Abstract
Ferrocene (FeCp2) was introduced as a non-magnetic guest molecule to activated carbon fibers (ACFs) as a nanographene-based host having localized spins originating from zigzag edges of graphene. The introduction of the guest molecule was confirmed by FTIR for ACFs-FeCp2 introduced at 55 (150) °C (FeCp2-ACFs-55(150)). The appearance of satellite Fe2p peaks and the increase in shake-up peak intensity of the C1s in the XPS spectrum proved the emergence of charge-transfer host–guest interaction in FeCp2-ACFs-150, supported by the red-shift of the G-band in the Raman spectrum. The six-times enhancement in the spin concentration in FeCp2-ACFs-150 compared with ACFs indicates the spin magnetism of the non-magnetic guest FeCp2+ molecule induced by a charge-transfer host–guest interaction in the nanographene host. The larger ESR linewidth than that expected from the dipolar interaction estimated by the localized spin concentration suggests the exchange interaction between the nanographene and FeCp2 spins. The narrowing of the ESR linewidth of FeCp2-ACFs-55 upon higher excitation microwave power suggests the inhomogeneity of the environment for FeCp2+ molecules in the nanographene host. The observed induction of spin magnetism by the interfacial interactions between the nanographene host and the guest molecules will be a promising strategy for developing a new class of molecular magnets.

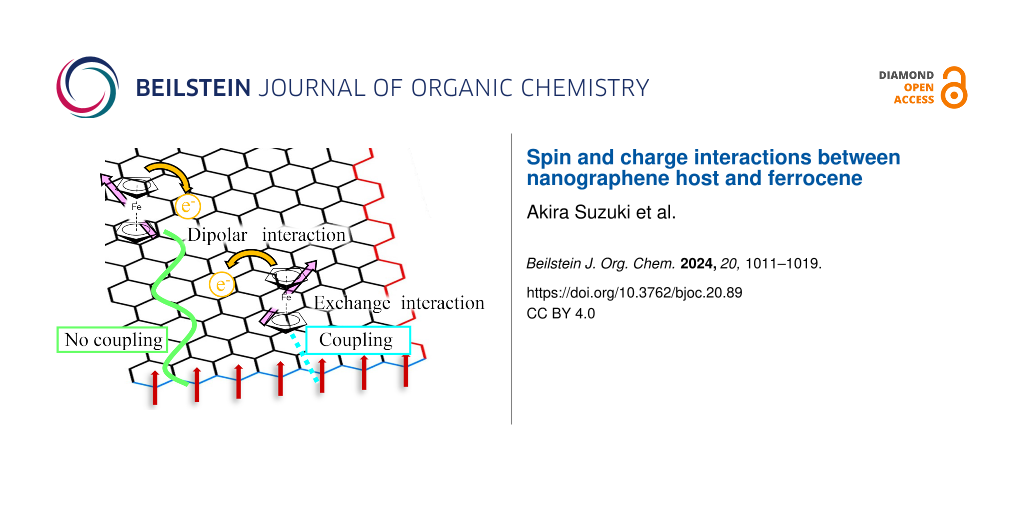
Graphical Abstract
Introduction
Nanocarbon host material, which is based on elements free from resource depletion, is attracting much attention due to its potential for creating a new class of functional materials with various guest molecules [1]. In particular, nanosized graphene called nanographene, the macroscopic limit of polycyclic aromatic hydrocarbon molecules, is a magnetic host material with spins localized at edges [2]. The presence of edges greatly modifies the electronic structure of nanographene, which strongly depends on the geometry of the edges [3-5]. Edges at the periphery of nanographene sheets consist of two kinds of geometry: zigzag edges and armchair edges. The presence of the zigzag part in the arbitrarily shaped edges results in the emergence of radical π-electron states called “edge states”, which are spatially localized at the edge site. The edge states appear at the Dirac point at which two linear conduction (anti-bonding) π*- and valence (bonding) π-bands touch each other in the electronic energy bands of graphene. Since the Fermi level is located at the Dirac point for neutral nanographene, edge states are half-filled like singly occupied molecular orbitals (SOMO) of radical states. Namely, nanographene sheets become magnetic and chemically active due to the edge states with localized spins of unpaired electrons [6]. Thus, it is interesting to introduce a magnetic guest molecule into a magnetic nanographene host regarding the development of a new class of magnetic materials.
Oxygen [7-11], nitrogen monoxide molecules [12,13], and potassium clusters having unpaired spins [14,15] have been introduced to nanographene hosts as magnetic guest molecules so far. However, the decomposition of molecules, the vanishment of guest magnetism, etc., after accommodation by the host material prevent magnetic interactions between the host and guest in these systems. The material design should be important in this viewpoint, especially in choosing appropriate guest molecules. Since π electrons extend to in-plane directions in nanographene, a guest molecule with an aromatic ring is promising for significant interaction with the nanographene host through π–π stacking.
Ferrocene (FeCp2) is a “sandwich” compound where the two cyclopentadienyl (Cp or C5H5-) rings sit above and below the Fe2+ ion [16]. The electronic structure of FeCp2 satisfies the 18-electron rule, so this compound is stable due to a closed L-shell structure in view of the atomic orbitals of Fe and it is a diamagnetic molecule (S = 0, no spin magnetism) compared with other metallocenes [17]. However, FeCp2 is easily oxidized to a monovalent cation, the electronic structure of which is magnetic (S = 1/2). Electron spin resonance (ESR) spectroscopy revealed the spin magnetism of cationic FeCp2 accommodated in mesoporous silica (MCM-41) [18]. So, ferrocene is expected to exhibit strong host–guest interactions with a nanographene host through π–π stacking.
