Abstract
Enantioselective desymmetrization of meso-aziridines and meso-epoxides with various nucleophiles by organocatalysis has emerged as a cutting-edge approach in recent years. This review summarizes the origin and recent developments of enantioselective desymmetrization of meso-aziridines and meso-epoxides in the presence of organocatalysts.

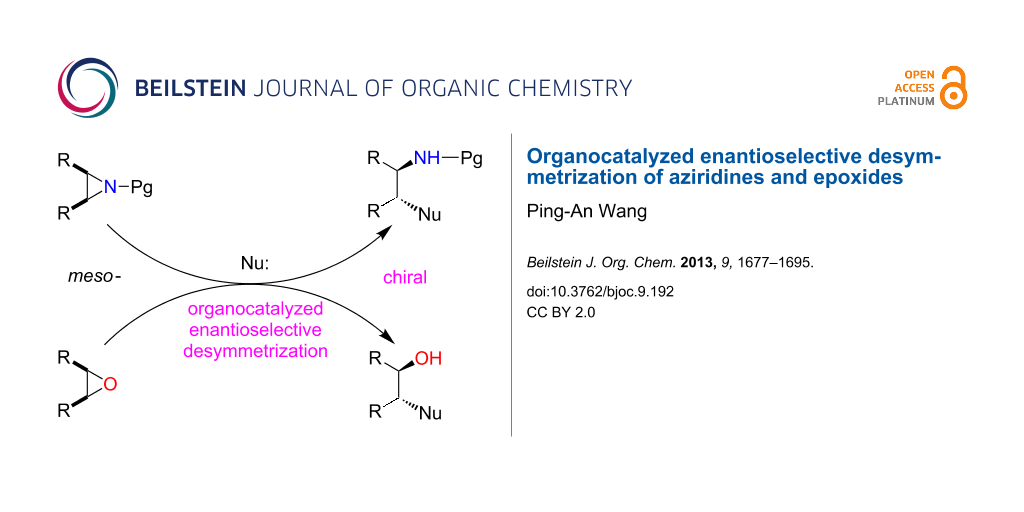
Graphical Abstract
Introduction
The high demand of enantiopure organic compounds in the fine chemical industry gives impetus to the development of chiral technologies. Among them, the catalytic asymmetric synthesis represents the state of the art in organic chemistry [1]. Over the past four decades, the asymmetric synthesis based on transition-metal catalyzed reactions has given rise to a variety of significant achievements both in academic and industrial chemistry [2,3]. Desymmetrization is the modification of a molecule which results in the loss of symmetry elements such as a mirror plane, a center of inversion or a rotation–reflection axis (Figure 1). Usually, a prochiral or meso-molecule can be converted into a chiral molecule in a single step [4-6] in the presence of chiral catalysts. Therefore, enantioselective desymmetrization is regarded as a very powerful strategy for producing a large amount of chiral compounds from readily available achiral substrates. The past decade witnessed the renaissance and the golden age of organocatalyzed reactions [7-9]. Aziridines and epoxides are quite reactive due to the large tension of their three-membered ring system. The enantioselective ring-opening of aziridines [10-12] or epoxides [13-15] provides facile access to various chiral amines and alcohols and their derivatives with two adjacent stereogenic centers, which are widely used as building blocks in pharmaceutical and organic synthesis. The enantioselective catalytic desymmetrization of meso-aziridines and meso-epoxides by metal-based Lewis acids cooperating with chiral ligands has been well established [16-28]. However, the enantioselective desymmetrization of meso-aziridines and meso-epoxides catalyzed by small chiral organic molecules has only emerged in recent years. The goal of this review is to give a comprehensive overview on newly developed strategies in the field of organocatalyzed enantioselective desymmetrization of meso-aziridines and meso-epoxides.
![[1860-5397-9-192-1]](/bjoc/content/figures/1860-5397-9-192-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The catalyzed enantioselective desymmetrization.
Figure 1: The catalyzed enantioselective desymmetrization.
Review
Organocatalyzed enantioselective desymmetrization of meso-aziridines
Many examples of the ring-opening of aziridines by various carbon [29,30], nitrogen [31,32], oxygen [33,34], sulfur [35,36] and halogen [37-39] nucleophilic reagents in the presence of catalytic amounts of small acids and bases such as BF3·Et2O, AcOH, TsOH, TFA, PBu3, TMEDA, TBAF, Bu4NHSO4, pyridine N-oxide, and N-heterocyclic carbenes were published. However, the enantioselective ring-opening of meso-aziridines in the presence of chiral organocatalysts (OC) has emerged as a research field only in recent years. The organocatalysts utilized in these processes are diverse in their structural features including cinchona alkaloid derivatives, chiral phosphoric acids, chiral amino alcohols, chiral thioureas, chiral guanidines, and chiral 1,2,3-triazolium chlorides. In this review, the research work of enantioselective desymmetrization of meso-aziridines is organized into sections according to the employed organocatalysts.
Cinchona alkaloid derivatives
The first organocatalytic enantioselective desymmetrization of meso-aziridines was discovered by Hou and co-workers in 2007 [40] with various arylthiols as nucleophiles in CCl4 at 0 °C in the presence of cinchonine-derived phase-transfer catalysts (PTCs, Figure 2, OC-1 to OC-6). The substituent on the bridgehead nitrogen of cinchona alkaloids has a great impact on the enantioselectivity of the reactions. The catalyst OC-2 with 9-anthracenylmethyl on the bridgehead nitrogen is more efficient than other cinchona alkaloid-derived catalysts for the desymmetrization of meso-N-tosylaziridine (tosyl = Ts) as it affords the corresponding chiral N-Ts thioamines (Scheme 1, 1 to 9,) in high yield (85%) and satisfactory enantioselectivity (73% ee). The OC-4 and OC-5 with a protecting group on 9-OH decreased the enantioselectivity dramatically. In the presence of OC-6, when p-nitrophenylsulfonylaziridine (Ns-aziridine) was employed as substrate, the corresponding N-Ns thioamine 8 was obtained in quantitative yield but with moderate enantioselectivity (55% ee). The need of two equivalents (equiv) of CsOH·H2O as a base in the procedure limited its application in organic synthesis.
![[1860-5397-9-192-2]](/bjoc/content/figures/1860-5397-9-192-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cinchona alkaloid-derived catalysts OC-1 to OC-11.
Figure 2: Cinchona alkaloid-derived catalysts OC-1 to OC-11.
![[1860-5397-9-192-i1]](/bjoc/content/inline/1860-5397-9-192-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The enantioselective desymmetrization of meso-aziridines in the presence of selected Cinchona alkaloid-derived catalysts.
Scheme 1: The enantioselective desymmetrization of meso-aziridines in the presence of selected Cinchona alkal...
Wu and co-workers have established the enantioselective ring-opening of meso-aziridines with arylthiols by using various quinine derivatives (OC-7 to OC-11) as organocatalysts (Scheme 1) [41]. It was found that the desymmetrization performed well in CHCl3 in the presence of 10 mol % of quinine (OC-7) at room temperature. The β-amino sulfides were obtained in high yields (up to 85%) and with moderate enantioselectivities (up to 72% ee). However, OC-10 and OC-11 afforded extremely disappointing results for desymmetrization and the corresponding β-amino sulfide was obtained nearly racemic (3% ee). The usage of Boc for the N-protected group in aziridine resulted in no enantioselectivity under the optimum conditions.
The organocatalytic enantioselective ring-opening of N-protected aziridines by β-ketoesters by means of cinchona alkaloid-derived PTCs (Figure 3, OC-12 to OC-19) for producing γ-amino acid derivatives with one stereogenic quaternary carbon has been independently developed by Dixon [42] and Jørgensen’s group [43]. Dixon and co-workers discovered that OC-16 bearing bulky substituents both on the 9-O atom and the bridgehead nitrogen afforded excellent enantioselectivity of desirable products (Scheme 2, 13 to 18, up to 97% ee) with 50% aqueous K2HPO4 as base. However, under the same conditions, the PTCs with a free 9-OH (OC-17) led to 13 in low yields and enantioselectivities (21% ee). The urea catalyst OC-19 proved to be very inefficient and afforded 13 in 10% yield with 6% ee.
![[1860-5397-9-192-3]](/bjoc/content/figures/1860-5397-9-192-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Cinchona alkaloid-derived catalysts OC-12 to OC-19.
Figure 3: Cinchona alkaloid-derived catalysts OC-12 to OC-19.
![[1860-5397-9-192-i2]](/bjoc/content/inline/1860-5397-9-192-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The enantioselective ring-opening of aziridines in the presence of OC-16.
Scheme 2: The enantioselective ring-opening of aziridines in the presence of OC-16.
By shifting the N-protecting group in aziridines from o-(trifluoromethyl)benzenesulfonyl to Ts, Jørgensen and colleagues found that excellent enantioselectivities (up to 97% ee) of the ring-opening products (Scheme 3, 19 to 27) were obtained by using OC-16 as a catalyst, 33 wt % aqueous K2CO3 as a base and a low reaction temperature of −20 °C. This afforded perfect enantioselectivity (99% ee) of compound 19, but at the sacrifice of the chemical yield (decrease to 21%). The N-Boc and N-Cbz protected meso-aziridines yielded the product in trace amounts under the optimum conditions.
![[1860-5397-9-192-i3]](/bjoc/content/inline/1860-5397-9-192-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: OC-16 catalyzed enantioselective ring-opening of aziridines.
Scheme 3: OC-16 catalyzed enantioselective ring-opening of aziridines.
