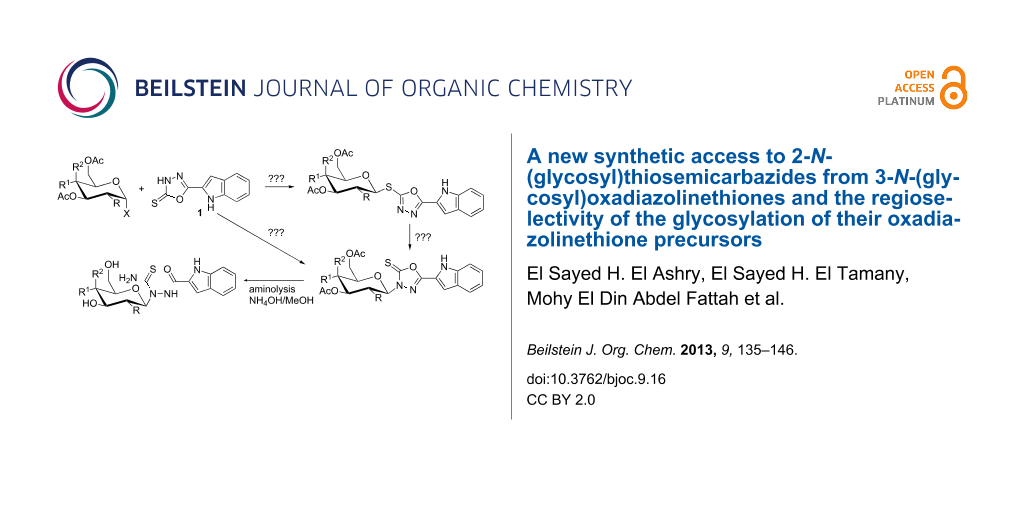
Abstract
Glycosylations of 5-(1H-indol-2-yl)-1,3,4-oxadiazoline-2(3H)-thione delivered various degrees of S- and/or N-glycosides depending on the reaction conditions. S-Glycosides were obtained regiospecifically by grinding oxadiazolinethiones with acylated α-D-glycosyl halides in basic alumina, whereas 3-N-(glycosyl)oxadiazolinethiones were selectively obtained by reaction with HgCl2 followed by heating the resultant chloromercuric salt with α-D-glycosyl halides in toluene under reflux. On using Et3N or K2CO3 as a base, mixtures of S- (major degree) and N-glycosides (minor degree) were obtained. Pure 3-N-(glycosyl)oxadiazolinethiones can also be selectively obtained from glycosylsulfanyloxadiazoles by the thermal S→N migration of the glycosyl moiety, which is proposed to occur by a tight-ion-pair mechanism. Thermal S→N migration of the glycosyl moiety can be used for purification of mixtures of S- or N-glycosides to obtain the pure N-glycosides. The aminolysis of the respective S- or N-glycosides with ammonia in aqueous methanol served as further confirmation of their structures. While in S-glycosides the glycosyl moiety was cleaved off again, 3-N-(glycosyl)oxadiazolinethiones showed a ring opening of the oxadiazoline ring (without affecting the glycosyl moiety) to give N-(glycosyl)thiosemicarbazides. Herewith, a new synthetic access to one of the four classes of glycosylthiosemicarbazides was found. The ultimate confirmation of new structures was achieved by X-ray crystallography. Finally, action of ammonia on benzylated 3-N-(galactosyl)oxadiazolinethione unexpectedly yielded 3-N-(galactosyl)triazolinethione. This represents a new path to the conversion of glycosyloxadiazolinethiones to new glycosyltriazolinethione nucleosides, which was until now unknown.

Graphical Abstract
Introduction
Modified nucleosides are versatile motifs for studying the relationship between the structure and functions of nucleic acids and problems of metabolism, besides their main potential in curing viral infections and cancer diseases [1]. The 1,3,4-oxadiazolines and 1,2,4-triazolines are potential inhibitors of physiologically relevant isoforms of the zinc enzyme carbonic anhydrase (CA, EC 4.2.2.1), i.e., cytosolic CA I and CA II, the tumor-associated transmembrane isoenzyme CA IX with inhibition constants in the low micromolar range [2]. Coupling of aglycones with relevant glycosyl donors is the common approach involved in the synthesis of most nucleosides. Another strategy for the synthesis of nucleoside analogues is the use of glycosylamines and related N-bonded glycosides [3-7], such as glycosylisothiocyanates, which afford glycosylthiosemicarbazides [8] or glycosyl 3-thioureidothiourea derivatives [9]. These 4-N-(glycosyl)thiosemicarbazides [8] were used for the synthesis of Schiff-like bases [10] and 4-N-glycosyl(thiosemicarbazido)phosphorothionates as precursors for the synthesis of the herbicidal and fungicidal agents thiazolidine-4-ones [11].
Thiosemicarbazide (TSC) and related amines were used to prepare modified amylase and amylopectin for biological studies [12]. Moreover, glycosylthiosemicarbazides are formed in vivo to modify cell-surface sialic acid. This is known as a metabolic cell-surface-engineering technique for cell-surface interactions and consequently shows the potential of these compounds for the development of anticancer agents [13-16]. Antituberculosis effects of glycosylthiosemicarbazides were also reviewed [17]. Glycosylamines are used also as enzyme inhibitors and vaccine precursors [18-21], and in glycopeptide synthesis [22,23] and in glycodendrimers and glycoclusters [24,25].