Regarding ferrocene as a guest molecule for nanocarbon hosts, carbon nanotubes (CNTs) have been used to accommodate guest ferrocene molecules, where the amount of the charge transfer from ferrocene to CNTs was estimated from the shift of peaks for van Hove singularities in the valence-band photoemission spectrum [19,20]. The magnetic properties of ferrocene encapsulated into CNTs have also been investigated by superconducting quantum interference devices (SQUID) [21,22] and X-ray magnetic circular dichroism (XMCD) spectroscopy [23]. However, only a tiny paramagnetic behavior of encapsulated ferrocene was observed, and no magnetic host–guest interactions were reported due to the diamagnetic nature of CNTs.
Activated carbon fibers (ACFs) consist of a three-dimensional disordered network of nanographite domains, each of which is a loose stack of 3–4 nanographene sheets with a mean in-plane size of 2–3 nm. ACFs have huge specific surface areas (about 2000 m2/g [24,25]) due to the presence of nanopores of ca. 1 nm in diameter between the nanographite domains, where various guest chemical species can be accommodated [2]. Thus, ACFs are widely used as nanographene host materials. Interestingly, a ferromagnetic behavior below 120 K was once mentioned for FeCp2-adsorbed ACFs, even though no data was shown in the report [26]. It is necessary to clarify the magnetic interactions between the nanographene host and FeCp2 guest molecules to achieve a ferromagnet using nanographene host–guest systems.
In this study, we introduced ferrocene to ACFs and investigated the magnetic interaction between the host ACFs and ferrocene as magnetic guest molecule using X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, Fourier-transform infrared (FTIR) spectroscopy, magnetic susceptibility, and electron-spin resonance (ESR).
Experimental
Commercially available ACFs (Kuraray, FR-20), of which the precursor was a phenol-resin, were pre-heat-treated in a glass tube at 200 °C for 24 hours under 2 × 10−4 Pa before the introduction of FeCp2 in order to eliminate ambient gas molecules adsorbed in ACFs. The introduction of FeCp2 was carried out by exposing ACFs to the vapor phase of FeCp2 in the evacuated glass without exposing samples to air after the pre-heat-treatment at temperatures 55 °C and 150 °C (FeCp2-ACFs-55, FeCp2-ACFs-150), for 18 to 24 hours. The vapor pressure of ferrocene corresponding to each temperature was previously reported (15 Pa for 55 °C, 5.7 × 103 Pa for 150 °C) [27]. In the case of introduction at 150 °C, excessive FeCp2 precipitated as crystals on the surface of ACFs, which were removed by heating the FeCp2-treated ACFs at 150 °C for 3 hours without FeCp2 vapor.
XPS spectra were recorded using a PHI-5600 (ULVAC-PHI) with an Al Kα X-ray source (1486.7 eV) for samples mounted on an indium film. Raman spectroscopy measurements were performed by LabRAM HR Evolution instruments (Horiba) with an excitation laser operated at 532 nm in the wavenumber range from 1000 to 2000 cm−1. FTIR spectra were obtained using an FT/IR-6600 (JASCO) in ATR method with a diamond prism. Magnetic susceptibility measurements were carried out by a superconducting quantum interference device (SQUID) magnetometer (Quantum Design, MPMS-XL) in the field of 1 T between 2 K and 300 K, where ca. 30 mg of the samples vacuum-sealed in glass tubes (for ACFs and FeCp2-ACFs-150), mounted inside a plastic straw (FeCp2) were used. The Weiss temperature Θ and the temperature-independent term of the magnetic susceptibility were obtained by least-square fitting the data of the temperature-dependence of the observed susceptibility χ with the following equation based on a model of the summation of the Curie–Weiss localized magnetism and temperature contribution,
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-89-i1.svg?max-width=637&scale=1.18182)
where C denotes the Curie constant. The spin concentration Nspin for each sample was calculated from the Curie constant.
ESR measurements were performed using a conventional ESR X-band spectrometer (JEOL, JES-FA300) at room temperature, where ca 1 mg of samples vacuum-sealed in glass tubes were used. In order to prevent the skin effect, ACFs were ground in a mortar before the measurement.
Results and Discussion
XPS spectra acquired in a wide binding energy region for ACFs and FeCp2-ACFs-150 are shown in Figure 1. Peaks of C1s and O1s were observed in ACFs, while C1s, O1s, and Fe2p peaks appeared in the spectrum for FeCp2-ACFs-150.
![[1860-5397-20-89-1]](/bjoc/content/figures/1860-5397-20-89-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: XPS spectra for ACFs and FeCp2-ACFs-150. Peaks without labels originate from the indium substrate used for mounting the samples. The base lines of the spectra are shifted vertically from each other for clarification.
Figure 1: XPS spectra for ACFs and FeCp2-ACFs-150. Peaks without labels originate from the indium substrate u...
Figure 2 shows the Fe2p spectrum for FeCp2-ACFs-150 in a narrow binding energy region. The binding energies of the Fe2p peaks are similar to the reported value for FeCp2 [16]. So, the Fe2p peaks observed in FeCp2-ACFs-150 indicate the successful introduction of the FeCp2 molecule into ACFs as nanographene host. In addition to the main peaks, satellite peaks clearly appear at the higher energy side (ca. +3 eV), which indicates that the FeCp2 molecules partially become cationized (positively charged) in FeCp2-ACFs-150.
![[1860-5397-20-89-2]](/bjoc/content/figures/1860-5397-20-89-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: XPS spectrum for FeCp2-ACFs-150 in the Fe2p region shown with fitting curves.
Figure 2: XPS spectrum for FeCp2-ACFs-150 in the Fe2p region shown with fitting curves.
Figure 3a and b show the C1s spectra for FeCp2-ACFs-150 and ACFs in a narrow binding energy region, respectively.
![[1860-5397-20-89-3]](/bjoc/content/figures/1860-5397-20-89-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: XPS spectra of (left) FeCp2-ACFs-150 and (right) ACFs in the C1s region with fitting curves.
Figure 3: XPS spectra of (left) FeCp2-ACFs-150 and (right) ACFs in the C1s region with fitting curves.