Chiral phosphoric acids
An elegant enantioselective desymmetrization of meso-aziridines catalyzed by chiral phosphoric acids OC-20 and OC-21 as catalysts was carried out by Antilla [44] and co-workers (Figure 4). When a bis(3,5-trifluoromethyl)benzoyl group was used for the N-activation of meso-aziridines, 1,2-azidoamides (Scheme 4, 28 to 36) were obtained in excellent yields (up to 97%) and enantioselectivities (up to 95% ee) by using TMSN3 as a nucleophile in the presence of 10 mol % of OC-20 or OC-21 in 1,2-dichloroethane (DCE) at room temperature. Interestingly, tetrabutylammonium azide or NaN3 were unreactive as a nucleophile under the same conditions, so the presence of the trimethylsilyl group is crucial to this ring-opening reaction. A proposed mechanism was based on NMR spectroscopic investigations as depicted in Figure 5. In the first step of the reaction, the active catalyst was formed by the displacement of the azide to produce HN3 and the chiral silane A. The latter activates the aziridine by means of coordinating to the carbonyl functionality of the aziridine to afford intermediate B. Species B was then attacked by HN3, resulting in C and the release of phosphoric acid. Compound C can be readily decomposed on silica gel to form the final product. Antilla and co-workers have also shown the enantioselective desymmetrization of meso-aziridines with functionalized mercaptans in the presence of (S)-VAPOL in ether at rt to afford β-(N-acylamino)phenyl thioethers in good yields and ee’s [45].
![[1860-5397-9-192-4]](/bjoc/content/figures/1860-5397-9-192-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The chiral phosphoric acids catalysts OC-20 and OC-21.
Figure 4: The chiral phosphoric acids catalysts OC-20 and OC-21.
![[1860-5397-9-192-i4]](/bjoc/content/inline/1860-5397-9-192-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: OC-20 and OC-21 catalyzed enantioselective desymmetrization of meso-aziridines.
Scheme 4: OC-20 and OC-21 catalyzed enantioselective desymmetrization of meso-aziridines.
![[1860-5397-9-192-5]](/bjoc/content/figures/1860-5397-9-192-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The proposed mechanism for chiral phosphorous acid-induced enantioselctive desymmetrization of meso-aziridines (chiral PA = OC-21).
Figure 5: The proposed mechanism for chiral phosphorous acid-induced enantioselctive desymmetrization of meso...
By using 10 mol % of commercially available chiral phosphoric acid OC-21 as a catalyst, Lattanzi [46] and colleagues have developed a facile desymmetrization of meso-N-acylaziridines with Me3SiSPh to produce β-(N-acylamino)phenyl thioethers (Scheme 5, 37 to 44) in high enantioselectivities (78–99% ee). Interestingly, the aziridines with a six-membered ring were desymmetrized to give the corresponding thioethers in excellent yields and enantioselectivities when the molar ratio of aziridine/nucleophile/catalyst is 1.0:1.5:0.1. For the unreactive acyclic and seven-membered ring aziridines, the good enantioselectivities were obtained under the reverse ratio of the reagents.
![[1860-5397-9-192-i5]](/bjoc/content/inline/1860-5397-9-192-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: OC-21 catalyzed enantioselective desymmetrization of meso-aziridines by Me3SiSPh.
Scheme 5: OC-21 catalyzed enantioselective desymmetrization of meso-aziridines by Me3SiSPh.
Della Sala’s group [47] has demonstrated the first example of a meso-aziridine desymmetrization with selenium nucleophiles to produce chiral β-aminoselenides in high enantioselectivities (up to 99% ee). Surprisingly, when the sterically hindered air-stable selenosilane t-BuMe2SiSePh was used as a sole nucleophile for desymmetrization of meso-N-acylaziridines in the presence of 10 mol % of chiral phosphoric acid OC-21, the good conversions and enantioselectivities were accomplished in toluene but with a very long reaction time (usually up to 4 days). The desymmetrization of meso-N-acylaziridines by the less sterically hindered selenosilane Me3SiSePh under the same conditions can be completed in several hours with good yields but moderate ee’s. According to the proposed mechanism, the formation of a small amount of PhSeH was involved in the induction step of the catalytic cycle and it acted as the actual nucleophile for the desymmetrization. In such a way a modified protocol based on the use of a combination of Me3SiSePh and PhseH as a nucleophile was developed. This modification proved to be very valuable and the desymmetrization can be accomplished within several hours to afford the chiral β-aminoselenides (Scheme 6, 45 to 52) in both high yields (up to 97%) and enantioselectivities (up to 97% ee).
![[1860-5397-9-192-i6]](/bjoc/content/inline/1860-5397-9-192-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: OC-21 catalyzed the enantioselective desymmetrization of meso-aziridines by Me3SiSePh/PhSeH.
Scheme 6: OC-21 catalyzed the enantioselective desymmetrization of meso-aziridines by Me3SiSePh/PhSeH.
Recently, Della Sala [48] has re-examined the enantioselective desymmetrization of meso-aziridines with various silylated reagents (Me3SiN3, Me3SiSMe, Me3SiSPh, Me3SiSBn, Me3SiNCS and Me3SiSePh/PhSeH) by chiral phosphoric acid OC-21. It was found that both purchased and synthetic OC-21 exhibited high catalytic activities and enantioselective inductions in the desymmetrization of meso-aziridines. Samples of OC-21 were washed with aq HCl to generate metal-free chiral phosphoric acid OC-21, which was reused as the catalyst, and the corresponding ring-opening products were obtained in moderate yields as racemates. By using a 1:1 mixture of calcium and magnesium phosphate salts of OC-21 as the catalyst, the corresponding ring-opening product 28 was produced in 94% yield and 91% ee. Therefore, calcium and magnesium phosphate salts of OC-21 proved to be the true catalysts in these processes. The author suggested that these metal phosphates may be generated through the purification of OC-21 on silica gel column. A dual Lewis-base activation mechanism of this desymmetrization was proposed.
Chiral amino alcohols
Lattanzi and colleagues [49] have also discovered the desymmetrization of meso-N-acylaziridines with benzenethiols in the presence of α,α-diaryl-L-prolinols (Figure 6, OC-22 to OC-25) to afford the products in good yields and moderate enantioselectivities (up to 61% ee, Scheme 7). The dual activation mode of α,α-diphenyl-L-prolinol (OC-23) for the desymmetrization of meso-N-acylaziridines is proposed in this context (Figure 7). The arylthiol was deprotonated by the pyrrolidinyl-N of the organocatalyst to produce a highly active nucleophile, while the meso-N-acylaziridine was activated by the tertiary-hydroxy group of the organocatalyst by hydrogen-bonding interaction with the carbonyl-O of the acyl group in meso-N-acylaziridine. The latter was attacked by the highly active thio anion to furnish chiral β-amino thioethers in good yields. Interestingly, meso-N-tosylaziridines reacted sluggishly in both chloroform and toluene as the solvents under the same catalytic system, and the reason of this phenomenon is unknown.
![[1860-5397-9-192-6]](/bjoc/content/figures/1860-5397-9-192-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: L-Proline and its derivatives OC-22 to OC-27.
Figure 6: L-Proline and its derivatives OC-22 to OC-27.
![[1860-5397-9-192-i7]](/bjoc/content/inline/1860-5397-9-192-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: OC-23 catalyzed enantioselective desymmetrization of meso-aziridines.
Scheme 7: OC-23 catalyzed enantioselective desymmetrization of meso-aziridines.
![[1860-5397-9-192-7]](/bjoc/content/figures/1860-5397-9-192-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Proposed bifunctional mode of action of OC-23.
Figure 7: Proposed bifunctional mode of action of OC-23.
Chiral thioureas
Jacobsen [50] and co-workers presented an enantioselective catalytic desymmetrization of meso-N-benzoylaziridines with HCl by a series of chiral thioureas (Figure 8, OC-28 to OC-44). In most of the cases, a diluted reaction mixture is necessary to obtain good enantioselectivities and OC-41 proved to be the best catalyst to provide β-chlorobenzamides (Scheme 8, 60 to 66) in high yields and enantioselectivities (up to 92% ee). The 31P NMR studies of the interaction between phosphinothiourea OC-41 and HCl indicated that the initial heterolysis of HCl affords a phosphonium chloride complex which is involved in the catalytic cycle. The desymmetrization of both cyclic and acyclic aziridines proceeded smoothly to give the corresponding β-chlorobenzamides in high yields and enantioselectivities.
![[1860-5397-9-192-8]](/bjoc/content/figures/1860-5397-9-192-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: The chiral thioureas OC-28 to OC-44 for the desymmetrization of meso-aziridines.
Figure 8: The chiral thioureas OC-28 to OC-44 for the desymmetrization of meso-aziridines.
![[1860-5397-9-192-i8]](/bjoc/content/inline/1860-5397-9-192-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Desymmetrization of meso-aziridines with OC-41.
Scheme 8: Desymmetrization of meso-aziridines with OC-41.
Chiral guanidines
Tan [51] and co-workers developed a series of chiral guanidines based on an amino indanol backbone (Figure 9, OC-45 to OC-48), which proved to be very efficient organocatalysts for the enantioselective desymmetrization of meso-aziridines (Scheme 9 and Scheme 10). It was found that meso-aziridines and arylthiols containing strong electron-withdrawing groups afforded the desired products with high yields and ee’s. The reaction of meso-N-3,5-dinitrobenzoylaziridine with 2,6-dichlorobenzenethiol provided the best enantioselectivity of the β-(N-acylamino)aryl thioether (up to 92% ee), with only 1 mol % of chiral guanidine OC-46 as a catalyst. Based on density functional theory (DFT) calculations, a plausible mechanism indicated that the hydrogen-bonding interaction between the chiral guanidine and the carbonyl group in meso-N-acylaziridine is crucial for the nucleophile attack and the enantioselectivity. It seems that the increase in steric repulsion between nucleophiles and aziridines is helpful to obtain high enantioselectivities of the corresponding products (Figure 10). The desymmetrization of meso-aziridines with the in situ generation of carbamodithioic acids from amine and carbon disulfide is also investigated to provide ring-opened products 76 to 80 in high yields and good enantioselectivities (Scheme 11).
