02659
Return to citation in text: [1] -
Lu, Y.; Zhou, Y.; Lin, L.; Zheng, H.; Fu, K.; Liua, X.; Feng, X. Chem. Commun. 2016, 52, 8255–8258. doi:10.1039/C6CC03346F
Return to citation in text: [1] -
Wei, H.-X.; Zhou, C.; Ham, S.; White, J. M.; Birney, D. M. Org. Lett. 2004, 6, 4289–4292. doi:10.1021/ol048197d
Return to citation in text: [1] -
George, L.; Bernhardt, P. V.; Netsch, K.-P.; Wentrup, C. Org. Biomol. Chem. 2004, 2, 3518–3523. doi:10.1039/b412530d
Return to citation in text: [1] -
Cobas, A.; Guitian, E.; Castedo, L. J. Org. Chem. 1993, 58, 3113–3117. doi:10.1021/jo00063a034
Return to citation in text: [1] -
Kappe, C. O.; Kollenz, G.; Wentrup, C. J. Chem. Soc., Chem. Commun. 1992, 485–486. doi:10.1039/c39920000485
Return to citation in text: [1] -
Ott, W.; Kollenz, G.; Ziegler, E. Synthesis 1976, 546–547. doi:10.1055/s-1976-24119
Return to citation in text: [1] -
Kollenz, G.; Penn, G.; Ott, W.; Peters, K.; Peters, E.-M.; von Schnering, H. G. Chem. Ber. 1984, 117, 1310–1329. doi:10.1002/cber.19841170406
Return to citation in text: [1] -
Dagoneau, D.; Xu, Z.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2016, 55, 760–763. doi:10.1002/anie.201508906
Return to citation in text: [1] -
Yoshida, Y.; Mohri, K.; Isobe, K.; Itoh, T.; Yamamoto, K. J. Org. Chem. 2009, 74, 6010–6015. doi:10.1021/jo9008645
Return to citation in text: [1] -
Nimgirawath, S.; Udomputtimekakul, P. Molecules 2009, 14, 917–924. doi:10.3390/molecules14030917
Return to citation in text: [1] -
Hosoi, S.; Nagao, M.; Tsuda, Y.; Isobe, K.; Sano, T.; Ohta, T. J. Chem. Soc., Perkin Trans. 1 2000, 1505–1511. doi:10.1039/b001906m
Return to citation in text: [1] -
Tsuda, Y.; Ohshima, T.; Hosoi, S.; Kaneuchi, S.; Kiuchi, F.; Jun, T.; Sano, T. Chem. Pharm. Bull. 1996, 44, 500–508. doi:10.1248/cpb.44.500
Return to citation in text: [1] -
Bannikova, Yu. N.; Maslivets, A. N. Chem. Heterocycl. Compd. 2004, 40, 118–119. doi:10.1023/B:COHC.0000023780.19123.5b
Return to citation in text: [1] -
Silaichev, P. S.; Chudinova, M. A.; Slepukhin, P. A.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 1718–1722. doi:10.1134/S107042801111011X
Return to citation in text: [1] -
Silaichev, P. S.; Filimonov, V. O.; Slepukhin, P. A.; Maslivets, A. N. Russ. J. Org. Chem. 2012, 48, 561–565. doi:10.1134/S1070428012040173
Return to citation in text: [1] -
Denislamova, E. S.; Maslivets, A. N. Russ. J. Org. Chem. 2010, 46, 389–393. doi:10.1134/S1070428010030152
Return to citation in text: [1] -
Dubovtsev, A. Yu.; Silaichev, P. S.; Nazarov, M. A.; Dmitriev, M. V.; Maslivets, A. N.; Rubin, M. RSC Adv. 2016, 6, 84730–84737. doi:10.1039/C6RA16889B
Return to citation in text: [1] -
Dudek, G. O.; Holm, R. H. J. Am. Chem. Soc. 1962, 84, 2691–2696. doi:10.1021/ja00873a009
Return to citation in text: [1] -
Zhuo, J.-C. Magn. Reson. Chem. 1997, 35, 21–29. doi:10.1002/(SICI)1097-458X(199701)35:1<21::AID-OMR28>3.0.CO;2-I
Return to citation in text: [1] -
Dubrovay, Z.; Szalay, P. G. J. Mol. Model. 2014, 20, 2293. doi:10.1007/s00894-014-2293-6
Return to citation in text: [1] -
Guda, R.; Narsimha, S.; Babu, R.; Muthadi, S.; Lingabathula, H.; Palabindela, R.; Yellu, N. R.; Kumar, G.; Kasula, M. Bioorg. Med. Chem. Lett. 2016, 26, 5517–5523. doi:10.1016/j.bmcl.2016.10.006