Table 1 shows peak positions for XPS C1s, O1s, and Fe2p peaks for ACFs and FeCp2-ACFs-150, where elemental abundances are obtained from the peak intensity. The amount of FeCp2 is calculated as 0.39 mmol in 1 g of FeCp2-ACFs-150 from the total intensity ratio of Fe2p. The amount of the cationized FeCp2 (FeCp2+) is obtained as 0.15 mmol/g of FeCp2-ACFs-150 according to the intensity ratio of satellite peaks to the main peaks. The O1s peaks mainly come from oxygen-containing functional groups bonded to nanographene because of oxidization by ambient gaseous species. The elemental abundance ratio O/C of 0.07 is the same for ACFs and FeCp2-ACFs-150, similar to the carbon atoms ratio at the nanographene's edge part with the in-plane size of 2–3 nm, where the ratio of edge atoms to total carbon atoms is ca. 0.1 with the assumption of a model circular nanographene C324H36. Thus, oxygen-containing functional groups in ACFs are mainly attached to the edge part of nanographene, being consistent with the higher chemical activity of the edges of graphene [2,6]. Furthermore, the almost same elemental abundance ratio O/C between ACFs and FeCp2-ACFs-150 indicates no additional oxidization occurred in the process of FeCp2 introduction to ACFs due to contaminated oxygen from ambient gaseous species.
Table 1: The peak positions for XPS C1s, O1s, and Fe2p spectra and abundances of the peak components for FeCp2-ACFs-150 and ACFs.
| XPS peak | Sample | Binding energy (eV) | Abundance (atom %) |
| C1s (C=C) | ACFs | 284.6 | 64 |
| FeCp2-ACFs-150 | 283.7 | 61 | |
| C1s (C-O) | ACFs | 286.4 | 16 |
| FeCp2-ACFs-150 | 285.4 | 16 | |
| C1s (C=O) | ACFs | 288.4 | 3.8 |
| FeCp2-ACFs-150 | 287.4 | 3.0 | |
| C1s (shake-up) | ACFs | 290.4 | 9.3 |
| FeCp2-ACFs-150 | 289.6 | 12 | |
| O1s | ACFs | 532.9 | 6.8 |
| FeCp2-ACFs-150 | 531.4 | 6.6 | |
| N1s | ACFs | 400.3 | 0.4 |
| FeCp2-ACFs-150 | – | – | |
| Fe2p | ACFs | – | – |
| FeCp2-ACFs-150 |
707.1, 719.9
710.5, 723.2 |
0.8
0.5 |
|
Peaks of C1s are assigned to sp2 carbon atoms (C=C) of nanographene sheets, carbon atoms in/near oxygen-containing functional groups bounded to edges of nanographene sheets (C–O, C=O), shake-up peak by π–π* transition of conduction π electrons (Shake-up) [28]. A more considerable contribution of the plasmon peak in C1s indicates an increase in π-electron carriers for FeCp2-ACFs-150. Indeed, the shift of the C=C peak of FeCp2-ACFs-150 to the lower energy side indicates an increment of screening effect on photoemission hole by increasing in conduction electrons. Increasing in conduction π electron of nanographene in FeCp2-ACFs-150 suggests the charge transfer from FeCp2 to ACFs. In this connection, the observed partial ionization of FeCp2 is well understood by the structure of ACFs. Nanopores between nanographene domains provide huge spaces for the adsorption of guest molecules inside ACFs [2,6], where only a part of introduced molecules directly face the nanographene with the interfacial host–guest interactions, and the rest is accommodated into the nanopores without significant influences by nanographene domains.
The Raman spectra for both ACFs and FeCp2–ACFs-150 shown in Figure 4 exhibit two broad peaks near 1350 and 1600 cm−1. The peak around 1600 cm−1 corresponds to the Raman-allowed E2g mode (G-band) in graphene. The D-band peak around 1350 cm−1 is forbidden in ideal graphene crystals but becomes Raman-active by an electron-scattering process due to impurities and edges in crystallites [29]. The G and D-bands were fitted with two Lorentzian curves, as shown in Figure 4. Although characteristic peaks of FeCp2 molecules around 1100 cm−1 are not obtained in the spectrum for FeCp2-ACFs-150 due to their tiny abundance, the G-band for FeCp2-ACFs-150 shifts by 3 cm−1 to the lower wavenumber side compared to ACFs. The red shift indicates the weakening of C=C bonding in nanographene caused by filling anti-bonding states (π* states) due to electron injection into nanographene. This is consistent with the increment of shake-up peak for C1s in XPS. The Raman D-band also supports charge transfer from FeCp2 to nanographene in FeCp2-ACFs-150. The intensity ratio of the D-peak to G-peak ID/IG increases from 2.3 for ACFs to 2.4 for FeCp2-ACFs-150. The larger ID/IG corresponds to the more significant carrier scattering by introducing FeCp2 as a positively charged impurity caused by charge transfer with nanographene in FeCp2-ACFs-150. This is also supported by the increase in the linewidth of the G-band from 28 cm−1 (ACFs) to 31 cm−1 (FeCp2-ACFs-150).
![[1860-5397-20-89-4]](/bjoc/content/figures/1860-5397-20-89-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Raman spectra for ACFs and FeCp2-ACFs-150. Each raw data (black) was fitted to the G-band (blue) and D-band (red) components, resulting in the total curve (green). The base lines of the spectra are shifted vertically from each other for clarify.
Figure 4: Raman spectra for ACFs and FeCp2-ACFs-150. Each raw data (black) was fitted to the G-band (blue) an...