![[1860-5397-9-192-9]](/bjoc/content/figures/1860-5397-9-192-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: The chiral guanidines (OC-45 to OC-48).
Figure 9: The chiral guanidines (OC-45 to OC-48).
![[1860-5397-9-192-i9]](/bjoc/content/inline/1860-5397-9-192-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: OC-46 catalyzed desymmetrization of meso-aziridines by arylthiols.
Scheme 9: OC-46 catalyzed desymmetrization of meso-aziridines by arylthiols.
![[1860-5397-9-192-i10]](/bjoc/content/inline/1860-5397-9-192-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Desymmetrization of cis-aziridine-2,3-dicarboxylate.
Scheme 10: Desymmetrization of cis-aziridine-2,3-dicarboxylate.
![[1860-5397-9-192-10]](/bjoc/content/figures/1860-5397-9-192-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: The proposed activation mode of OC-46.
Figure 10: The proposed activation mode of OC-46.
![[1860-5397-9-192-i11]](/bjoc/content/inline/1860-5397-9-192-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: The enantioselective desymmetrization of meso-aziridines by amine/CS2 in the presence of OC-46.
Scheme 11: The enantioselective desymmetrization of meso-aziridines by amine/CS2 in the presence of OC-46.
Chiral 1,2,3-triazolium chlorides
Most recently, Ooi [52] and colleagues have described a desymmetrization of meso-N-p-tert-butylphenylsulfonylaziridines with trimethylsilyl halides by using novel chiral 1,2,3-triazolium chlorides (OC-49 to OC-55) as catalysts (Figure 11). The treatment of meso-aziridine with 1 equiv of chiral 1,2,3-triazolium chloride OC-49·Cl or Me3SiCl in toluene at −40 °C for 12 h did not result in a product formation, but the combined use of a catalytic amount of OC-49·Cl (5 mol %) and a stoichiometric quantity of Me3SiCl led to the desired ring-opening product in low yield (35%) and moderate enantioselectivity (65% ee). The control experiments indicated that the hypervalent silicate ions were involved in the reaction pathway. The enantioselectivities of the desired products were improved by modifying the substituents of catalysts and additives. It was found that the catalysts of chiral 1,2,3-triazolium chlorides containing electron-withdrawing groups usually afforded the high enantioselectivities. The more sterically hindered groups such as iPr and cyclohexyl in catalysts OC-54 and OC-55 demonstrated better enantioselective inductions than the phenyl in OC-53. By using 10 mol % of Me3SiOH as additive, both high yields (up to 99%) and enantioselectivities (up to 95% ee) of the corresponding β-chloro-N-arylsulfonylamines 81 to 87 were obtained (Scheme 12). The additive in the desymmetrization was proposed to facilitate the regeneration of the active chiral triazolium chlorosilicate.
![[1860-5397-9-192-11]](/bjoc/content/figures/1860-5397-9-192-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: The chiral 1,2,3-triazolium chlorides OC-49 to OC-55.
Figure 11: The chiral 1,2,3-triazolium chlorides OC-49 to OC-55.
![[1860-5397-9-192-i12]](/bjoc/content/inline/1860-5397-9-192-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: The enantioselective desymmetrization of meso-aziridines by Me3SiX (X = Cl or Br) in the presence of OC-54 or OC-55.
Scheme 12: The enantioselective desymmetrization of meso-aziridines by Me3SiX (X = Cl or Br) in the presence o...
Organocatalyzed enantioselective desymmetrization of meso-epoxides
Compared to aziridines, the epoxides are much more active in their transformations because of the stronger electronegativity of the oxygen atom compared to the nitrogen atom. Some meso-epoxides are attacked by various nucleophiles to afford β-functionalized alcohols in the absence of a catalysts [53-60], so the stereo-controlling of the ring-opening of epoxides is quite challenging, and the background reaction should be suppressed to achieve high enantioselectivities. However, the catalytic asymmetric ring-opening of meso-epoxides gained much attention by its unique advantages of good atom-economy and versatile transformations. On the one hand, the desymmetrization of meso-epoxides has been successfully performed with various nucleophiles in the presence of metal-based chiral Lewis acid catalysts [61-72]. In recent years, expensive and toxic metals were more and more replaced by cheap and environmentally benign elements [73-77] such as Mg, Fe, Ti, Sc and Ni. On the other hand, the enantioselective desymmetrization of meso-epoxides by organocatalysts received much less attention, and only a few examples provided the desired products in both high yields and high enantioselectivities. In the presence of some achiral phase-transfer catalysts or phosphines such as TBAB and P(t-Bu)3, the vicinal chlorohydrins were obtained in high yields. Up to now, three classes of organocatalysts including chiral phosphoramides, chiral phosphine oxides, and chiral pyridine N-oxides were used in the enantioselective desymmetrization of meso-epoxides.
Organocatalyzed enantioselective ring-opening of epoxides in early stage
In 1981, Andrews [78] and co-workers have found that the ring-opening of cyclohexene and cyclopentene oxides with silicon halides was facilitated by the addition of catalytic amounts of tetra-n-butylammonium chloride or triphenylphosphine to afford trans O-protected vicinal chlorohydrins in quantitative yields. This pioneering work opened the possibility for desymmetrization of meso-epoxides by small organic molecules, but the exploration of organocatalysts for the asymmetric ring-opening of epoxides remained stagnant for sixteen years.
In 1997, Fu [79] and colleagues have developed a phosphaferrocene OC-56 for the organocatalytic ring-opening of epoxides with trimethylsilyl chloride (TMSCl) to provide trans O-TMS protected vicinal chlorohydrins in quantitative yields under very mild conditions (Figure 12).
![[1860-5397-9-192-12]](/bjoc/content/figures/1860-5397-9-192-12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Early organocatalysts for enantioselective desymmetrization of meso-epoxides.
Figure 12: Early organocatalysts for enantioselective desymmetrization of meso-epoxides.
In 2003, Kim [80] and co-workers have prepared a new class of 1,2-ferrocenediylazaphosphinines (OC-59 to OC-61), which served as organocatalysts for the ring-opening of a series of meso-epoxides to produce chlorohydrins with high yields and good regioselectivities. However, in the presence of enantiopure 1,2-ferrocenediylazaphosphinines OC-59, the desired chlorohydrins were obtained from corresponding meso-epoxides in excellent yields but with quite disappointing enantiomeric excesses (Scheme 13).
![[1860-5397-9-192-i13]](/bjoc/content/inline/1860-5397-9-192-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Attempts of enantioselective desymmetrization of meso-epoxides in the presence of OC-58 or OC-60.
Scheme 13: Attempts of enantioselective desymmetrization of meso-epoxides in the presence of OC-58 or OC-60.
Pietrusiewicz [81] and researchers have discovered the enantioselective desymmetrization of a phospholene meso-epoxide by cinchona alkaloids to P,C-chirogenic 3-hydroxy-2-phospholene derivatives 93 and 94. Among the four main components of cinchona alkaloids, quinidine (OC-57) proved to be the most effective base in the enantioselective rearrangement of epoxide, and in the presence of 0.5 equiv of quinidine, the corresponding rearranged product 94 was obtained in 41% yield with 52% ee. The other P,C-chirogenic phospholene derivative 93 was produced by using 100 mol % cinchonidine as a promoter (Scheme 14). But this desymmetrization process is extremely sluggish and the reaction time is up to 90 days!
![[1860-5397-9-192-i14]](/bjoc/content/inline/1860-5397-9-192-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: The enantioselective desymmetrization of a meso-epoxide containing one P atom.
Scheme 14: The enantioselective desymmetrization of a meso-epoxide containing one P atom.
Chiral phosphoramides and phosphine oxides
The first efficient organocatalyzed enantioselective desymmetrization of meso-epoxides was realized by Denmark [82] and colleagues in 1998 (Figure 13). In their initial experiments, various epoxides were attacked with 1.1 equiv of freshly distilled SiCl4 by using 10 mol % of HMPA as a catalyst in CH2Cl2 at −78 °C to afford the corresponding trans-vicinal halohydrins in high yields (up to 96%). This exciting discovery provides an opportunity to enantioselectively desymmetrize meso-epoxides by chiral phosphoramides. A series of chiral phosphoramides were tested. Among these organocatalysts, the chiral HMPA analogue OC-62 is most efficient to produce enantiomeric chlorohydrins in good to excellent yields but low enantioselectivities except with acyclic meso-epoxides (Scheme 15). The highest enantioselectivity was 87% ee. The proposed reaction mechanism is depicted in Figure 14. The first step of the catalytic cycle is the activation of SiCl4 by chiral phosphoramide OC-62 to form a complex, which was ionized to produce a highly reactive silicon cation and a chloride ion. The epoxide was activated by the chiral complexation of the phosphorus/silicon cation, and then followed by an attack with the chloride ion in an SN2 fashion to furnish chlorohydrin enantioselectively (Figure 14).
![[1860-5397-9-192-13]](/bjoc/content/figures/1860-5397-9-192-13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 13: Some chiral phosphoramide and chiral phosphine oxides.