There are four structural isomers of glycosyl-thiosemicarbazides according to the location of the glycosyl residue on the thiosemicarbazide; the 1-N-, 2-N-, S- and 4-N- (glycosyl)thiosemicarbazides (Scheme 1). To the best of our knowledge, examples of 1-N-(I)-type [26] and 4-N-(III)-type glycosylthiosemicarbazides [27-31] are known. While we had focused on simple alkylations and glycosylations of 1,3,4-oxadiazolethione [32,33] and on 2-N-(glycosyl)thiosemicarbazides II obtained from the aminolysis of 3-N-(glycosyl)oxadiazolinethione precursors, the growing interest in glycosylthiosemicarbazides stimulated the development of direct regioselective formations of the corresponding 3-N-(glycosyl)oxadiazolinethiones, which are shown in this work. Earlier literature showed that 3-N-(glycosyl)oxadiazolinethiones could be obtained from the chloromercuric salts of 1,3,4-oxadiazolethiones by heating at high temperatures in dry nitromethane under reflux using dry calcium sulfate [34] or toluene [35-37]. In continuation of our previous work [33] in which the synthesis of compounds 5 to 13 and 20 were published in a preliminary manner without the experimental details, this paper introduces more examples for the thermal rearrangement of S-glycosides to the corresponding N-glycosides, which deliver excellent yields without solvent or a catalyst in a short reaction time. Moreover, the thiosemicarbazide II precursors can also be selectively obtained from 3-N-(glycosyl)oxadiazolinethiones.
![[1860-5397-9-16-i1]](/bjoc/content/inline/1860-5397-9-16-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structures of the different glycosylthiosemicarbazides.
Scheme 1: Structures of the different glycosylthiosemicarbazides.
In addition to our previous work [33], we investigated the potential of the benzylated N-glycosides as precursors for the respective thiosemicarbazides. Surprisingly, the formation of the galactosyl-1,2,4-triazoline-3-thione from the galactosyl-1,3,4-oxadiazole-2(3H)-thione was discovered for the first time. This is a new method for the conversion of an 1,3,4-oxadiazole-2(3H)-thione ring into a 1,2,4-triazoline-3-thione, because formerly, only conversions of 1,3,4-oxadiazolethione (by reaction of hydrazine hydrate) to 4-amino-1,2,4-triazolinethiones were reported [38-40]. The products of the new reactions are verified by the X-ray single-crystal analysis of the galactosyl-1,2,4-triazoline-3-thione, and a representative glucopyranosylsulfanyl-1,3,4-oxadiazole and glucopyranosylthiosemicarbazide, each.
Results and Discussion
The regiospecificity of glycosylations of 1,3,4-oxadiazolinethiones was tested by reacting 5-(1H-indol-2-yl)-1,3,4-oxadiazoline-2(3H)-thione (1) with a set of α-D-glycosyl halides 2–4 under different conditions. Glycosylations in the presence of either Et3N or K2CO3 yielded a mixture of both the S- (5–7) and N-linked (8–10) glycosides in varying yields of 42–71% and 12–35%, respectively (Scheme 2). Generally, both bases could be considered more regioselective towards S-glycosides than towards the N-glycosyl analogues. Grinding the reactants with basic alumina afforded regiospecifically S-linked glycosides 5–7 in 52–63% yields. However, if glycosylations were carried out on the chloromercuric salt of 1 in toluene under reflux, 3-N-linked glycosides 8–10 were regiospecifically obtained in 48–60% yields.
![[1860-5397-9-16-i2]](/bjoc/content/inline/1860-5397-9-16-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Glycosylations of oxadiazolinethione.
Scheme 2: Glycosylations of oxadiazolinethione.
Anomeric β-configurations of the S-linked 5–7 and N-linked 8–10 glycosides were deduced from the 1H NMR spectra, which revealed large J1,2 values of 10.3–10.6 and 9.2–9.5 Hz, respectively, for the anomeric protons. The chemical shifts of the anomeric protons of S-glycosides were at lower values (δ 5.45–5.56 ppm) than those of N-glycosides (δ 5.92–6.10 ppm). The differentiation between S- and N-glycosides was supported by the presence or absence of the signal of the carbon atom of the C=S moiety in the 13C NMR spectra. In other words, the 13C NMR spectra of the N-glycosides 8–10 revealed signals at δc 176.10–177.40 ppm. Anomeric carbons in both types were observed at δ 83.20–84.00 ppm.
Thermal rearrangement of the S-glycosides 5–7 under solvent-free and atmospheric conditions afforded the corresponding 3-N-glycosides 8–10. The conversion was achieved in a few minutes with good to excellent yields (60–90%). Therefore, the thermal rearrangement from S- to N-glycosides may also serve as a rapid and economic (free of solvents) purification step for crude mixtures of S- and 3-N-glycosides obtained from glycosylations mediated by either Et3N or K2CO3. Additional experiments on crude mixtures of S- and 3-N-glycosides successfully afforded pure N-glycosides.
The mechanism of this rearrangement is presumably proceeding by an ionization–recombination pathway in which a thermally induced heterolysis of the thioglycosidic bond results in a tight ion pair generated upon ionization of this bond. An intramolecular reaction of the tight ion pair results in migration of the glycosyl moiety from sulfur to nitrogen, which proceeds with complete retention of configuration. As a result, a tight-ion-pair mechanism in which the migrating group retains chirality is suggested (Scheme 2).
Aminolysis of the N-glycosides 8–10 (Scheme 3) mediated with ammonia in aqueous methanolic solution led to de-O-acetylation of the glycan moieties along with aminolysis of the oxadiazole ring affording 2-N-(glycosyl)thiosemicarbazides 11–13 instead of the corresponding nucleosides 14–16. The oxadiazole ring cleavage combined with de-O-acetylation of 8–10 is proven by three facts. First, the molecular weights derived from the mass spectra of the products 11–13 are higher by seventeen atomic mass units than would be those of the deacetylated products 14–16. Second, the IR spectra of 11–13 show new amide absorption bands that do not appear in the IR spectra of their precursors. Finally, the 13C NMR spectra of 11–13 show signals for NC=O groups at δC 159.40–162.90 ppm in addition to the NC=S groups at δC 183.10–184.50 ppm. Deacetylation of 10 is only confined to the O-acetyl groups, but the N-acetyl group survived under these conditions. This was confirmed by an extra 13C NMR signal at δC 172.50 ppm for the NHCOCH3 group of 13. The presence of 1H NMR signals at δH 6.46–6.55 ppm as doublets with coupling constants of J1,2 = 8.5–9.0 Hz indicated the stability of the pyranose ring and its β-anomeric configuration under these conditions.