Figure 5 shows IR spectra for FeCp2, and the infrared spectra differences from that of ACFs for FeCp2-ACFs-150 (Δ([FeCp2-ACFs-150]-ACFs) and FeCp2-ACFs-55 (Δ([FeCp2-ACFs-55]-ACFs). The difference spectra exhibit peaks for vibration modes of Cp–Fe (ν), C–C (ν), C–H (γ), C–H (δ), Cp-breathing (ν) typical for FeCp2 molecular vibration [30]. These spectra also indicate the successful introduction of FeCp2 to ACFs and that most FeCp2 maintains its molecular structure inside the nanographene host in both of FeCp2-ACFs-55 and FeCp2-ACFs-150. Moreover, the higher peak intensities of FeCp2 molecular vibrations in the spectrum for FeCp2-ACFs-150 than FeCp2-ACFs-55 suggests that more guest molecules are introduced in FeCp2-ACFs-150. This is quite reasonable, taking the much higher vapor pressure of FeCp2 (5.7 × 103 Pa) into account in the process of guest molecular adsorption into ACFs for FeCp2-ACFs-150 than FeCp2-ACFs-55 (15 Pa). Here, it should be noted that the vibrational spectra are more distorted due to electromagnetic shielding effects by the conductive nature of graphene-based materials upon IR excitation. Thus, the “apparent” negative absorption peak in the spectrum of FeCp2-ACFs-55 is caused by the phase shift of the IR electromagnetic wave by shielding effects of the conductive nanographene assembly. Interestingly, the FeCp2 molecular vibrational peaks appear as typical positive peaks for FeCp2-ACFs-150, indicating that most molecules are present inside the nanopores of ACFs without significant interactions such as charge transfer and electromagnetic shielding. This is well consistent with the observed partial cationization of the guest molecules for FeCp2-ACFs-150 in XPS due to nanopore structure of the nanographene network in ACFs.
![[1860-5397-20-89-5]](/bjoc/content/figures/1860-5397-20-89-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The raw infrared spectrum for FeCp2 (black) and differential absorbance spectra for FeCp2-ACFs-55 (red) and FeCp2-ACFs-150 (green) after subtracting the ACFs spectrum as a background signal. The subtracted spectra are magnified five times, where the base lines are shifted vertically from each other for clarity. Raw spectra for FeCp2-ACFs-55 and FeCp2-ACFs-150 are shown in Figure S1 in Supporting Information File 1.
Figure 5: The raw infrared spectrum for FeCp2 (black) and differential absorbance spectra for FeCp2-ACFs-55 (...
The host–guest interaction between guest FeCp2 and host ACFs is most pronounced in the magnetic susceptibility measurements. The magnetic susceptibility for each sample shows the Curie–Weiss-type temperature dependence with temperature-independence susceptibility χconst, mainly composed of the orbital diamagnetism by core and π electrons. Regarding FeCp2-doped ACFs, ferromagnetism has been reported [26], but all of our samples showed only paramagnetism, and no ferromagnetism was observed in the present study.
The temperature-independent term of the magnetic susceptibility χconst for ACFs and FeCp2-ACFs-150 were obtained as −10 × 10−6 and −0.8 × 10−6 emu g−1, respectively. The reduction in the absolute value of χconst suggests the upshift of the Fermi energy from the Dirac point in the electronic band of nanographene in ACFs, being consistent with charge transfer from FeCp2 observed in XPS. The decrease in the absolute value of the Weiss temperature Θ from −6.4 K for ACFs to −0.1 K for ACFs-FeCp2-150 indicates the change in the character of the observed spins. The temperature dependences of the magnetic susceptibility χ multiplied by temperature T for FeCp2-ACFs-150, ACFs, and plain FeCp2 are shown in Figure 6, where χconst was subtracted. The quantity χT tells us an estimation for the effective spin concentration modified by spin-exchange interactions at each temperature. The χT for ACFs remains constant in the temperature region above 50 K. However, it becomes decreasing below 10 K as temperature decreases. This is featured as the localized spin paramagnetism with antiferromagnetic interaction. ACFs exhibit the localized spin paramagnetism by edge-states of nanographene, as reported [2]. As expected from the diamagnetic (no spin magnetism) electronic structure of FeCp2, plain FeCp2 shows only tiny paramagnetism caused by impurities. On the other hand, after FeCp2-introduction to ACFs, the χT remarkably increases for FeCp2-ACFs-150, as shown in Figure 6. Indeed, Nspin for FeCp2-ACFs-150 is about six times larger than that for non-doped ACFs (0.39 × 1020 g−1 for ACFs and 2.2 × 1020 g−1 for FeCp2-ACFs-150). The results indicate that FeCp2 in FeCp2-ACFs-150 becomes cationized (FeCp2+) and magnetic (S = 1/2) by the charge-transfer interaction with nanographene.
![[1860-5397-20-89-6]](/bjoc/content/figures/1860-5397-20-89-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The temperature (T) dependence of the magnetic susceptibility χ for FeCp2-ACFs-150, ACFs, and FeCp2 measured at 1 T, where the vertical axis denotes the χ multiplied by T. The temperature-independent diamagnetic contribution to the magnetic susceptibility is subtracted.
Figure 6: The temperature (T) dependence of the magnetic susceptibility χ for FeCp2-ACFs-150, ACFs, and FeCp2...
Here, we quantitatively discuss the observed spin magnetism induced by charge-transfer interactions between host and guest with the results of XPS. The additional spin concentration by FeCp2 introduction into ACFs is 1.8 × 1020 g−1 for FeCp2-ACFs-150, being equivalent to 0.30 mmol for 1 g of FeCp2-ACFs-150. The ratio of the satellite peak to the main peak of the XPS Fe2p spectrum tells us 0.15 mmol of FeCp2+ for 1 g of FeCp2-ACFs-150, which is in the same order as the observed spins induced by charge-transfer host–guest interactions. Considering the accuracy of elemental abundance by XPS (≈0.2 atom %), this is enough reasonable coincidence.