Figure 13: Some chiral phosphoramide and chiral phosphine oxides.
![[1860-5397-9-192-i15]](/bjoc/content/inline/1860-5397-9-192-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: OC-62 catalyzed enantioselective desymmetrization of meso-epoxides by SiCl4.
Scheme 15: OC-62 catalyzed enantioselective desymmetrization of meso-epoxides by SiCl4.
![[1860-5397-9-192-14]](/bjoc/content/figures/1860-5397-9-192-14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: The proposed mechanism of the chiral HMPA-catalyzed desymmetrization of meso-epoxides.
Figure 14: The proposed mechanism of the chiral HMPA-catalyzed desymmetrization of meso-epoxides.
From this description, the activation of SiCl4 was depending on the formation of the chiral complexation of the phosphorus/silicon cation through the oxygen atom of P=O and the coordination with the silicon atom of the nucleophiles. It can be deduced that the chiral phosphine oxides can also play a role for the activation of SiCl4 and serve as catalysts for the desymmetrization of meso-epoxides. The first example of a chiral phosphine oxide-catalyzed asymmetric ring-opening of meso-epoxides has been realized by Nakajima [83] and co-workers and proved to be very effective. They have prepared a chiral phosphine oxide BINAPO (OC-63) based on a binaphthyl-skeleton, which was utilized as an organocatalyst for the enantioselective desymmetrization of meso-epoxides by SiCl4. In the presence of 1.5 equiv of iPr2NEt, various acyclic and cyclic meso-epoxides were desymmetrized to enantioenriched chlorohydrins (96 to 101) in high yields (Scheme 16). When meso-stilbene oxide was used as a substrate, the best enantioselectivity was up to 90% ee under optimum conditions. However, the other substrates, including the meso-epoxides derived from butenediol, pyrroline and dihydrofuran, gave quite disappointing enantioselectivities. Interestingly, OC-65 significantly reduced the enantioselectivities under the optimized conditions, but the chiral phosphine oxides OC-66 and OC-67 afforded enantioselectivities similar to that of BINAPO (OC-63). This indicated that the substituents on the phenyl ring had a remarkable effect on the enantioselectivities in the desymmetrization of meso-epoxides [84]. Benaglia [85] and colleagues have developed chiral phosphine oxides OC-64 and OC-69 for the enantioselective desymmetrization of meso-stilbene oxide to provide the corresponding vicinal chlorohydrin 96 in up to 82% ee.
![[1860-5397-9-192-i16]](/bjoc/content/inline/1860-5397-9-192-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: The enantioselective desymmetrization of meso-epoxides in the presence of OC-63.
Scheme 16: The enantioselective desymmetrization of meso-epoxides in the presence of OC-63.
More recently, Ready [86] and co-workers have synthesized a series of chiral phosphine oxides (OC-70 to OC-77) based on an allene backbone (Figure 15). These novel chiral mono- and bisphosphine oxides were investigated as organocatalysts for the efficient enantioselective desymmetrization of meso-epoxides. Due to the lack of C2 symmetry of mono-phosphine oxides, they displayed lower reactivities and enantioselectivities than the bisphosphine oxides. The variation of the substituents on the allene itself was tolerated in the desymmetrization processes, but both the yields and the enantioselectivities are sensitive to changes in the aryl rings on the phosphine oxide. It was found that the diphenyl-substituted catalyst OC-73 is a highly reactive and enantioselective catalyst to produce the chlorohydrins in up to 94% ee with only 0.1 mol % catalyst-loading. The cis-stilbene oxides with substituent in the meta or para position are tolerated, the yields and enantioselectivities of desired chlorohydrins are satisfying, but ortho-substituted stilbene oxides are not reactive in the desymmetrization under the same reaction conditions. Unsatisfactorily, the desymmetrization of linear, cyclic, or bicyclic aliphatic meso-epoxides resulted in corresponding chlorohydrins in high yield but with low enantiomeric excesses (Scheme 17).
![[1860-5397-9-192-15]](/bjoc/content/figures/1860-5397-9-192-15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: The Chiral phosphine oxides (OC-70 to OC-77) based on an allene backbone.
Figure 15: The Chiral phosphine oxides (OC-70 to OC-77) based on an allene backbone.
![[1860-5397-9-192-i17]](/bjoc/content/inline/1860-5397-9-192-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: OC-73 catalyzed enantioselective desymmetrization of meso-epoxides by SiCl4.
Scheme 17: OC-73 catalyzed enantioselective desymmetrization of meso-epoxides by SiCl4.
Chiral pyridine N-oxides
The breakthrough of the organocatalyzed enantioselective desymmetrization of meso-epoxides was the employment of chiral pyridine N-oxides as catalysts. In 2001, Fu [87] and colleagues demonstrated the utilization of selected chiral pyridine N-oxides in the enantioselective desymmetrization of meso-epoxides. These novel chiral pyridine N-oxides possess a plane of chirality in a ferrocenyl backbone. The steric hindrance of the Fe(η5-C5Ar5) group on catalysts (Figure 16, OC-78 to OC-80) is very crucial to the enantioselectivities of desymmetrization of meso-epoxides. The increase of steric hindrance of the Fe(η5-C5Ar5) group can indeed lead to increased enantioselectivities of the desired chlorohydrins. For example, by shifting the substituents on Fe(η5-C5Ar5) from phenyl (OC-79) to 3,5-dimethylphenyl (OC-80), the enantioselectivity of desymmetrization of cis-stilbene oxide with SiCl4 at room-temperature was improved significantly from 25 to 68% ee. The highest enantiomeric excess of chlorohydrin (92% ee) was obtained by simple decreasing the reaction temperature to −78 °C. A series of substituted cis-stilbene oxides were desymmetrized in very good yield and high stereoselection, and the substrates with electron-withdrawing group such as F or CF3 at the aryl ring can produce chlorohydrins with excellent enantioselectivities (up to 98% ee). Further investigation indicated that the catalyst-loading can be decreased to be 1 mol % and OC-80 can be recovered nearly quantitatively (>90%) with high ee’s.
![[1860-5397-9-192-16]](/bjoc/content/figures/1860-5397-9-192-16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 16: Chiral pyridine N-oxides used in enantioselective desymmetrization of meso-epoxides.
Figure 16: Chiral pyridine N-oxides used in enantioselective desymmetrization of meso-epoxides.
After Fu’s work, two axially-chiral bipyridine N,N’-dioxides OC-81 and OC-82 were selected by Nakajima [88] and co-workers for the enantioselective desymmetrization of meso-epoxides (Scheme 18). When SiCl4 was used as a nucleophile, the desymmetrization of cis-stilbene oxide was performed smoothly in the presence of 10 mol % of catalyst OC-81 in dichloromethane at −78 °C with iPr2NEt as a base to give chlorohydrin in 94% yield and 56% ee. The ee of chlorohydrin was increased to 90% by using OC-82 as a catalyst under the same conditions, but the use of methyl-, allyl-, or phenyltrichlorosilane as a nucleophile gave racemic products. The choice of a proper solvent is very crucial to the stereoselection. For example, THF or toluene as a solvent gave the chlorohydrin in almost racemic form. The substituted cis-stilbene oxides can provide the corresponding chlorohydrins in high chemical yields and ee’s, but meso-cyclohexene oxide resulted in a racemic product under the optimal conditions. Because of the low catalytic activities of axially-chiral pyridine N-oxides OC-83 or OC-84 in the desymmetrization process, a hexacoordinate silicate intermediate between SiCl4 and chiral bipyridine N,N’-dioxide OC-82 was envisaged to explain the good enantioselectivity of catalyst OC-82.
![[1860-5397-9-192-i18]](/bjoc/content/inline/1860-5397-9-192-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Catalyzed enantioselective desymmetrization of meso-epoxides in the presence of OC-80 or OC-82.
Scheme 18: Catalyzed enantioselective desymmetrization of meso-epoxides in the presence of OC-80 or OC-82.
In 2008, a set of enantiomerically pure C2-symmetric bipyridine mono-N-oxides and N,N’-dioxides (OC-85 to OC-94) derived from naturally occurring monoterpenes have been synthesized by Benaglia [89] and colleagues (Figure 17). These chiral bipyridine N,N’-dioxides were utilized in the enantioselective desymmetrization of cis-stilbene oxide and cyclooctene epoxide with SiCl4. Some showed high catalytic activities but with low to moderate enantioselectivities. Among them, chiral bipyridine N,N’-dioxide OC-87 proved to be the most efficient to produce the corresponding chlorohydrins in quantitative yields and good enantioselectivities (up to 70% ee). Unfortunately, chiral bipyridine mono-N-oxides OC-89 and OC-90 provided racemic chlorohydrins (Scheme 19).
![[1860-5397-9-192-17]](/bjoc/content/figures/1860-5397-9-192-17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 17: Chiral pyridine N-oxides OC-85 to OC-94.
Figure 17: Chiral pyridine N-oxides OC-85 to OC-94.
![[1860-5397-9-192-i19]](/bjoc/content/inline/1860-5397-9-192-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Enantioselective desymmetrization of cis-stilbene oxide by using OC-85 to OC-92 as catalysts.
Scheme 19: Enantioselective desymmetrization of cis-stilbene oxide by using OC-85 to OC-92 as catalysts.
Takenaka’s group [90] has developed a novel family of helical chiral pyridine N-oxides (Figure 18, OC-95 to OC-97) which were employed as catalysts for the enantioselective desymmetrization of meso-epoxides with SiCl4. All catalysts demonstrated high activities in the desymmetrization process but with different enantioselectivities. The substrates containing aromatic substituents can provide the corresponding chlorohydrins in higher enantioselectivities than those bearing alkyl groups. Unfortunately, the cyclic epoxides produced the chlorohydrins 115 and 117 in low ee’s (Scheme 20).