![[1860-5397-9-16-i3]](/bjoc/content/inline/1860-5397-9-16-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of glycosylthiosemicarbazides by solvolysis.
Scheme 3: Synthesis of glycosylthiosemicarbazides by solvolysis.
Aminolysis of the S-glycosides 5–7 (Scheme 4) under the same conditions (ammonia in aqueous methanolic solution) generally led to splitting of the thioglycosyl moiety as a result of hydrolysis of the bond between the oxadiazole (C2) and the glycosidic sulfur atom. Hence, the indolyloxadiazolone 20 was formed [33] whereas the expected deacetylated glycosides 17–19 were not obtained. Structure elucidation of 20 yielded a melting point of 271–273 °C [33], while literature reports give a value of 102 °C [41] or 285 °C [42]. Therefore, compound 20 was prepared in another reaction sequence [33] to prove its structure and the correctness of the physical and structural data obtained. Hence, the indol-2-carbohydrazide (21) was reacted with methyl chloroformate followed by cyclization of the resulting ester 22. As a result, 20 was obtained in high yield, and its structural analytical data were identical with those of the aminolysis product (Scheme 4).
![[1860-5397-9-16-i4]](/bjoc/content/inline/1860-5397-9-16-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Solvolysis of glycosylsulfanyloxadiazoles.
Scheme 4: Solvolysis of glycosylsulfanyloxadiazoles.
For ultimate confirmation of the structures of S- and N-glycosides 5–10, the benzylation of the indole was chosen, because these derivatives can serve to prove the glycosyl rearrangement and glycosyl TSC formation. Moreover, crystalline glycosides are obtained that are suitable for X-ray analysis. In the presence of K2CO3, the S- and N-glycosides 23–28 (Scheme 5) were obtained. Disappearance of the NH-signal of the indole in the 1H NMR spectra of all products and appearance of the benzyl methylene protons at δH 5.76–5.97 ppm and the methylene carbon at δC 48.4–48.8 ppm in the 13C NMR spectra support a successful benzylation. Additional NMR signals of the phenyl protons and carbon atoms of the phenyl ring were another strong evidence for the N-benzylation of the indole units. The different (yet large) values of the coupling constants (J1,2) of the anomeric protons of S-glycosides 23–25 (10–10.4 Hz) and of the N-glycosides 26–28 (9.5–9.6 Hz), respectively, indicate that all benzylated glycosides still have the β-configuration. Successful thermal S→N glycosyl migration (and the concomitant change of structural analytical data of the products) served as a final proof that N-glycosylated products were obtained from their former pure S-analogues.
![[1860-5397-9-16-i5]](/bjoc/content/inline/1860-5397-9-16-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Benzylation of the S- and N-glycosides 5–10.
Scheme 5: Benzylation of the S- and N-glycosides 5–10.
Aminolysis of the benzylated N-glycosides 26 and 27 was done with ammonia in aqueous methanolic solution to obtain additional new thiosemicarbazide derivatives. Although, the reaction conditions remained unchanged compared to the cleavage of 8–10, derivatives 26 and 27 (Scheme 6) yielded different products under these conditions. Oxadiazole ring cleavage combined with de-O-acetylation converted 26 into the corresponding 2-N-(glycosyl)thiosemicarbazide 29 while the galactonucleoside 27 was converted into the galactosyltriazole 30. We propose that it is formed by a cyclization with the elimination of water from a thiosemicarbazide as intermediate. The structure of 30 (and other key compounds) could be confirmed by X-ray crystallography. The large values of the coupling constants (J1,2) of the anomeric protons of the N-glycosides 29, 30 indicate that β-configuration is still retained here.
![[1860-5397-9-16-i6]](/bjoc/content/inline/1860-5397-9-16-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Aminolysis of benzylated indolyl-3-N-(glucosyl)- and (galactosyl)oxadiazolinethiones.
Scheme 6: Aminolysis of benzylated indolyl-3-N-(glucosyl)- and (galactosyl)oxadiazolinethiones.
X-ray analysis
Single-crystal X-ray diffraction experiments yielded unambiguous confirmations of the structural assignments of the S-glycoside 23, the 2-N-(glycosyl)thiosemicarbazide 29, and the galactosyltriazole 30. Single crystals were slowly grown in EtOH. 23 crystallized in the monoclinic space group C2 with the following unit cell parameters: a = 25.7078 Å, α = 90º, b = 7.1500 Å, β = 105.2576º, c = 17.8411 Å, γ = 90º and V = 3163.81 Å3. The crystallographic data of 23 are shown in Table 1. The whole molecule is nonplanar; the phenyl group is located perpendicular to the plane of the indole ring by making a torsion angle of C(8)N(1)C(1)C(2) = 95.9º while the oxadiazole ring is located in the plane of the indole ring by making torsion angles of N(1)C(15)C(16)O(1) = −171.48°, C(14)C(15)C(16)O(1) = 10.6º and N(1)C(15)C(16)N(2) = 9.2°, respectively. The crystallographic analysis revealed that the sugar molecule has the glucopyranose form and has 4C1 conformation. The anomeric β-configuration is derived from the bond lengths of O(2)–C(18) and O(2)–C(19), which are 1.416 and 1.442 Å, and all substituents have equatorial orientation. Moreover, the crystal data revealed that the S(1)–C(17) bond length is 1.743 Å suggesting a certain degree of conjugation with the oxadiazole ring, whereas the S(1)–C(18) bond length is 1.816 Å, which is typical for single bonds of this kind [43]. The crystal structure and molecular conformation is stabilized by three intramolecular C–H···N hydrogen bonds, three intramolecular C–H···O hydrogen bonds, and five intermolecular C–H···O hydrogen bonds in the crystal network (Figure 1, Figure 2 and Table 1).