The Weiss temperature also supports the emergence of FeCp2+ spin magnetism. The absolute value of Θ decreases from −6 K to −0.09 K after FeCp2 introduction to ACFs. The wavefunction of the edge-state is coupled to each other through the π-electron systems in the nanographene sheet, resulting in antiferromagnetic interactions. In contrast, the wavefunction (molecular orbital) of FeCp2+ has a more isolated nature, and the exchange interactions between cation spins are less than those for edge-state spins. The apparent reduction in Θ for FeCp2-ACFs-150 is attributed to the contribution of FeCp2+ spins having less exchange interaction in the observed magnetic susceptibility. So, the spin magnetism of the guest molecule is induced by host–guest interactions in the nanographene host.
Despite the less interacting nature of FeCp2+ spins than that of edge-state spins, the ESR measurement proves the presence of the magnetic interaction between spins of the ACFs host and the guest FeCp2 molecule. Figure 7a shows the ESR spectra for ACFs and FeCp2-ACFs-55 at the excitation microwave powers of 1, 9, and 100 mW. The ESR linewidth of the spectrum for FeCp2-ACFs-150 was extremely broad to analyze the spectra, such as estimation of the linewidths and intensities, where the spectra are merged with the baseline contribution on the wider field range, being hard to distinguish from each other (Figure S2 in Supporting Information File 1).
![[1860-5397-20-89-7]](/bjoc/content/figures/1860-5397-20-89-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: a) ESR spectra of ACFs and FeCp2-ACFs-55 with different excitation microwave power. Each spectrum was magnified for clarity, where the base lines are shifted vertically from each other for clarity. b) The excitation microwave power dependence of ESR linewidth (ΔHpp) for ACFs and FeCp2-ACFs-55.
Figure 7: a) ESR spectra of ACFs and FeCp2-ACFs-55 with different excitation microwave power. Each spectrum w...
ESR for ACFs and FeCp2–ACFs-55 gives a g-value of 2.0019 for ACFs and a g-value of 2.003 for FeCp2-ACFs-55, being in good agreement with the reported value for edge-state spins of nanographene in ACFs (g = 2.002) [26]. These g-values are almost constant within the error bar in the excitation microwave power-dependence measurement. Figure 7b shows the excitation microwave power dependence of the ESR linewidth ΔHpp for ACFs and FeCp2-ACFs-55. If we only consider the magnetic dipole interaction, ΔHpp is proportional to Nspin [26], so ΔHpp for FeCp2-ACFs-150 should be six times larger than that for ACFs, according to the magnetic susceptibility results. However, the ESR of FeCp2-ACFs-150 results in a broad linewidth undistinguishable from the baseline (Figure S2 in Supporting Information File 1). Even ACFs-FeCp2-55 shown in Figure 7a, where FeCp2 was introduced at 1/300 lower pressure gives ΔHpp about seven times larger than ACFs in ESR. The observed ”excess” broadening factor for FeCp2-ACFs-150 and FeCp2-ACFs-55 in ESR is attributed to the exchange interaction between spins. Generally, the exchange interaction between identical spins results in the narrowing of the ESR peak (exchange narrowing). However, the exchange between non-identical spins broadens the ESR spectrum. In FeCp2-ACFs-55 and FeCp2-ACFs-150, the exchange interaction between nanographene spin and FeCp2+ spin (non-identical spins) contributes in addition to the magnetic dipolar interaction.
Figure 8 shows the square root of excitation microwave power dependence of relative intensities for ACFs and FeCp2-ACFs-55. At higher excitation power conditions, the relative intensities of ESR decrease because of a larger excitation rate than the spin relaxation rate (saturation), accompanied by linewidth broadening (saturation broadening). ACFs show moderate saturation phenomena with simple saturation broadening as the excitation power increases, where the coupling with conduction electrons in nanographene sheets is the primary path for spin energy relaxation. In Figure 8, the relative intensity for FeCp2-ACFs-55 suddenly decreases in the lower excitation power region. It shows a more saturated nature than ACFs at the same power region despite the larger conduction carrier than ACFs. The more saturating nature for FeCp2-ACFs-55 is well explained by the contribution of FeCp2+ spin having a more isolated nature than edge-state spins, consistent with the magnetic susceptibility results. However, the ΔHpp of FeCp2-ACFs-55 suddenly decreases even at the lower excitation power similar to the relative intensity and remains a decreasing trend despite its easily saturated nature. These behaviors suggest that spins have an inhomogeneous environment for spin relaxation. In FeCp2-ACFs-55, the adsorption site of FeCp2 is not unique, and each FeCp2 interacts with edge-state spins at the edges and π-electron carriers on nanographene sheets in different manners in ACFs as illustratively shown as the inset of Figure 8.
![[1860-5397-20-89-8]](/bjoc/content/figures/1860-5397-20-89-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: The square root of the excitation microwave power dependence of the relative ESR intensities for ACFs and FeCp2-ACFs-55. Each intensity is normalized by that at microwave power of 0.04 mW. The insert is an illustrative sketch to explain spin and charge interactions between the nanographene host and FeCp2 guest molecules.
Figure 8: The square root of the excitation microwave power dependence of the relative ESR intensities for AC...
Conclusion
Non-magnetic guest molecules with aromatic moiety were successfully introduced into the nanographene host. The charge-transfer interaction with the nanographene host in FeCp2-ACFs induces the localized spin magnetism of the guest molecule (cationized FeCp2). The presence of the exchange interaction by hybridization between FeCp2+ orbitals and edge-state orbitals is suggested in addition to the magnetic dipolar interaction. The observed induction and modulation of the spin magnetism by the interfacial interactions between magnetic nanographene host and guest molecules will give insight into a new class of developing methods of molecular magnets.
Supporting Information
| Supporting Information File 1: Supporting figures. | ||
| Format: PDF | Size: 130.6 KB | Download |
Funding
This work was partially supported by JSPS KAKENHI Grant Nos. 19K05410, 22K05056, 23K19256 and 26107532. This work was partially supported by the Research Center for Micro-nano Technology, the Research Center of Ion Beam Technology, and the Center for Instrumental analysis in Hosei University.