![[1860-5397-9-192-18]](/bjoc/content/figures/1860-5397-9-192-18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 18: A novel family of helical chiral pyridine N-oxides OC-95 to OC-97.
Figure 18: A novel family of helical chiral pyridine N-oxides OC-95 to OC-97.
![[1860-5397-9-192-i20]](/bjoc/content/inline/1860-5397-9-192-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Desymmetrization of meso-epoxides catalyzed by OC-95 to OC-97.
Scheme 20: Desymmetrization of meso-epoxides catalyzed by OC-95 to OC-97.
Kočovský [91] and colleagues have introduced a chiral bipyridine mono-N-oxide PINDOX OC-98 for the enantioselective ring-opening of cyclic meso-epoxides with SiCl4 to produce chlorohydrins in moderate to good enantiomeric excess (Scheme 21). The stereoselectivities are very sensitive to the ring size of the cyclic oxides. For example, when cyclohexene oxide was used as a substrate, the corresponding chlorohydrin was obtained in nearly racemic form. The greater the ring sizes of the cyclic oxides, the higher the enantioselectivities. It was found that the catalyst OC-98 is unique for the desymmetrization of meso-cyclooctene oxide, which was proved to be a very challenging substrate for desymmetrization with other reported organocatalysts, and the chlorohydrin 94 was obtained in 90% ee in the presence of 10 mol % of OC-98 in dichloromethane at −90 °C. Interestingly, the tricyclic exo-norbornene oxide afforded the syn-exo-chloroalcohol 123 as the major product in 53% yield and 90% ee by a Wagner–Meerwein rearrangement. The mechanism of the enantioselective desymmetrization of meso-epoxides by chiral pyridine N-oxides is not fully understood at the time of this writing.
![[1860-5397-9-192-i21]](/bjoc/content/inline/1860-5397-9-192-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: OC-98 catalyzed enantioselective desymmetrization of meso-epoxides by SiCl4.
Scheme 21: OC-98 catalyzed enantioselective desymmetrization of meso-epoxides by SiCl4.
Conclusion
In this review, we summarized the recent advances of the organocatalyzed enantioselective desymmetrization of meso-aziridines and meso-epoxides. Undoubtedly, these synthetic methodologies will continue to demonstrate their versatile utilities in organic synthesis. However, there are some difficulties that should be pointed out.
Organocatalysts for the enantioselective desymmetrization of meso-aziridines are plentiful and can be found in a diverse set of privileged structures, including cinchona alkaloids-based PTCs, L-proline-derived amino alcohols, chiral phosphorous acids, chiral thioureas, chiral guanidines, and chiral 1,2,3-triazolium chlorides. But for the desymmetrization of meso-epoxides efficient catalysts are presently limited to chiral phosphine oxides and chiral pyridine N-oxides. Obviously, there is still a need for the exploration of more efficient organocatalysts with high enantioselectivities in this field. In most cases, the catalyst-loading is up to 10 mol %, and some catalysts are not readily accessible and tunable. Therefore, the search for practical methods for the facile synthesis of organocatalysts, which exhibit a plethora of applications, is still in high demand.
Various C-, N-, S-, Se- and halo-nucleophiles are involved in the organocatalyzed enantioselective desymmetrization of meso-aziridines to afford β-functional amine derivatives with high yields and enantioselectivities, while the organocatalyzed enantioselective desymmetrization of meso-epoxides is restricted to chloro-nucleophiles. More specifically, SiCl4 was used in most cases to produce enantioenriched chlorohydrins. It might be worthwhile to extend research activities toward nucleophiles which are able to produce chiral β-functional alcohols.
The acyclic meso-aziridines and meso-epoxides usually showed disappointing results except for a few examples. With some further mechanism studies, new and more efficient organocatalysts for the enantioselective desymmetrization of meso-aziridines and meso-epoxides with a broader substrate scope and milder reaction conditions may be discovered [92-95].
References
-
Ojima, I. Catalytic Asymmetric Synthesis, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2010.
Return to citation in text: [1] -
Bates, R. Organic Synthesis Using Transition Metals, 2nd ed.; John Wiley & Sons: West Sussex, U.K., 2012.
Return to citation in text: [1] -
Crawley, M. L.; Trost, B. M., Eds. Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective; John Wiley & Sons: Hoboken, NJ, U.S.A., 2012.
Return to citation in text: [1] -
Enríquez-García, Á.; Kündig, E. P. Chem. Soc. Rev. 2012, 41, 7803–7831. doi:10.1039/C2CS35049A
Return to citation in text: [1] -
Díaz-de-Villegas, M. D.; Gálvez, J. A.; Badorrey, R.; López-Ram-de-Víu, M. P. Chem.–Eur. J. 2012, 18, 13920–13935. doi:10.1002/chem.201202264
Return to citation in text: [1] -
Willis, M. C. J. Chem. Soc., Perkin Trans. 1 1999, 1765–1784. doi:10.1039/A906269B
Return to citation in text: [1] -
Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Chem. Soc. Rev. 2012, 41, 2406–2447. doi:10.1039/C1CS15206H
Return to citation in text: [1] -
Renzi, P.; Bella, M. Chem. Commun. 2012, 48, 6881–6896. doi:10.1039/C2CC31599H
Return to citation in text: [1] -
Dondoni, A.; Massi, A. Angew. Chem., Int. Ed. 2008, 47, 4638–4660. doi:10.1002/anie.200704684
Return to citation in text: [1] -
Hu, X. E. Tetrahedron 2004, 60, 2701–2743. doi:10.1016/j.tet.2004.01.042
Return to citation in text: [1] -
Lu, P. Tetrahedron 2010, 66, 2549–2560. doi:10.1016/j.tet.2010.01.077
Return to citation in text: [1] -
Stanković, S.; D'hooghe, M.; Catak, S.; Eum, H.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N.; Ha, H.-J. Chem. Soc. Rev. 2012, 41, 643–665. doi:10.1039/C1CS15140A
Return to citation in text: [1] -
Pastor, I. M.; Yus, M. Curr. Org. Chem. 2005, 9, 1–29. doi:10.2174/1385272053369385
Return to citation in text: [1] -
Dhakshinamoorthy, A.; Alvaro, M.; Concepción, P.; Fornés, V.; Garcia, H. Chem. Commun. 2012, 48, 5443–5445. doi:10.1039/C2CC31385E
Return to citation in text: [1] -
Bonollo, S.; Lanari, D.; Vaccaro, L. Eur. J. Org. Chem. 2011, 2587–2598. doi:10.1002/ejoc.201001693
Return to citation in text: [1] -
Nakamura, S.; Hayashi, M.; Kamada, Y.; Sasaki, R.; Hiramatsu, Y.; Shibata, N.; Toru, T. Tetrahedron Lett. 2010, 51, 3820–3823. doi:10.1016/j.tetlet.2010.05.065
Return to citation in text: [1] -
Arai, K.; Lucarini, S.; Salter, M. M.; Ohta, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 8103–8111. doi:10.1021/ja0708666
Return to citation in text: [1] -
Fukuta, Y.; Mita, T.; Fukuda, N.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 6312–6313. doi:10.1021/ja061696k
Return to citation in text: [1] -
Wu, B.; Gallucci, J. C.; Parquette, J. R.; RajanBabu, T. V. Angew. Chem., Int. Ed. 2009, 48, 1126–1129. doi:10.1002/anie.200804415
Return to citation in text: [1] -
Fujimori, I.; Mita, T.; Maki, K.; Shiro, M.; Sato, A.; Furusho, S.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 16438–16439. doi:10.1021/ja067003h
Return to citation in text: [1] -
Schaus, S. E.; Brandes, B. D.; Larrow, J. F.; Tokunaga, M.; Hansen, K. B.; Gould, A. E.; Furrow, M. E.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 1307–1315. doi:10.1021/ja016737l
Return to citation in text: [1] -
Schneider, C.; Sreekanth, A. R.; Mai, E. Angew. Chem., Int. Ed. 2004, 43, 5691–5694. doi:10.1002/anie.200460786
Return to citation in text: [1] -
Li, Z.; Fernández, M.; Jacobsen, E. N. Org. Lett. 1999, 1, 1611–1613. doi:10.1021/ol990992h