Table 1: Crystal data, instrumental and refinement data for 23.
| Crystal data | |
|---|---|
| Empirical formula | C31H31N3O10S |
| Formula weight | 637.66 |
| Crystal size | 0.2241 × 0.0456 × 0.0328 mm |
| Crystal description | Stick |
| Crystal color | Colorless |
| Crystal system | Monoclinic |
| Space group | C2 |
| Unit-cell dimensions |
a = 25.7078 Å; α = 90°
b = 7.15002 Å; β = 105.2576° c = 17.8411 Å; γ = 90° |
| Volume | 3163.81(8) Å3 |
| Z | 4 |
| Calculated density | 1.339 Mg/m3 |
| Absorption coefficient | 1.433 mm−1 |
| F(000) | 1336 |
| Data collection | |
| Measurement device type | SuperNova, single source at offset, Atlas |
| Measurement method | w Scans |
| Temperature | 123 K |
| Wavelength | 1.54184 Å |
| Monochromator | Graphite |
| Theta range for data collection | 3.56 to 73.15° |
| Index ranges | −31<=h<=31, −8<=k<=7, −22<=l<=22 |
| Reflections collected/unique | 11616/4707 [R(int) = 0.0199] |
| Reflections greater | I>2\s(I);4572 |
| Absorption correction | Analytical |
| Max. and min. transmission | 0.960 and 0.833 |
| Refinement | |
| Refinement method | Full-matrix least-squares on F2 |
|
Hydrogen treatment;
Data/restraints/parameters |
4707/1/406 |
| Goodness-of-fit on F2 | 1.040 |
| Final R indices [I>2sigma(I)] | R1 = 0.0305, wR2 = 0.0817 |
| R indices (all data) | R1 = 0.0314, wR2 = 0.0827 |
| Absolute structure parameter | 0.024(14) |
| Largest diff. peak and hole | 0.286 and −0.282 e·Å−3 |
![[1860-5397-9-16-1]](/bjoc/content/figures/1860-5397-9-16-1.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-16-2]](/bjoc/content/figures/1860-5397-9-16-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Compound 29 (Figure 3, Figure 4 and Table 2) crystallized as a cyclic dimer and contains two independent molecules in the unit cell. The dimer is stabilized by an intermolecular hydrogen bond N(8)–H(8M)···S(1). Moreover, S(1)–C(17) and S(2)–C(40) displayed bond lengths of 1.699 and 1.660 Å, respectively, reflecting the double-bond character of the thiocarbonyl unit of the TSC group. This also supports the cleavage of the oxadiazole ring in the reaction of 26 to 29. The bond lengths O(6)–C(18) and O(6)–C(22) are 1.414 and 1.433 Å (similar to those found in 23), respectively, which shows that the sugar moiety still has the glucopyranose form with 4C1 anomeric β-configuration. In addition, the crystal structure of 29 shows that the whole molecule is nonplanar. The phenyl group makes a dihedral angle of C(1)N(1)C(9)C(10) = −100.3°, which means that it is perpendicular to the indole ring.
![[1860-5397-9-16-3]](/bjoc/content/figures/1860-5397-9-16-3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-16-4]](/bjoc/content/figures/1860-5397-9-16-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Table 2: Crystal data, instrumental and refinement data for 29.
| Crystal data | |
|---|---|
| Empirical formula | C23H26N4O6S |
| Formula weight | 486.55 |
| Crystal size | 0.3575 × 0.0513 × 0.0329 mm |
| Crystal description | Needle |
| Crystal color | Colorless |
| Crystal system | Monoclinic |
| Space group | P21 |
| Unit-cell dimensions |
a = 6.25643 Å; α = 90°
b = 41.019 Å; β= 96.092° c = 9.1898 Å; γ = 90° |
| Volume | 2345.06 Å3 |
| Z | 4 |
| Calculated density | 1.378 Mg/m3 |
| Absorption coefficient | 1.632 mm−1 |
| F(000) | 1024 |
| Data collection | |
| Measurement device type | SuperNova, single source at offset, Atlas |
| Measurement method | w Scans |
| Temperature | 293 K |
| Wavelength | 1.54184 Å |
| Monochromator | Graphite |
| Theta range for data collection | 4.31 to 73.83° |
| Index ranges | −4<=h<=7, −50<=k<=50, −11<=l<=10 |
| Reflections collected/unique | 8866/7227 [R(int) = 0.0371] |
| Reflections greater | I>2\s(I) 6676 |
| Absorption correction | Analytical |
| Max. and min. transmission | 0.953 and 0.749 |
| Refinement | |
| Refinement method | Full-matrix least-squares on F2 |
|
Hydrogen treatment;
Data/restraints/parameters |
7227/1/633 |
| Goodness-of-fit on F2 | 1.009 |
| Final R indices [I>2sigma(I)] | R1 = 0.0431, wR2 = 0.1113 |
| R indices (all data) | R1 = 0.0463, wR2 = 0.1132 |
| Absolute structure parameter | 0.054(15) |
| Largest diff. peak and hole | 0.798 and −0.275 e·Å−3 |
Single-crystal diffraction analysis of 30 (Figure 5, Figure 6 and Table 3) showed that the S(1)–C(17) bond length of 1.694 Å reflects double-bond character (similar to the bond lengths of S(1)–C(17) and S(2)–C(40) with 1.699(3) and 1.660(3) Å, respectively, of 29) and suggests the thione form. O(5)–C(18) and O(5)–C(22) bond lengths are 1.411 and 1.439 Å, respectively (again similar to those found in 23 and 29), which shows that, first, the compound still has the cyclic galactopyranose structure, and second, the sugar moiety is stable, even if the oxadiazole moiety has opened and the TSC formed has cyclized to form a triazole. The whole structure is nonplanar and the phenyl group still oriented perpendicular to the indole ring making torsion angles of C(8)–N(1)–C(7)–C(1) = 85.2° and C(15)–N(1)–C(7)–C(1) = −88.4°. On the other hand, the triazole ring is located in the plane of the indole ring making only small torsion angles of C(14)–C(15)–C(16)–N(4) = 3.6(4)° and N(1)–C(15)–C(16)–N(2) = 2.2(4)°. The molecular conformation is stabilized by an intramolecular hydrogen bond O(4)–H(4)···O(3) and a water molecule links two molecules in the crystal lattice through an intermolecular hydrogen bond O(6)–H(6P)···S(1).