Data Availability Statement
The data that supports the findings of this study is available from the corresponding author upon reasonable request.
References
-
Hebard, A. F.; Rosseinsky, M. J.; Haddon, R. C.; Murphy, D. W.; Glarum, S. H.; Palstra, T. T. M.; Ramirez, A. P.; Kortan, A. R. Nature 1991, 350, 600–601. doi:10.1038/350600a0
Return to citation in text: [1] -
Takai, K.; Tsujimura, S.; Kang, F.; Inagaki, M. Graphene: Preparations, Properties, Applications, and Prospects; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2019-0-00212-9
Return to citation in text: [1] [2] [3] [4] [5] -
Fujita, M.; Wakabayashi, K.; Nakada, K.; Kusakabe, K. J. Phys. Soc. Jpn. 1996, 65, 1920–1923. doi:10.1143/jpsj.65.1920
Return to citation in text: [1] -
Kobayashi, Y.; Fukui, K.-i.; Enoki, T.; Kusakabe, K.; Kaburagi, Y. Phys. Rev. B 2005, 71, 193406. doi:10.1103/physrevb.71.193406
Return to citation in text: [1] -
Sakai, K.-i.; Takai, K.; Fukui, K.-i.; Nakanishi, T.; Enoki, T. Phys. Rev. B 2010, 81, 235417. doi:10.1103/physrevb.81.235417
Return to citation in text: [1] -
Enoki, T.; Takai, K. Solid State Commun. 2009, 149, 1144–1150. doi:10.1016/j.ssc.2009.02.054
Return to citation in text: [1] [2] [3] -
Kobayashi, N.; Enoki, T.; Ishii, C.; Kaneko, K.; Endo, M. J. Chem. Phys. 1998, 109, 1983–1990. doi:10.1063/1.476774
Return to citation in text: [1] -
Takahara, K.; Takai, K.; Enoki, T.; Sugihara, K. Phys. Rev. B 2007, 76, 035442. doi:10.1103/physrevb.76.035442
Return to citation in text: [1] -
Sevinçli, H.; Cuniberti, G. Phys. Rev. B 2010, 81, 113401. doi:10.1103/physrevb.81.113401
Return to citation in text: [1] -
Osipov, V. Y.; Shames, A. I.; Enoki, T.; Takai, K.; Endo, M.; Kaburagi, Y.; Vul', A. Y. Diamond Relat. Mater. 2010, 19, 492–495. doi:10.1016/j.diamond.2010.01.027
Return to citation in text: [1] -
Sumanasekera, G. U.; Chen, G.; Takai, K.; Joly, J.; Kobayashi, N.; Enoki, T.; Eklund, P. C. J. Phys.: Condens. Matter 2010, 22, 334208. doi:10.1088/0953-8984/22/33/334208
Return to citation in text: [1] -
Hao, S.-J.; Takai, K.; Joly, V. L. J.; Yokota, K.; Kiguchi, M.; Enoki, T. Bull. Chem. Soc. Jpn. 2012, 85, 376–388. doi:10.1246/bcsj.20110300
Return to citation in text: [1] -
Hao, S.; Takai, K.; Kang, F.; Enoki, T. Carbon 2008, 46, 110–116. doi:10.1016/j.carbon.2007.10.037
Return to citation in text: [1] -
Takai, K.; Eto, S.; Inaguma, M.; Enoki, T.; Ogata, H.; Tokita, M.; Watanabe, J. Phys. Rev. Lett. 2007, 98, 017203. doi:10.1103/physrevlett.98.017203
Return to citation in text: [1] -
Takai, K.; Suzuki, T.; Enoki, T.; Nishihara, H.; Kyotani, T. Phys. Rev. B 2010, 81, 205420. doi:10.1103/physrevb.81.205420
Return to citation in text: [1] -
Woodbridge, C. M.; Pugmire, D. L.; Johnson, R. C.; Boag, N. M.; Langell, M. A. J. Phys. Chem. B 2000, 104, 3085–3093. doi:10.1021/jp993235+
Return to citation in text: [1] [2] -
Barber, M.; Connor, J. A.; Derrick, L. M. R.; Hall, M. B.; Hillier, I. H. J. Chem. Soc., Faraday Trans. 2 1973, 69, 559–562. doi:10.1039/f29736900559
Return to citation in text: [1] -
Toda, Y.; Ishimaru, S.; Ikeda, R.; Mitani, T.; Kitao, S.; Seto, M. J. Phys. Chem. Solids 2004, 65, 471–473. doi:10.1016/j.jpcs.2003.09.018
Return to citation in text: [1] -
Shiozawa, H.; Pichler, T.; Grüneis, A.; Pfeiffer, R.; Kuzmany, H.; Liu, Z.; Suenaga, K.; Kataura, H. Adv. Mater. (Weinheim, Ger.) 2008, 20, 1443–1449. doi:10.1002/adma.200701466
Return to citation in text: [1] -
Sauer, M.; Shiozawa, H.; Ayala, P.; Ruiz-Soria, G.; Liu, X.; Chernov, A.; Krause, S.; Yanagi, K.; Kataura, H.; Pichler, T. Carbon 2013, 59, 237–245. doi:10.1016/j.carbon.2013.03.014
Return to citation in text: [1] -
Li, Y.; Kaneko, T.; Ogawa, T.; Takahashi, M.; Hatakeyama, R. Jpn. J. Appl. Phys. 2008, 47, 2048–2055. doi:10.1143/jjap.47.2048
Return to citation in text: [1] -
Li, Y.; Kaneko, T.; Ogawa, T.; Takahashi, M.; Hatakeyama, R. Chem. Commun. 2007, 254–256. doi:10.1039/b611256k