Return to citation in text: [1] -
Schneider, C. Angew. Chem., Int. Ed. 2009, 48, 2082–2084. doi:10.1002/anie.200805542
Return to citation in text: [1] -
Xu, Y.; Lin, L.; Kanai, M.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2011, 133, 5791–5793. doi:10.1021/ja201492x
Return to citation in text: [1] -
Peruncheralathan, S.; Henze, M.; Schneider, C. Tetrahedron Lett. 2007, 48, 6743–6746. doi:10.1016/j.tetlet.2007.07.082
Return to citation in text: [1] -
Kalow, J. A.; Doyle, A. G. J. Am. Chem. Soc. 2010, 132, 3268–3269. doi:10.1021/ja100161d
Return to citation in text: [1] -
Peruncheralathan, S.; Aurich, S.; Teller, H.; Schneider, C. Org. Biomol. Chem. 2013, 11, 2787–2803. doi:10.1039/C3OB40222C
Return to citation in text: [1] -
Blyumin, E. V.; Gallon, H. J.; Yudin, A. K. Org. Lett. 2007, 9, 4677–4680. doi:10.1021/ol7015302
Return to citation in text: [1] -
Minakata, S.; Okada, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2005, 7, 3509–3512. doi:10.1021/ol051186f
Return to citation in text: [1] -
Fan, R.-H.; Hou, X.-L. J. Org. Chem. 2003, 68, 726–730. doi:10.1021/jo025983s
Return to citation in text: [1] -
Wu, J.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2000, 65, 1344–1348. doi:10.1021/jo9913816
Return to citation in text: [1] -
Hou, X.-L.; Fan, R.-H.; Dai, L.-X. J. Org. Chem. 2002, 67, 5295–5300. doi:10.1021/jo016230t
Return to citation in text: [1] -
Liu, Y.-K.; Li, R.; Yue, L.; Li, B.-J.; Chen, Y. -C.; Wu, Y.; Ding, L.-S. Org. Lett. 2006, 8, 1521–1524. doi:10.1021/ol0529905
Return to citation in text: [1] -
Alagiri, K.; Prabhu, K. R. Chem.–Eur. J. 2011, 17, 6922–6925. doi:10.1002/chem.201100817
Return to citation in text: [1] -
Minakata, S.; Hotta, T.; Oderaotoshi, Y.; Komatsu, M. J. Org. Chem. 2006, 71, 7471–7472. doi:10.1021/jo061239m
Return to citation in text: [1] -
Fan, R.-H.; Zhou, Y.-G.; Zhang, W.-X.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2004, 69, 335–338. doi:10.1021/jo034895k
Return to citation in text: [1] -
Kalow, J. A.; Schmitt, D. E.; Doyle, A. G. J. Org. Chem. 2012, 77, 4177–4183. doi:10.1021/jo300433a
Return to citation in text: [1] -
Narender, M.; Surendra, K.; Srilakshmi Krishnaveni, N.; Somi Reddy, M.; Rama Rao, K. Tetrahedron Lett. 2004, 45, 7995–7997. doi:10.1016/j.tetlet.2004.09.014
Return to citation in text: [1] -
Luo, Z.-B.; Hou, X.-L.; Dai, L.-X. Tetrahedron: Asymmetry 2007, 18, 443–446. doi:10.1016/j.tetasy.2007.02.017
Return to citation in text: [1] -
Wang, Z.; Sun, X.; Ye, S.; Wang, W.; Wang, B.; Wu, J. Tetrahedron: Asymmetry 2008, 19, 964–969. doi:10.1016/j.tetasy.2008.03.017
Return to citation in text: [1] -
Moss, T. A.; Fenwick, D. R.; Dixon, D. J. J. Am. Chem. Soc. 2008, 130, 10076–10077. doi:10.1021/ja8036965
Return to citation in text: [1] -
Paixão, M. W.; Nielsen, M.; Jacobsen, C. B.; Jørgensen, K. A. Org. Biomol. Chem. 2008, 6, 3467–3470. doi:10.1039/B812369A
Return to citation in text: [1] -
Rowland, E. B.; Rowland, G. B.; Rivera-Otero, E.; Antilla, J. C. J. Am. Chem. Soc. 2007, 129, 12084–12085. doi:10.1021/ja0751779
Return to citation in text: [1] -
Larson, S. E.; Baso, J. C.; Li, G.; Antilla, J. C. Org. Lett. 2009, 11, 5186–5189. doi:10.1021/ol902123h
Return to citation in text: [1] -
Della Sala, G.; Lattanzi, A. Org. Lett. 2009, 11, 3330–3333. doi:10.1021/ol901209n
Return to citation in text: [1] -
Senatore, M.; Lattanzi, A.; Santoro, S.; Santi, C.; Della Sala, G. Org. Biomol. Chem. 2011, 9, 6205–6207. doi:10.1039/C1OB05837A
Return to citation in text: [1] -
Della Sala, G. Tetrahedron 2013, 69, 50–56. doi:10.1016/j.tet.2012.10.068
Return to citation in text: [1] -
Lattanzi, A.; Della Sala, G. Eur. J. Org. Chem. 2009, 1845–1848. doi:10.1002/ejoc.200900095
Return to citation in text: [1] -
Mita, T.; Jacobsen, E. N. Synlett 2009, 1680–1684. doi:10.1055/s-0029-1217344
Return to citation in text: [1] -
Zhang, Y.; Kee, C. W.; Lee, R.; Fu, X.; Soh, J. Y.-T.; Loh, E. M. F.; Huang, K.-W.; Tan, C.-H. Chem. Commun. 2011, 47, 3897–3899. doi:10.1039/C0CC05840H
Return to citation in text: [1] -
Ohmatsu, K.; Hamajima, Y.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 8794–8797. doi:10.1021/ja3028668
Return to citation in text: [1] -
Chen, J.; Wu, H.; Jin, C.; Zhang, X.; Xie, Y.; Su, W. Green Chem. 2006, 8, 330–332. doi:10.1039/B600620E
Return to citation in text: [1] -
Wang, Z.; Cui, Y.-T.; Xu, Z.-B.; Qu, J. J. Org. Chem. 2008, 73, 2270–2274. doi:10.1021/jo702401t
Return to citation in text: [1] -
Azizi, N.; Saidi, M. R. Org. Lett. 2005, 7, 3649–3651. doi:10.1021/ol051220q
Return to citation in text: [1] -
Torregrosa, R.; Pastor, I. M.; Yus, M. Tetrahedron 2007, 63, 469–473. doi:10.1016/j.tet.2006.10.055
Return to citation in text: [1] -
Das, B.; Krishnaiah, M.; Thirupathi, P.; Laxminarayana, K. Tetrahedron Lett. 2007, 48, 4263–4265. doi:10.1016/j.tetlet.2007.04.062
Return to citation in text: [1] -
Lau, E. Y.; Newby, Z. E.; Bruice, T. C. J. Am. Chem. Soc. 2001, 123, 3350–3357. doi:10.1021/ja0037724
Return to citation in text: [1] -
Das, B.; Reddy, V. S.; Tehseen, F.; Krishnaiah, M. Synthesis 2007, 666–668. doi:10.1055/s-2007-965921
Return to citation in text: [1] -
Vilotijevic, I.; Jamison, T. F. Science 2007, 317, 1189–1192. doi:10.1126/science.1146421
Return to citation in text: [1] -
Azoulay, S.; Manabe, K.; Kobayashi, S. Org. Lett. 2005, 7, 4593–4595. doi:10.1021/ol051546z
Return to citation in text: [1] -
Kureshy, R. I.; Singh, S.; Khan, N.-u. H.; Abdi, S. H. R.; Suresh, E.; Jasra, R. V. Eur. J. Org. Chem. 2006, 1303–1309. doi:10.1002/ejoc.200500765
Return to citation in text: [1] -
Wu, J.; Hou, X.-L.; Dai, L.-X.; Xia, L.-J.; Tang, M.-H. Tetrahedron: Asymmetry 1998, 9, 3431–3436. doi:10.1016/S0957-4166(98)00358-9
Return to citation in text: [1] -
Kokubo, M.; Naito, T.; Kobayashi, S. Tetrahedron 2010, 66, 1111–1118. doi:10.1016/j.tet.2009.11.018
Return to citation in text: [1] -
Gao, B.; Xie, M.; Sun, A.; Hu, X.; Ding, X.; Liu, X.; Lin, L.; Feng, X. Adv. Synth. Catal. 2012, 354, 1509–1518. doi:10.1002/adsc.201200142
Return to citation in text: [1] -
Boudou, M.; Ogawa, C.; Kobayashi, S. Adv. Synth. Catal. 2006, 348, 2585–2589. doi:10.1002/adsc.200600290
Return to citation in text: [1] -
Yang, M.; Zhu, C.; Yuan, F.; Huang, Y.; Pan, Y. Org. Lett. 2005, 7, 1927–1930. doi:10.1021/ol0503034
Return to citation in text: [1] -
Matsunaga, S.; Das, J.; Roels, J.; Vogl, E. M.; Yamamoto, N.; Lida, T.; Yamaguchi, K.; Shibasaki, M. J. Am. Chem. Soc. 2000, 122, 2252–2260. doi:10.1021/ja993650f
Return to citation in text: [1] -
Arai, K.; Salter, M. M.; Yamashita, Y.; Kobayashi, S. Angew. Chem., Int. Ed. 2007, 46, 955–957. doi:10.1002/anie.200603787
Return to citation in text: [1] -
Chen, Y.-J.; Chen, C. Tetrahedron: Asymmetry 2007, 18, 1313–1319. doi:10.1016/j.tetasy.2007.04.030
Return to citation in text: [1] -
Gansäuer, A.; Fan, C.-A.; Keller, F.; Karbaum, P. Chem.–Eur. J. 2007, 13, 8084–8090. doi:10.1002/chem.200701021
Return to citation in text: [1] -
Belokon, Y. N.; Chusov, D.; Peregudov, A. S.; Yashkina, L. V.; Timofeeva, G. I.; Maleev, V. I.; North, M.; Kagan, H. B. Adv. Synth. Catal. 2009, 351, 3157–3167. doi:10.1002/adsc.200900523
Return to citation in text: [1] -
Bao, H.; Wu, J.; Li, H.; Wang, Z.; You, T.; Ding, K. Eur. J. Org. Chem. 2010, 35, 6722–6726. doi:10.1002/ejoc.201001222
See for the Mg-catalyzed enantioselective desymmetrization of meso-epoxides.
Return to citation in text: [1] -
Plancq, B.; Ollevier, T. Chem. Commun. 2012, 48, 3806–3808. doi:10.1039/C2CC18032D
See for the Fe-catalyzed enantioselective desymmetrization of meso-epoxides.