![[1860-5397-9-16-5]](/bjoc/content/figures/1860-5397-9-16-5.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-16-6]](/bjoc/content/figures/1860-5397-9-16-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Table 3: Crystal data, instrumental and refinement data for 30.
| Crystal data | |
|---|---|
| Empirical formula | C23H24N4O5S·H2O |
| Formula weight | 486.55 |
| Crystal size | 0.5666 × 0.0262 × 0.0131 mm |
| Crystal description | Needle |
| Crystal color | Colorless |
| Crystal system | Monoclinic |
| Space group | P21 |
| Unit cell dimensions |
a = 9.7950 Å; α = 90°
b = 4.93845 Å; β = 90.813° c = 22.3598 Å; γ = 90° |
| Volume | 1081.48 Å3 |
| Z | 2 |
| Calculated density | 1.491 Mg/m3 |
| Absorption coefficient | 1.770 mm−1 |
| F(000) | 510 |
| Data collection | |
| Measurement device type | SuperNova, single source at offset, Atlas |
| Measurement method | w Scans |
| Temperature | 123 K |
| Wavelength | 1.54184 Å |
| Monochromator | Graphite |
| Theta range for data collection | 3.95 to 72.89° |
| Index ranges | −11<=h<=11, −5<=k<=3, −25<=l<=27 |
| Reflections collected/unique | 3935/3005 [R(int) = 0.0286] |
| Reflections greater | I>2\s(I) 2785 |
| Absorption correction | Analytical |
| Max. and min. transmission | 1.00000 and 0.86469 |
| Refinement | |
| Refinement method | Full-matrix least-squares on F2 |
|
Hydrogen treatment;
Data/restraints/parameters |
3005/1/327 |
| Goodness-of-fit on F2 | 1.027 |
| Final R indices [I>2sigma(I)] | R1 = 0.0378, wR2 = 0.0933 |
| R indices (all data) | R1 = 0.0422, wR2 = 0.0959 |
| Absolute structure parameter | 0.00(2) |
| Largest diff. peak and hole | 0.372 and −0.231 e·Å−3 |
Conclusion
In conclusion, 2-N-(glycosyl)thiosemicarbazides of type II (from the four glycosylthiosemicarbazide structural isomers I–IV shown in Scheme 1) were synthesized from 3-N-(glycosyl)oxadiazolinethiones, which were accessed by new regioselective glycosylations. Additionally, 3-N-(glycosyl)oxadiazolinethiones may be prepared by a mild solvent-free thermal S→N migration of the glycosyl moiety in glycosylsulfanyloxadiazoles. (Benzylindolyl)glycosylsulfanyl-1,3,4-oxadiazoles could be thermally rearranged into the corresponding N-glycosides. These may either be converted into the corresponding (benzylindolyl)-2-N-(glycosyl)thiosemicarbazides (of type II) or into the galactosyl triazolinethione from the galactosyl oxadiazolinethione, as confirmed by X-ray single-crystal analysis and from further common structural analytical data.
Supporting Information
Complete crystallographic data of the structural analysis of compounds 23, 29 and 30 have been deposited with the Cambridge Crystallographic Data Centre, CCDC 867245–867247. Copies of this information may be obtained free of charge from the Director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK. (fax: +44-1223-336033, e-mail: deposit@ccdc.cam.ac.uk or via http://www.ccdc.cam.ac.uk).
| Supporting Information File 1: Complete experimental section with full characterization data of all compounds. | ||
| Format: PDF | Size: 331.6 KB | Download |
| Supporting Information File 2: Chemical information files (cif) of compounds 23, 29 and 30. | ||
| Format: RAR | Size: 188.9 KB | Download |
Acknowledgements
The authors thank Prof. Atta-Ur-Rahman and Prof. M. I. Choudhary for making available the facilities for research at ICCBS. We would like also to thank Dr. Manfred Zabel from the University of Regensburg for performing the X-ray analyses, and Dr. Gasser M. Khairy and Prof. Dr. Hassan A. Azab from Suez Canal University for their help with the X-ray measurements. The support from the Higher Education Commission (HEC) Pakistan (Project No. 20-697/R&D/06/38) is highly appreciated.
References
-
García Fernández, J. M.; Ortiz Mellet, C. Adv. Carbohydr. Chem. Biochem. 2000, 55, 35–135. doi:10.1016/S0065-2318(00)55004-5
Return to citation in text: [1] -
Almajan, G. L.; Barbuceanu, S.-F.; Innocenti, A.; Scozzafava, A.; Supuran, C. T. J. Enzyme Inhib. Med. Chem. 2008, 23, 101–107. doi:10.1080/14756360701342417
Return to citation in text: [1] -
Schmidt, R. R.; Guilliard, W.; Heermann, D.; Hoffmann, M. J. Heterocycl. Chem. 1983, 20, 1447–1451.
Return to citation in text: [1] -
El Ashry, E. S. H.; El Kilany, Y.; Nahas, N. M. Manipulation of Carbohydrate Carbon Atoms for the Synthesis of Heterocycles. In Heterocycles from Carbohydrate Precursors; El Ashry, E. S. H., Ed.; Topics in Heterocyclic Chemistry, Vol. 7; Springer: Berlin, 2007; pp 1–30.