Return to citation in text: [1] -
Weigert, F. J. J. Fluorine Chem. 1972, 1, 445–462. doi:10.1016/S0022-1139(00)82966-8
Return to citation in text: [1]
| 1. | Raghavan, S.; Ravi, A. Org. Biomol. Chem. 2016, 14, 10222–10229. doi:10.1039/C6OB01966H |
| 2. | Zhong, S.; Sauter, P. F.; Nieger, M.; Bräse, S. Chem. – Eur. J. 2015, 21, 11219–11225. doi:10.1002/chem.201501199 |
| 3. | Zhang, W.; Ma, L.; Li, S.; Liu, Z.; Chen, Y.; Zhang, H.; Zhang, G.; Zhang, Q.; Tian, X.; Yuan, C.; Zhang, S.; Zhang, W.; Zhang, C. J. Nat. Prod. 2014, 77, 1887–1892. doi:10.1021/np500362p |
| 4. | Diethelm, S.; Schindler, C. S.; Carreira, E. M. Chem. – Eur. J. 2014, 20, 6071–6080. doi:10.1002/chem.201400046 |
| 5. | Hostetler, G.; Dunn, D.; McKenna, B. A.; Kopec, K.; Chatterjee, S. Chem. Biol. Drug Des. 2014, 83, 149–153. doi:10.1111/cbdd.12240 |
| 6. | Zhang, F.-M.; Tu, Y.-Q.; Liu, J.-D.; Fan, X.-H.; Shi, L.; Hu, X.-D.; Wang, S.-H.; Zhang, Y.-Q. Tetrahedron 2006, 62, 9446–9455. doi:10.1016/j.tet.2006.07.027 |
| 7. | Wipf, P.; Rector, S. R.; Takahashi, H. J. Am. Chem. Soc. 2002, 124, 14848–14849. doi:10.1021/ja028603t |
| 8. | Padwa, A.; Brodney, M. A.; Dimitroff, M.; Liu, B.; Wu, T. J. Org. Chem. 2001, 66, 3119–3128. doi:10.1021/jo010020z |
| 9. | Rigby, J. H.; Cavezza, A.; Heeg, M. J. J. Am. Chem. Soc. 1998, 120, 3664–3670. doi:10.1021/ja974317j |
| 10. | Martin, S. F.; Davidsen, S. K. J. Am. Chem. Soc. 1984, 106, 6431–6433. doi:10.1021/ja00333a061 |
| 11. | Antúnez, D.-J. B.; Greenhalgh, M. D.; Fallan, C.; Slawin, A. M. Z.; Smith, A. D. Org. Biomol. Chem. 2016, 14, 7268–7274. doi:10.1039/C6OB01326K |
| 12. | He, Z.-T.; Tian, B.; Fukui, Y.; Tong, X.; Tian, P.; Lin, G.-Q. Angew. Chem., Int. Ed. 2013, 52, 5314–5318. doi:10.1002/anie.201300137 |
| 13. | Takeda, N.; Ueda, M.; Kagehira, S.; Komei, H.; Tohnai, N.; Miyata, M.; Naito, T.; Miyata, O. Org. Lett. 2013, 15, 4382–4385. doi:10.1021/ol401897u |
| 14. | Saliu, F.; Tolppa, E.-L.; Zoia, L.; Orlandi, M. Tetrahedron Lett. 2011, 52, 3856–3860. doi:10.1016/j.tetlet.2011.05.072 |
| 15. | Bertolini, F.; Pineschi, M. Org. Prep. Proced. Int. 2009, 41, 385–418. doi:10.1080/00304940903240836 |
| 16. | Barradas, S.; Carreño, M. C.; González-Lopez, M.; Latorre, A.; Urbano, A. Org. Lett. 2007, 9, 5019–5022. doi:10.1021/ol702236e |
| 17. | Liu, Q.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 2552–2553. doi:10.1021/ja058337u |
| 18. | Crimmins, M. T.; Brown, B. H.; Plake, H. R. J. Am. Chem. Soc. 2006, 128, 1371–1378. doi:10.1021/ja056334b |
| 19. | Ogino, T.; Kurihara, C.; Baba, Y.; Kanematsu, K. J. Chem. Soc., Chem. Commun. 1994, 1979–1980. doi:10.1039/c39940001979 |
| 20. | Zambias, R. A.; Caldwell, C. G.; Kopka, I. E.; Hammond, M. L. J. Org. Chem. 1988, 53, 4135–4137. doi:10.1021/jo00252a056 |
| 39. | Bannikova, Yu. N.; Maslivets, A. N. Chem. Heterocycl. Compd. 2004, 40, 118–119. doi:10.1023/B:COHC.0000023780.19123.5b |
| 40. | Silaichev, P. S.; Chudinova, M. A.; Slepukhin, P. A.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 1718–1722. doi:10.1134/S107042801111011X |
| 41. | Silaichev, P. S.; Filimonov, V. O.; Slepukhin, P. A.; Maslivets, A. N. Russ. J. Org. Chem. 2012, 48, 561–565. doi:10.1134/S1070428012040173 |