Return to citation in text: [1] -
Bao, H.; Zhou, J.; Wang, Z.; Guo, Y.; You, T.; Zhang, H.; Ding, K. J. Am. Chem. Soc. 2008, 130, 10116–10127. doi:10.1021/ja801847r
See for the Ti-catalyzed enantioselective desymmetrization of meso-epoxides.
Return to citation in text: [1] -
Hu, X.; Gao, B.; Chu, Y.; Li, W.; Liu, X.; Lin, L.; Feng, X. Chem.–Eur. J. 2012, 18, 3473–3477. doi:10.1002/chem.201103792
See for the Sc-catalyzed enantioselective desymmetrization of meso-epoxides.
Return to citation in text: [1] -
Beaver, M. G.; Jamison, T. F. Org. Lett. 2011, 13, 4140–4143. doi:10.1021/ol201702a
See for the Ni-catalyzed ring-opening of meso-epoxides.
Return to citation in text: [1] -
Andrews, G. C.; Crawford, T. C.; Contillo, L. G., Jr. Tetrahedron Lett. 1981, 22, 3803–3806. doi:10.1016/S0040-4039(01)91312-7
Return to citation in text: [1] -
Garrett, C. E.; Fu, G. C. J. Org. Chem. 1997, 62, 4534–4535. doi:10.1021/jo970419g
Return to citation in text: [1] -
Paek, S. H.; Shim, S. C.; Cho, C. S.; Kim, T.-J. Synlett 2003, 849–851. doi:10.1055/s-2003-38758
Return to citation in text: [1] -
Pietrusiewicz, K. M.; Koprowski, M.; Pakulski, Z. Tetrahedron: Asymmetry 2002, 13, 1017–1019. doi:10.1016/S0957-4166(02)00245-8
Return to citation in text: [1] -
Denmark, S. E.; Barsanti, P. A.; Wong, K.-T.; Stavenger, R. A. J. Org. Chem. 1998, 63, 2428–2429. doi:10.1021/jo9801420
Return to citation in text: [1] -
Tokuoka, E.; Kotani, S.; Matsunaga, H.; Ishizuka, T.; Hashimoto, S.; Nakajima, M. Tetrahedron: Asymmetry 2005, 16, 2391–2392. doi:10.1016/j.tetasy.2005.06.021
Return to citation in text: [1] -
Kotani, S.; Furusho, H.; Sugiura, M.; Nakajima, M. Tetrahedron 2013, 69, 3075–3081. doi:10.1016/j.tet.2013.01.066
Return to citation in text: [1] -
Simonini, V.; Benaglia, M.; Benincori, T. Adv. Synth. Catal. 2008, 350, 561–564. doi:10.1002/adsc.200700564
Return to citation in text: [1] -
Pu, X.; Qi, X.; Ready, J. M. J. Am. Chem. Soc. 2009, 131, 10364–10365. doi:10.1021/ja9041127
Return to citation in text: [1] -
Tao, B.; Lo, M. M.-C.; Fu, G. C. J. Am. Chem. Soc. 2001, 123, 353–354. doi:10.1021/ja003573k
Return to citation in text: [1] -
Nakajima, M.; Saito, M.; Uemura, M.; Hashimoto, S. Tetrahedron Lett. 2002, 43, 8827–8829. doi:10.1016/S0040-4039(02)02229-3
Return to citation in text: [1] -
Chelucci, G.; Baldino, S.; Pinna, G. A.; Benaglia, M.; Buffa, L.; Guizzettie, S. Tetrahedron 2008, 64, 7574–7582. doi:10.1016/j.tet.2008.05.105
Return to citation in text: [1] -
Takenaka, N.; Sarangthem, R. S.; Captain, B. Angew. Chem., Int. Ed. 2008, 47, 9708–9710. doi:10.1002/anie.200803338
Return to citation in text: [1] -
Malkov, A. V.; Gordon, M. R.; Stončius, S.; Hussain, J.; Kočovský, P. Org. Lett. 2009, 11, 5390–5393. doi:10.1021/ol902148s
Return to citation in text: [1] -
Hayashi, M.; Shiomi, N.; Funahashi, Y.; Nakamura, S. J. Am. Chem. Soc. 2012, 134, 19366–19369. doi:10.1021/ja309963w
Return to citation in text: [1] -
Wang, Z.; Chen, Z.; Sun, J. Angew. Chem., Int. Ed. 2013, 52, 6685–6688. doi:10.1002/anie.201300188
Return to citation in text: [1] -
Chawla, R.; Singh, A. K.; Yadav, L. D. S. RSC Adv. 2013, 3, 11385–11403. doi:10.1039/C3RA00175J
Return to citation in text: [1] -
Cao, Y.-M.; Zhang, F.-T.; Shen, F.-F.; Wang, R. Chem.–Eur. J. 2013, 19, 9476–9480. doi:10.1002/chem.201300297
Return to citation in text: [1]
| 73. |
Bao, H.; Wu, J.; Li, H.; Wang, Z.; You, T.; Ding, K. Eur. J. Org. Chem. 2010, 35, 6722–6726. doi:10.1002/ejoc.201001222
See for the Mg-catalyzed enantioselective desymmetrization of meso-epoxides. |
| 74. |
Plancq, B.; Ollevier, T. Chem. Commun. 2012, 48, 3806–3808. doi:10.1039/C2CC18032D
See for the Fe-catalyzed enantioselective desymmetrization of meso-epoxides. |
| 75. |
Bao, H.; Zhou, J.; Wang, Z.; Guo, Y.; You, T.; Zhang, H.; Ding, K. J. Am. Chem. Soc. 2008, 130, 10116–10127. doi:10.1021/ja801847r
See for the Ti-catalyzed enantioselective desymmetrization of meso-epoxides. |
| 76. |
Hu, X.; Gao, B.; Chu, Y.; Li, W.; Liu, X.; Lin, L.; Feng, X. Chem.–Eur. J. 2012, 18, 3473–3477. doi:10.1002/chem.201103792
See for the Sc-catalyzed enantioselective desymmetrization of meso-epoxides. |
| 77. |
Beaver, M. G.; Jamison, T. F. Org. Lett. 2011, 13, 4140–4143. doi:10.1021/ol201702a
See for the Ni-catalyzed ring-opening of meso-epoxides. |
| 78. | Andrews, G. C.; Crawford, T. C.; Contillo, L. G., Jr. Tetrahedron Lett. 1981, 22, 3803–3806. doi:10.1016/S0040-4039(01)91312-7 |
| 79. | Garrett, C. E.; Fu, G. C. J. Org. Chem. 1997, 62, 4534–4535. doi:10.1021/jo970419g |
| 1. | Ojima, I. Catalytic Asymmetric Synthesis, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2010. |
| 10. | Hu, X. E. Tetrahedron 2004, 60, 2701–2743. doi:10.1016/j.tet.2004.01.042 |
| 11. | Lu, P. Tetrahedron 2010, 66, 2549–2560. doi:10.1016/j.tet.2010.01.077 |
| 12. | Stanković, S.; D'hooghe, M.; Catak, S.; Eum, H.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N.; Ha, H.-J. Chem. Soc. Rev. 2012, 41, 643–665. doi:10.1039/C1CS15140A |
| 42. | Moss, T. A.; Fenwick, D. R.; Dixon, D. J. J. Am. Chem. Soc. 2008, 130, 10076–10077. doi:10.1021/ja8036965 |
| 86. | Pu, X.; Qi, X.; Ready, J. M. J. Am. Chem. Soc. 2009, 131, 10364–10365. doi:10.1021/ja9041127 |
| 7. | Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Chem. Soc. Rev. 2012, 41, 2406–2447. doi:10.1039/C1CS15206H |
| 8. | Renzi, P.; Bella, M. Chem. Commun. 2012, 48, 6881–6896. doi:10.1039/C2CC31599H |
| 9. | Dondoni, A.; Massi, A. Angew. Chem., Int. Ed. 2008, 47, 4638–4660. doi:10.1002/anie.200704684 |
| 43. | Paixão, M. W.; Nielsen, M.; Jacobsen, C. B.; Jørgensen, K. A. Org. Biomol. Chem. 2008, 6, 3467–3470. doi:10.1039/B812369A |
| 87. | Tao, B.; Lo, M. M.-C.; Fu, G. C. J. Am. Chem. Soc. 2001, 123, 353–354. doi:10.1021/ja003573k |
| 4. | Enríquez-García, Á.; Kündig, E. P. Chem. Soc. Rev. 2012, 41, 7803–7831. doi:10.1039/C2CS35049A |
| 5. | Díaz-de-Villegas, M. D.; Gálvez, J. A.; Badorrey, R.; López-Ram-de-Víu, M. P. Chem.–Eur. J. 2012, 18, 13920–13935. doi:10.1002/chem.201202264 |
| 6. | Willis, M. C. J. Chem. Soc., Perkin Trans. 1 1999, 1765–1784. doi:10.1039/A906269B |
| 40. | Luo, Z.-B.; Hou, X.-L.; Dai, L.-X. Tetrahedron: Asymmetry 2007, 18, 443–446. doi:10.1016/j.tetasy.2007.02.017 |
| 84. | Kotani, S.; Furusho, H.; Sugiura, M.; Nakajima, M. Tetrahedron 2013, 69, 3075–3081. doi:10.1016/j.tet.2013.01.066 |
| 2. | Bates, R. Organic Synthesis Using Transition Metals, 2nd ed.; John Wiley & Sons: West Sussex, U.K., 2012. |