Return to citation in text: [1] -
Pleuss, N.; Kunz, H. Synthesis 2005, 122–130. doi:10.1055/s-2004-834911
Return to citation in text: [1] -
Fernandez-Bolanos, J. G.; Lopez, O. Synthesis of Heterocycles from Glycosylamines and Glycosyl Azides. In Heterocycles from Carbohydrate Precursors; El Ashry, E. S. H., Ed.; Topics in Heterocyclic Chemistry, Vol. 7; Springer: Berlin, 2007; pp 31–66.
Return to citation in text: [1] -
Fuentes, J.; Angulo, M.; Pradera, M. A. J. Org. Chem. 2002, 67, 2577–2587. doi:10.1021/jo0110303
Return to citation in text: [1] -
Ogura, H.; Takahashi, H.; Sakaguchi, M. Nucleic Acids Res. 1978, 1 (Suppl. 2), s251–s256. doi:10.1093/nar/1.suppl_2.s251
Return to citation in text: [1] [2] -
Sun, W.; Yao, J.; Bai, L.; Wang, X. Acta Crystallogr., Sect. E 2009, 65, o242. doi:10.1107/S1600536808043833
Return to citation in text: [1] -
Wieniawski, W.; Gmernicka-haftek, C. Diss. Pharm. Pharmacol. 1968, 20, 411–417.
Return to citation in text: [1] -
Li, Y. X.; Wang, H. A.; Yang, X. P.; Cheng, H. Y.; Wang, Z. H.; Li, Y. M.; Wang, S. H.; Yan, D. W. Carbohydr. Res. 2009, 344, 1248–1253. doi:10.1016/j.carres.2009.03.027
Return to citation in text: [1] -
El Khadem, H. S.; Fatiadi, A. J. Adv. Carbohydr. Chem. Biochem. 2004, 59, 135–173. doi:10.1016/S0065-2318(04)59004-2
Return to citation in text: [1] -
Marcaurelle, L. A.; Bertozzi, C. R. Glycobiology 2002, 12, 69R–77R. doi:10.1093/glycob/12.6.69R
Return to citation in text: [1] -
Davis, B. G. Chem. Rev. 2002, 102, 579–602. doi:10.1021/cr0004310
Return to citation in text: [1] -
Langenhan, J. M.; Thorson, J. S. Curr. Org. Synth. 2005, 2, 59–81. doi:10.2174/1570179052996937
Return to citation in text: [1] -
Alviano, C. S.; Farbiarz, S. R.; De Souza, W.; Angluster, J.; Travassos, L. R. J. Gen. Microbiol. 1991, 137, 837–841. doi:10.1099/00221287-137-4-837
Return to citation in text: [1] -
Tripathi, R. P.; Tewari, N.; Dwivedi, N.; Tiwari, V. K. Med. Res. Rev. 2005, 25, 93–131. doi:10.1002/med.20017
Return to citation in text: [1] -
Fernández-Bolaños, J. G.; Zafra, E.; López, O.; Robina, I.; Fuentes, J. Tetrahedron: Asymmetry 1999, 10, 3011–3023. doi:10.1016/S0957-4166(99)00314-6
Return to citation in text: [1] -
Fuentes, J. J.; Molina, J. L.; Pradera, M. A. Tetrahedron: Asymmetry 1998, 9, 2517–2532. doi:10.1016/S0957-4166(98)00243-2
Return to citation in text: [1] -
Fujita, Y.; Abdel-Aal, A.-B. M.; Wimmer, N.; Batzloff, M. R.; Good, M. F.; Toth, I. Bioorg. Med. Chem. 2008, 16, 8907–8913. doi:10.1016/j.bmc.2008.08.064
Return to citation in text: [1] -
El Ashry, E. S. H.; Rashed, N.; Shobier, H. S. Pharmazie 2000, 55, 251–262.
Return to citation in text: [1] -
Taylor, C. M. Tetrahedron 1998, 54, 11317–11362. doi:10.1016/S0040-4020(98)00477-3
Return to citation in text: [1] -
Günther, W.; Kunz, H. Angew. Chem., Int. Ed. Engl. 1990, 29, 1050–1051. doi:10.1002/anie.199010501
Return to citation in text: [1] -
Dubber, M.; Lindhorst, T. K. Org. Lett. 2001, 3, 4019–4022. doi:10.1021/ol016717o
Return to citation in text: [1] -
Lindhorst, T. K.; Kieburg, C. Angew. Chem., Int. Ed. Engl. 1996, 35, 1953–1956. doi:10.1002/anie.199619531
Return to citation in text: [1] -
Belkadi, M.; Othman, A. A. Trends Appl. Sci. Res. 2011, 6, 19–33. doi:10.3923/tasr.2011.19.33
Return to citation in text: [1] -
Ghosh, S.; Misra, A. K.; Bhatia, G.; Khan, M. M.; Khanna, A. K. Bioorg. Med. Chem. Lett. 2009, 19, 386–389. doi:10.1016/j.bmcl.2008.11.070
Return to citation in text: [1] -
Alexacou, K.-M.; Tenchiu, A.-C.; Chrysina, E. D.; Charavgi, M.-D.; Kostas, I. D.; Zographos, S. E.; Oikonomakos, N. G.; Leonidas, D. D. Bioorg. Med. Chem. 2010, 18, 7911–7922. doi:10.1016/j.bmc.2010.09.039
Return to citation in text: [1] -
Tao, J.; Wang, D.-Z.; Cao, L.-H. J. Chin. Chem. Soc. 2007, 54, 1287–1292.
Return to citation in text: [1] -
Thanh, N. D.; Giang, N. T. K.; Hoai, L. T. E-J. Chem. 2010, 7, 899–907.
Return to citation in text: [1] -
Tao, J.; Wang, D.-Z.; Cao, L.-H. J. Chin. Chem. Soc. 2010, 57, 1077–1080.