Return to citation in text: [1] -
Briones‐Leon, A.; Liu, X.; Ayala, P.; Kataura, H.; Yanagi, K.; Weschke, E.; Pichler, T.; Shiozawa, H. Phys. Status Solidi B 2012, 249, 2424–2427. doi:10.1002/pssb.201200165
Return to citation in text: [1] -
Oshida, K.; Kogiso, K.; Matsubayashi, K.; Takeuchi, K.; Kobayashi, S.; Endo, M.; Dresselhaus, M. S.; Dresselhaus, G. J. Mater. Res. 1995, 10, 2507–2517. doi:10.1557/jmr.1995.2507
Return to citation in text: [1] -
El-Merraoui, M.; Tamai, H.; Yasuda, H.; Kanata, T.; Mondori, J.; Nadai, K.; Kaneko, K. Carbon 1998, 36, 1769–1776. doi:10.1016/s0008-6223(98)00122-5
Return to citation in text: [1] -
Nakayama, A.; Ishii, C.; Takayama, T.; Watanabe, M.; Zanma, A.; Kaneko, K.; Sugihara, K. Synth. Met. 1997, 86, 2335–2336. doi:10.1016/s0379-6779(97)81149-6
Return to citation in text: [1] [2] [3] [4] -
Fulem, M.; Růžička, K.; Červinka, C.; Rocha, M. A. A.; Santos, L. M. N. B. F.; Berg, R. F. J. Chem. Thermodyn. 2013, 57, 530–540. doi:10.1016/j.jct.2012.07.023
Return to citation in text: [1] -
Chiang, Y.-C.; Lee, C.-Y.; Lee, H.-C. Mater. Chem. Phys. 2007, 101, 199–210. doi:10.1016/j.matchemphys.2006.03.007
Return to citation in text: [1] -
Cançado, L. G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y. A.; Mizusaki, H.; Jorio, A.; Coelho, L. N.; Magalhães-Paniago, R.; Pimenta, M. A. Appl. Phys. Lett. 2006, 88, 163106. doi:10.1063/1.2196057
Return to citation in text: [1] -
Lippincott, E. R.; Nelson, R. D. Spectrochim. Acta 1958, 10, 307–329. doi:10.1016/0371-1951(58)80097-1
Return to citation in text: [1]
| 1. | Hebard, A. F.; Rosseinsky, M. J.; Haddon, R. C.; Murphy, D. W.; Glarum, S. H.; Palstra, T. T. M.; Ramirez, A. P.; Kortan, A. R. Nature 1991, 350, 600–601. doi:10.1038/350600a0 |
| 7. | Kobayashi, N.; Enoki, T.; Ishii, C.; Kaneko, K.; Endo, M. J. Chem. Phys. 1998, 109, 1983–1990. doi:10.1063/1.476774 |
| 8. | Takahara, K.; Takai, K.; Enoki, T.; Sugihara, K. Phys. Rev. B 2007, 76, 035442. doi:10.1103/physrevb.76.035442 |
| 9. | Sevinçli, H.; Cuniberti, G. Phys. Rev. B 2010, 81, 113401. doi:10.1103/physrevb.81.113401 |
| 10. | Osipov, V. Y.; Shames, A. I.; Enoki, T.; Takai, K.; Endo, M.; Kaburagi, Y.; Vul', A. Y. Diamond Relat. Mater. 2010, 19, 492–495. doi:10.1016/j.diamond.2010.01.027 |
| 11. | Sumanasekera, G. U.; Chen, G.; Takai, K.; Joly, J.; Kobayashi, N.; Enoki, T.; Eklund, P. C. J. Phys.: Condens. Matter 2010, 22, 334208. doi:10.1088/0953-8984/22/33/334208 |
| 2. | Takai, K.; Tsujimura, S.; Kang, F.; Inagaki, M. Graphene: Preparations, Properties, Applications, and Prospects; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2019-0-00212-9 |
| 6. | Enoki, T.; Takai, K. Solid State Commun. 2009, 149, 1144–1150. doi:10.1016/j.ssc.2009.02.054 |
| 26. | Nakayama, A.; Ishii, C.; Takayama, T.; Watanabe, M.; Zanma, A.; Kaneko, K.; Sugihara, K. Synth. Met. 1997, 86, 2335–2336. doi:10.1016/s0379-6779(97)81149-6 |
| 3. | Fujita, M.; Wakabayashi, K.; Nakada, K.; Kusakabe, K. J. Phys. Soc. Jpn. 1996, 65, 1920–1923. doi:10.1143/jpsj.65.1920 |
| 4. | Kobayashi, Y.; Fukui, K.-i.; Enoki, T.; Kusakabe, K.; Kaburagi, Y. Phys. Rev. B 2005, 71, 193406. doi:10.1103/physrevb.71.193406 |
| 5. | Sakai, K.-i.; Takai, K.; Fukui, K.-i.; Nakanishi, T.; Enoki, T. Phys. Rev. B 2010, 81, 235417. doi:10.1103/physrevb.81.235417 |
| 23. | Briones‐Leon, A.; Liu, X.; Ayala, P.; Kataura, H.; Yanagi, K.; Weschke, E.; Pichler, T.; Shiozawa, H. Phys. Status Solidi B 2012, 249, 2424–2427. doi:10.1002/pssb.201200165 |
| 2. | Takai, K.; Tsujimura, S.; Kang, F.; Inagaki, M. Graphene: Preparations, Properties, Applications, and Prospects; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2019-0-00212-9 |
| 24. | Oshida, K.; Kogiso, K.; Matsubayashi, K.; Takeuchi, K.; Kobayashi, S.; Endo, M.; Dresselhaus, M. S.; Dresselhaus, G. J. Mater. Res. 1995, 10, 2507–2517. doi:10.1557/jmr.1995.2507 |