| 34. | Dagoneau, D.; Xu, Z.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2016, 55, 760–763. doi:10.1002/anie.201508906 |
| 35. | Yoshida, Y.; Mohri, K.; Isobe, K.; Itoh, T.; Yamamoto, K. J. Org. Chem. 2009, 74, 6010–6015. doi:10.1021/jo9008645 |
| 36. | Nimgirawath, S.; Udomputtimekakul, P. Molecules 2009, 14, 917–924. doi:10.3390/molecules14030917 |
| 37. | Hosoi, S.; Nagao, M.; Tsuda, Y.; Isobe, K.; Sano, T.; Ohta, T. J. Chem. Soc., Perkin Trans. 1 2000, 1505–1511. doi:10.1039/b001906m |
| 38. | Tsuda, Y.; Ohshima, T.; Hosoi, S.; Kaneuchi, S.; Kiuchi, F.; Jun, T.; Sano, T. Chem. Pharm. Bull. 1996, 44, 500–508. doi:10.1248/cpb.44.500 |
| 26. | Wang, C.; Jia, H.; Zhang, C.; Gao, Z.; Zhou, L.; Yuan, C.; Xiao, Y.; Guo, H. J. Org. Chem. 2017, 82, 633–641. doi:10.1021/acs.joc.6b02659 |
| 27. | Lu, Y.; Zhou, Y.; Lin, L.; Zheng, H.; Fu, K.; Liua, X.; Feng, X. Chem. Commun. 2016, 52, 8255–8258. doi:10.1039/C6CC03346F |
| 28. | Wei, H.-X.; Zhou, C.; Ham, S.; White, J. M.; Birney, D. M. Org. Lett. 2004, 6, 4289–4292. doi:10.1021/ol048197d |
| 29. | George, L.; Bernhardt, P. V.; Netsch, K.-P.; Wentrup, C. Org. Biomol. Chem. 2004, 2, 3518–3523. doi:10.1039/b412530d |
| 30. | Cobas, A.; Guitian, E.; Castedo, L. J. Org. Chem. 1993, 58, 3113–3117. doi:10.1021/jo00063a034 |
| 31. | Kappe, C. O.; Kollenz, G.; Wentrup, C. J. Chem. Soc., Chem. Commun. 1992, 485–486. doi:10.1039/c39920000485 |
| 32. | Ott, W.; Kollenz, G.; Ziegler, E. Synthesis 1976, 546–547. doi:10.1055/s-1976-24119 |
| 33. | Kollenz, G.; Penn, G.; Ott, W.; Peters, K.; Peters, E.-M.; von Schnering, H. G. Chem. Ber. 1984, 117, 1310–1329. doi:10.1002/cber.19841170406 |
| 21. | Li, J.-L.; Li, Q.; Yang, K.-C.; Li, Y.; Zhou, L.; Han, B.; Peng, C.; Gou, X.-J. RSC Adv. 2016, 6, 38875–38879. doi:10.1039/C6RA06441H |
| 22. | Zhang, M.-L.; Yue, D.-F.; Wang, Z.-H.; Luo, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Beilstein J. Org. Chem. 2016, 12, 295–300. doi:10.3762/bjoc.12.31 |
| 23. | Bhanushali, M.; Zhao, C.-G. Tetrahedron Lett. 2012, 53, 359–362. doi:10.1016/j.tetlet.2011.11.056 |
| 24. | Abd El-Nabi, H. A. Tetrahedron 2002, 58, 135–141. doi:10.1016/S0040-4020(01)01029-8 |
| 25. | Fabian, W. M. F. J. Org. Chem. 2002, 67, 7475–7482. doi:10.1021/jo026142o |
| 46. | Dubrovay, Z.; Szalay, P. G. J. Mol. Model. 2014, 20, 2293. doi:10.1007/s00894-014-2293-6 |
| 47. | Guda, R.; Narsimha, S.; Babu, R.; Muthadi, S.; Lingabathula, H.; Palabindela, R.; Yellu, N. R.; Kumar, G.; Kasula, M. Bioorg. Med. Chem. Lett. 2016, 26, 5517–5523. doi:10.1016/j.bmcl.2016.10.006 |
| 48. | Weigert, F. J. J. Fluorine Chem. 1972, 1, 445–462. doi:10.1016/S0022-1139(00)82966-8 |
| 44. | Dudek, G. O.; Holm, R. H. J. Am. Chem. Soc. 1962, 84, 2691–2696. doi:10.1021/ja00873a009 |
| 45. | Zhuo, J.-C. Magn. Reson. Chem. 1997, 35, 21–29. doi:10.1002/(SICI)1097-458X(199701)35:1<21::AID-OMR28>3.0.CO;2-I |
| 43. | Dubovtsev, A. Yu.; Silaichev, P. S.; Nazarov, M. A.; Dmitriev, M. V.; Maslivets, A. N.; Rubin, M. RSC Adv. 2016, 6, 84730–84737. doi:10.1039/C6RA16889B |
| 42. | Denislamova, E. S.; Maslivets, A. N. Russ. J. Org. Chem. 2010, 46, 389–393. doi:10.1134/S1070428010030152 |
© 2017 Dubovtsev et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)