Return to citation in text: [1] -
El Ashry, E. S. H.; El Tamany, E. S. H.; Abd El Fattah, M. E. D.; Aly, M. R. E.; Boraei, A. T. A. Lett. Org. Chem. 2009, 6, 462–469. doi:10.2174/157017809789124902
Return to citation in text: [1] -
El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ahmad, R.; Iqbal, R.; Akhtar, H.; Zia-ul-Haq; Duddeck, H.; Stefaniak, L.; Sitkowski, J. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1671–1682. doi:10.1081/NCN-100105903
Return to citation in text: [1] -
Wagner, G.; Dietzsch, B.; Krake, U. Pharmazie 1975, 30, 694–698.
Return to citation in text: [1] -
Abdel-Megeid, F. M. E.; Elkaschef, M. A.-F.; Abdel-Bary, H. M. A. Carbohydr. Res. 1977, 59, 95–102. doi:10.1016/S0008-6215(00)83296-6
Return to citation in text: [1] -
Wagner, G.; Dietzsch, B. Pharmazie 1976, 31, 153–154.
Return to citation in text: [1] -
Khanum, S. A.; Shashikanth, S.; Umesha, S.; Kavitha, R. Eur. J. Med. Chem. 2005, 40, 1156–1162. doi:10.1016/j.ejmech.2005.04.005
Return to citation in text: [1] -
Farghaly, A.-R.; El-Kashef, H. ARKIVOC 2006, xi, 76–90.
Return to citation in text: [1] -
Mekuskiene, G.; Burbuliene, M. M.; Jakubkiene, V.; Udrenaite, E.; Gaidelis, P.; Vainilavicius, P. Chem. Heterocycl. Compd. 2003, 39, 1364–1368. doi:10.1023/B:COHC.0000010654.66587.4a
Return to citation in text: [1] -
Hirmath, S. P.; Hirmath, D. M.; Purohit, M. G. Indian J. Chem. 1983, 22B, 571–576.
Return to citation in text: [1] -
Robba, M.; Maume, D.; Lancelot, J. C. Bull. Soc. Chim. Fr. 1977, 3–4, 333–336.
Return to citation in text: [1] -
El Ashry, E. S. H.; Aly, A. A.; Amer, M. R.; Shah, M. R.; Ng, S. W. Carbohydr. Res. 2011, 346, 169–176. doi:10.1016/j.carres.2010.11.005
Return to citation in text: [1]
| 43. | El Ashry, E. S. H.; Aly, A. A.; Amer, M. R.; Shah, M. R.; Ng, S. W. Carbohydr. Res. 2011, 346, 169–176. doi:10.1016/j.carres.2010.11.005 |
| 1. | García Fernández, J. M.; Ortiz Mellet, C. Adv. Carbohydr. Chem. Biochem. 2000, 55, 35–135. doi:10.1016/S0065-2318(00)55004-5 |
| 9. | Sun, W.; Yao, J.; Bai, L.; Wang, X. Acta Crystallogr., Sect. E 2009, 65, o242. doi:10.1107/S1600536808043833 |
| 26. | Belkadi, M.; Othman, A. A. Trends Appl. Sci. Res. 2011, 6, 19–33. doi:10.3923/tasr.2011.19.33 |
| 8. | Ogura, H.; Takahashi, H.; Sakaguchi, M. Nucleic Acids Res. 1978, 1 (Suppl. 2), s251–s256. doi:10.1093/nar/1.suppl_2.s251 |
| 27. | Ghosh, S.; Misra, A. K.; Bhatia, G.; Khan, M. M.; Khanna, A. K. Bioorg. Med. Chem. Lett. 2009, 19, 386–389. doi:10.1016/j.bmcl.2008.11.070 |
| 28. | Alexacou, K.-M.; Tenchiu, A.-C.; Chrysina, E. D.; Charavgi, M.-D.; Kostas, I. D.; Zographos, S. E.; Oikonomakos, N. G.; Leonidas, D. D. Bioorg. Med. Chem. 2010, 18, 7911–7922. doi:10.1016/j.bmc.2010.09.039 |
| 29. | Tao, J.; Wang, D.-Z.; Cao, L.-H. J. Chin. Chem. Soc. 2007, 54, 1287–1292. |
| 30. | Thanh, N. D.; Giang, N. T. K.; Hoai, L. T. E-J. Chem. 2010, 7, 899–907. |
| 31. | Tao, J.; Wang, D.-Z.; Cao, L.-H. J. Chin. Chem. Soc. 2010, 57, 1077–1080. |
| 3. | Schmidt, R. R.; Guilliard, W.; Heermann, D.; Hoffmann, M. J. Heterocycl. Chem. 1983, 20, 1447–1451. |
| 4. | El Ashry, E. S. H.; El Kilany, Y.; Nahas, N. M. Manipulation of Carbohydrate Carbon Atoms for the Synthesis of Heterocycles. In Heterocycles from Carbohydrate Precursors; El Ashry, E. S. H., Ed.; Topics in Heterocyclic Chemistry, Vol. 7; Springer: Berlin, 2007; pp 1–30. |
| 5. | Pleuss, N.; Kunz, H. Synthesis 2005, 122–130. doi:10.1055/s-2004-834911 |
| 6. | Fernandez-Bolanos, J. G.; Lopez, O. Synthesis of Heterocycles from Glycosylamines and Glycosyl Azides. In Heterocycles from Carbohydrate Precursors; El Ashry, E. S. H., Ed.; Topics in Heterocyclic Chemistry, Vol. 7; Springer: Berlin, 2007; pp 31–66. |
| 7. | Fuentes, J.; Angulo, M.; Pradera, M. A. J. Org. Chem. 2002, 67, 2577–2587. doi:10.1021/jo0110303 |
| 22. | Taylor, C. M. Tetrahedron 1998, 54, 11317–11362. doi:10.1016/S0040-4020(98)00477-3 |