| 25. | El-Merraoui, M.; Tamai, H.; Yasuda, H.; Kanata, T.; Mondori, J.; Nadai, K.; Kaneko, K. Carbon 1998, 36, 1769–1776. doi:10.1016/s0008-6223(98)00122-5 |
| 17. | Barber, M.; Connor, J. A.; Derrick, L. M. R.; Hall, M. B.; Hillier, I. H. J. Chem. Soc., Faraday Trans. 2 1973, 69, 559–562. doi:10.1039/f29736900559 |
| 19. | Shiozawa, H.; Pichler, T.; Grüneis, A.; Pfeiffer, R.; Kuzmany, H.; Liu, Z.; Suenaga, K.; Kataura, H. Adv. Mater. (Weinheim, Ger.) 2008, 20, 1443–1449. doi:10.1002/adma.200701466 |
| 20. | Sauer, M.; Shiozawa, H.; Ayala, P.; Ruiz-Soria, G.; Liu, X.; Chernov, A.; Krause, S.; Yanagi, K.; Kataura, H.; Pichler, T. Carbon 2013, 59, 237–245. doi:10.1016/j.carbon.2013.03.014 |
| 16. | Woodbridge, C. M.; Pugmire, D. L.; Johnson, R. C.; Boag, N. M.; Langell, M. A. J. Phys. Chem. B 2000, 104, 3085–3093. doi:10.1021/jp993235+ |
| 21. | Li, Y.; Kaneko, T.; Ogawa, T.; Takahashi, M.; Hatakeyama, R. Jpn. J. Appl. Phys. 2008, 47, 2048–2055. doi:10.1143/jjap.47.2048 |
| 22. | Li, Y.; Kaneko, T.; Ogawa, T.; Takahashi, M.; Hatakeyama, R. Chem. Commun. 2007, 254–256. doi:10.1039/b611256k |
| 14. | Takai, K.; Eto, S.; Inaguma, M.; Enoki, T.; Ogata, H.; Tokita, M.; Watanabe, J. Phys. Rev. Lett. 2007, 98, 017203. doi:10.1103/physrevlett.98.017203 |
| 15. | Takai, K.; Suzuki, T.; Enoki, T.; Nishihara, H.; Kyotani, T. Phys. Rev. B 2010, 81, 205420. doi:10.1103/physrevb.81.205420 |
| 12. | Hao, S.-J.; Takai, K.; Joly, V. L. J.; Yokota, K.; Kiguchi, M.; Enoki, T. Bull. Chem. Soc. Jpn. 2012, 85, 376–388. doi:10.1246/bcsj.20110300 |
| 13. | Hao, S.; Takai, K.; Kang, F.; Enoki, T. Carbon 2008, 46, 110–116. doi:10.1016/j.carbon.2007.10.037 |
| 18. | Toda, Y.; Ishimaru, S.; Ikeda, R.; Mitani, T.; Kitao, S.; Seto, M. J. Phys. Chem. Solids 2004, 65, 471–473. doi:10.1016/j.jpcs.2003.09.018 |
| 2. | Takai, K.; Tsujimura, S.; Kang, F.; Inagaki, M. Graphene: Preparations, Properties, Applications, and Prospects; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2019-0-00212-9 |
| 6. | Enoki, T.; Takai, K. Solid State Commun. 2009, 149, 1144–1150. doi:10.1016/j.ssc.2009.02.054 |
| 27. | Fulem, M.; Růžička, K.; Červinka, C.; Rocha, M. A. A.; Santos, L. M. N. B. F.; Berg, R. F. J. Chem. Thermodyn. 2013, 57, 530–540. doi:10.1016/j.jct.2012.07.023 |
| 16. | Woodbridge, C. M.; Pugmire, D. L.; Johnson, R. C.; Boag, N. M.; Langell, M. A. J. Phys. Chem. B 2000, 104, 3085–3093. doi:10.1021/jp993235+ |
| 26. | Nakayama, A.; Ishii, C.; Takayama, T.; Watanabe, M.; Zanma, A.; Kaneko, K.; Sugihara, K. Synth. Met. 1997, 86, 2335–2336. doi:10.1016/s0379-6779(97)81149-6 |
| 26. | Nakayama, A.; Ishii, C.; Takayama, T.; Watanabe, M.; Zanma, A.; Kaneko, K.; Sugihara, K. Synth. Met. 1997, 86, 2335–2336. doi:10.1016/s0379-6779(97)81149-6 |
| 26. | Nakayama, A.; Ishii, C.; Takayama, T.; Watanabe, M.; Zanma, A.; Kaneko, K.; Sugihara, K. Synth. Met. 1997, 86, 2335–2336. doi:10.1016/s0379-6779(97)81149-6 |
| 2. | Takai, K.; Tsujimura, S.; Kang, F.; Inagaki, M. Graphene: Preparations, Properties, Applications, and Prospects; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2019-0-00212-9 |
| 29. | Cançado, L. G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y. A.; Mizusaki, H.; Jorio, A.; Coelho, L. N.; Magalhães-Paniago, R.; Pimenta, M. A. Appl. Phys. Lett. 2006, 88, 163106. doi:10.1063/1.2196057 |
| 30. | Lippincott, E. R.; Nelson, R. D. Spectrochim. Acta 1958, 10, 307–329. doi:10.1016/0371-1951(58)80097-1 |
| 28. | Chiang, Y.-C.; Lee, C.-Y.; Lee, H.-C. Mater. Chem. Phys. 2007, 101, 199–210. doi:10.1016/j.matchemphys.2006.03.007 |
| 2. | Takai, K.; Tsujimura, S.; Kang, F.; Inagaki, M. Graphene: Preparations, Properties, Applications, and Prospects; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2019-0-00212-9 |
| 6. | Enoki, T.; Takai, K. Solid State Commun. 2009, 149, 1144–1150. doi:10.1016/j.ssc.2009.02.054 |
© 2024 Suzuki et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.