| 23. | Günther, W.; Kunz, H. Angew. Chem., Int. Ed. Engl. 1990, 29, 1050–1051. doi:10.1002/anie.199010501 |
| 2. | Almajan, G. L.; Barbuceanu, S.-F.; Innocenti, A.; Scozzafava, A.; Supuran, C. T. J. Enzyme Inhib. Med. Chem. 2008, 23, 101–107. doi:10.1080/14756360701342417 |
| 24. | Dubber, M.; Lindhorst, T. K. Org. Lett. 2001, 3, 4019–4022. doi:10.1021/ol016717o |
| 25. | Lindhorst, T. K.; Kieburg, C. Angew. Chem., Int. Ed. Engl. 1996, 35, 1953–1956. doi:10.1002/anie.199619531 |
| 12. | El Khadem, H. S.; Fatiadi, A. J. Adv. Carbohydr. Chem. Biochem. 2004, 59, 135–173. doi:10.1016/S0065-2318(04)59004-2 |
| 17. | Tripathi, R. P.; Tewari, N.; Dwivedi, N.; Tiwari, V. K. Med. Res. Rev. 2005, 25, 93–131. doi:10.1002/med.20017 |
| 11. | Li, Y. X.; Wang, H. A.; Yang, X. P.; Cheng, H. Y.; Wang, Z. H.; Li, Y. M.; Wang, S. H.; Yan, D. W. Carbohydr. Res. 2009, 344, 1248–1253. doi:10.1016/j.carres.2009.03.027 |
| 18. | Fernández-Bolaños, J. G.; Zafra, E.; López, O.; Robina, I.; Fuentes, J. Tetrahedron: Asymmetry 1999, 10, 3011–3023. doi:10.1016/S0957-4166(99)00314-6 |
| 19. | Fuentes, J. J.; Molina, J. L.; Pradera, M. A. Tetrahedron: Asymmetry 1998, 9, 2517–2532. doi:10.1016/S0957-4166(98)00243-2 |
| 20. | Fujita, Y.; Abdel-Aal, A.-B. M.; Wimmer, N.; Batzloff, M. R.; Good, M. F.; Toth, I. Bioorg. Med. Chem. 2008, 16, 8907–8913. doi:10.1016/j.bmc.2008.08.064 |
| 21. | El Ashry, E. S. H.; Rashed, N.; Shobier, H. S. Pharmazie 2000, 55, 251–262. |
| 10. | Wieniawski, W.; Gmernicka-haftek, C. Diss. Pharm. Pharmacol. 1968, 20, 411–417. |
| 8. | Ogura, H.; Takahashi, H.; Sakaguchi, M. Nucleic Acids Res. 1978, 1 (Suppl. 2), s251–s256. doi:10.1093/nar/1.suppl_2.s251 |
| 13. | Marcaurelle, L. A.; Bertozzi, C. R. Glycobiology 2002, 12, 69R–77R. doi:10.1093/glycob/12.6.69R |
| 14. | Davis, B. G. Chem. Rev. 2002, 102, 579–602. doi:10.1021/cr0004310 |
| 15. | Langenhan, J. M.; Thorson, J. S. Curr. Org. Synth. 2005, 2, 59–81. doi:10.2174/1570179052996937 |
| 16. | Alviano, C. S.; Farbiarz, S. R.; De Souza, W.; Angluster, J.; Travassos, L. R. J. Gen. Microbiol. 1991, 137, 837–841. doi:10.1099/00221287-137-4-837 |
| 35. | Wagner, G.; Dietzsch, B.; Krake, U. Pharmazie 1975, 30, 694–698. |
| 36. | Abdel-Megeid, F. M. E.; Elkaschef, M. A.-F.; Abdel-Bary, H. M. A. Carbohydr. Res. 1977, 59, 95–102. doi:10.1016/S0008-6215(00)83296-6 |
| 37. | Wagner, G.; Dietzsch, B. Pharmazie 1976, 31, 153–154. |
| 32. | El Ashry, E. S. H.; El Tamany, E. S. H.; Abd El Fattah, M. E. D.; Aly, M. R. E.; Boraei, A. T. A. Lett. Org. Chem. 2009, 6, 462–469. doi:10.2174/157017809789124902 |
| 33. | El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024 |
| 34. | Ahmad, R.; Iqbal, R.; Akhtar, H.; Zia-ul-Haq; Duddeck, H.; Stefaniak, L.; Sitkowski, J. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1671–1682. doi:10.1081/NCN-100105903 |
| 42. | Robba, M.; Maume, D.; Lancelot, J. C. Bull. Soc. Chim. Fr. 1977, 3–4, 333–336. |
| 33. | El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024 |
| 33. | El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024 |
| 41. | Hirmath, S. P.; Hirmath, D. M.; Purohit, M. G. Indian J. Chem. 1983, 22B, 571–576. |
| 38. | Khanum, S. A.; Shashikanth, S.; Umesha, S.; Kavitha, R. Eur. J. Med. Chem. 2005, 40, 1156–1162. doi:10.1016/j.ejmech.2005.04.005 |
| 39. | Farghaly, A.-R.; El-Kashef, H. ARKIVOC 2006, xi, 76–90. |
| 40. | Mekuskiene, G.; Burbuliene, M. M.; Jakubkiene, V.; Udrenaite, E.; Gaidelis, P.; Vainilavicius, P. Chem. Heterocycl. Compd. 2003, 39, 1364–1368. doi:10.1023/B:COHC.0000010654.66587.4a |
| 33. | El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024 |
| 33. | El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024 |
| 33. | El Ashry, E. S. H.; El Tamany, E. H.; Aly, M. R. E.; Boraei, A. T. A. Synlett 2010, 2247–2250. doi:10.1055/s-0030-1258024 |
© 2013 El Ashry et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)