| 3. | Crawley, M. L.; Trost, B. M., Eds. Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective; John Wiley & Sons: Hoboken, NJ, U.S.A., 2012. |
| 41. | Wang, Z.; Sun, X.; Ye, S.; Wang, W.; Wang, B.; Wu, J. Tetrahedron: Asymmetry 2008, 19, 964–969. doi:10.1016/j.tetasy.2008.03.017 |
| 85. | Simonini, V.; Benaglia, M.; Benincori, T. Adv. Synth. Catal. 2008, 350, 561–564. doi:10.1002/adsc.200700564 |
| 31. | Fan, R.-H.; Hou, X.-L. J. Org. Chem. 2003, 68, 726–730. doi:10.1021/jo025983s |
| 32. | Wu, J.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2000, 65, 1344–1348. doi:10.1021/jo9913816 |
| 35. | Alagiri, K.; Prabhu, K. R. Chem.–Eur. J. 2011, 17, 6922–6925. doi:10.1002/chem.201100817 |
| 36. | Minakata, S.; Hotta, T.; Oderaotoshi, Y.; Komatsu, M. J. Org. Chem. 2006, 71, 7471–7472. doi:10.1021/jo061239m |
| 82. | Denmark, S. E.; Barsanti, P. A.; Wong, K.-T.; Stavenger, R. A. J. Org. Chem. 1998, 63, 2428–2429. doi:10.1021/jo9801420 |
| 29. | Blyumin, E. V.; Gallon, H. J.; Yudin, A. K. Org. Lett. 2007, 9, 4677–4680. doi:10.1021/ol7015302 |
| 30. | Minakata, S.; Okada, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2005, 7, 3509–3512. doi:10.1021/ol051186f |
| 37. | Fan, R.-H.; Zhou, Y.-G.; Zhang, W.-X.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2004, 69, 335–338. doi:10.1021/jo034895k |
| 38. | Kalow, J. A.; Schmitt, D. E.; Doyle, A. G. J. Org. Chem. 2012, 77, 4177–4183. doi:10.1021/jo300433a |
| 39. | Narender, M.; Surendra, K.; Srilakshmi Krishnaveni, N.; Somi Reddy, M.; Rama Rao, K. Tetrahedron Lett. 2004, 45, 7995–7997. doi:10.1016/j.tetlet.2004.09.014 |
| 83. | Tokuoka, E.; Kotani, S.; Matsunaga, H.; Ishizuka, T.; Hashimoto, S.; Nakajima, M. Tetrahedron: Asymmetry 2005, 16, 2391–2392. doi:10.1016/j.tetasy.2005.06.021 |
| 16. | Nakamura, S.; Hayashi, M.; Kamada, Y.; Sasaki, R.; Hiramatsu, Y.; Shibata, N.; Toru, T. Tetrahedron Lett. 2010, 51, 3820–3823. doi:10.1016/j.tetlet.2010.05.065 |
| 17. | Arai, K.; Lucarini, S.; Salter, M. M.; Ohta, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 8103–8111. doi:10.1021/ja0708666 |
| 18. | Fukuta, Y.; Mita, T.; Fukuda, N.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 6312–6313. doi:10.1021/ja061696k |
| 19. | Wu, B.; Gallucci, J. C.; Parquette, J. R.; RajanBabu, T. V. Angew. Chem., Int. Ed. 2009, 48, 1126–1129. doi:10.1002/anie.200804415 |
| 20. | Fujimori, I.; Mita, T.; Maki, K.; Shiro, M.; Sato, A.; Furusho, S.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 16438–16439. doi:10.1021/ja067003h |
| 21. | Schaus, S. E.; Brandes, B. D.; Larrow, J. F.; Tokunaga, M.; Hansen, K. B.; Gould, A. E.; Furrow, M. E.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 1307–1315. doi:10.1021/ja016737l |
| 22. | Schneider, C.; Sreekanth, A. R.; Mai, E. Angew. Chem., Int. Ed. 2004, 43, 5691–5694. doi:10.1002/anie.200460786 |
| 23. | Li, Z.; Fernández, M.; Jacobsen, E. N. Org. Lett. 1999, 1, 1611–1613. doi:10.1021/ol990992h |
| 24. | Schneider, C. Angew. Chem., Int. Ed. 2009, 48, 2082–2084. doi:10.1002/anie.200805542 |
| 25. | Xu, Y.; Lin, L.; Kanai, M.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2011, 133, 5791–5793. doi:10.1021/ja201492x |
| 26. | Peruncheralathan, S.; Henze, M.; Schneider, C. Tetrahedron Lett. 2007, 48, 6743–6746. doi:10.1016/j.tetlet.2007.07.082 |
| 27. | Kalow, J. A.; Doyle, A. G. J. Am. Chem. Soc. 2010, 132, 3268–3269. doi:10.1021/ja100161d |
| 28. | Peruncheralathan, S.; Aurich, S.; Teller, H.; Schneider, C. Org. Biomol. Chem. 2013, 11, 2787–2803. doi:10.1039/C3OB40222C |
| 80. | Paek, S. H.; Shim, S. C.; Cho, C. S.; Kim, T.-J. Synlett 2003, 849–851. doi:10.1055/s-2003-38758 |
| 13. | Pastor, I. M.; Yus, M. Curr. Org. Chem. 2005, 9, 1–29. doi:10.2174/1385272053369385 |
| 14. | Dhakshinamoorthy, A.; Alvaro, M.; Concepción, P.; Fornés, V.; Garcia, H. Chem. Commun. 2012, 48, 5443–5445. doi:10.1039/C2CC31385E |
| 15. | Bonollo, S.; Lanari, D.; Vaccaro, L. Eur. J. Org. Chem. 2011, 2587–2598. doi:10.1002/ejoc.201001693 |
| 33. | Hou, X.-L.; Fan, R.-H.; Dai, L.-X. J. Org. Chem. 2002, 67, 5295–5300. doi:10.1021/jo016230t |
| 34. | Liu, Y.-K.; Li, R.; Yue, L.; Li, B.-J.; Chen, Y. -C.; Wu, Y.; Ding, L.-S. Org. Lett. 2006, 8, 1521–1524. doi:10.1021/ol0529905 |
| 81. | Pietrusiewicz, K. M.; Koprowski, M.; Pakulski, Z. Tetrahedron: Asymmetry 2002, 13, 1017–1019. doi:10.1016/S0957-4166(02)00245-8 |
| 46. | Della Sala, G.; Lattanzi, A. Org. Lett. 2009, 11, 3330–3333. doi:10.1021/ol901209n |
| 44. | Rowland, E. B.; Rowland, G. B.; Rivera-Otero, E.; Antilla, J. C. J. Am. Chem. Soc. 2007, 129, 12084–12085. doi:10.1021/ja0751779 |
| 88. | Nakajima, M.; Saito, M.; Uemura, M.; Hashimoto, S. Tetrahedron Lett. 2002, 43, 8827–8829. doi:10.1016/S0040-4039(02)02229-3 |
| 45. | Larson, S. E.; Baso, J. C.; Li, G.; Antilla, J. C. Org. Lett. 2009, 11, 5186–5189. doi:10.1021/ol902123h |
| 89. | Chelucci, G.; Baldino, S.; Pinna, G. A.; Benaglia, M.; Buffa, L.; Guizzettie, S. Tetrahedron 2008, 64, 7574–7582. doi:10.1016/j.tet.2008.05.105 |
| 90. | Takenaka, N.; Sarangthem, R. S.; Captain, B. Angew. Chem., Int. Ed. 2008, 47, 9708–9710. doi:10.1002/anie.200803338 |
| 53. | Chen, J.; Wu, H.; Jin, C.; Zhang, X.; Xie, Y.; Su, W. Green Chem. 2006, 8, 330–332. doi:10.1039/B600620E |
| 54. | Wang, Z.; Cui, Y.-T.; Xu, Z.-B.; Qu, J. J. Org. Chem. 2008, 73, 2270–2274. doi:10.1021/jo702401t |
| 55. | Azizi, N.; Saidi, M. R. Org. Lett. 2005, 7, 3649–3651. doi:10.1021/ol051220q |
| 56. | Torregrosa, R.; Pastor, I. M.; Yus, M. Tetrahedron 2007, 63, 469–473. doi:10.1016/j.tet.2006.10.055 |
| 57. | Das, B.; Krishnaiah, M.; Thirupathi, P.; Laxminarayana, K. Tetrahedron Lett. 2007, 48, 4263–4265. doi:10.1016/j.tetlet.2007.04.062 |
| 58. | Lau, E. Y.; Newby, Z. E.; Bruice, T. C. J. Am. Chem. Soc. 2001, 123, 3350–3357. doi:10.1021/ja0037724 |
| 59. | Das, B.; Reddy, V. S.; Tehseen, F.; Krishnaiah, M. Synthesis 2007, 666–668. doi:10.1055/s-2007-965921 |
| 60. | Vilotijevic, I.; Jamison, T. F. Science 2007, 317, 1189–1192. doi:10.1126/science.1146421 |
| 61. | Azoulay, S.; Manabe, K.; Kobayashi, S. Org. Lett. 2005, 7, 4593–4595. doi:10.1021/ol051546z |
| 62. | Kureshy, R. I.; Singh, S.; Khan, N.-u. H.; Abdi, S. H. R.; Suresh, E.; Jasra, R. V. Eur. J. Org. Chem. 2006, 1303–1309. doi:10.1002/ejoc.200500765 |
| 63. | Wu, J.; Hou, X.-L.; Dai, L.-X.; Xia, L.-J.; Tang, M.-H. Tetrahedron: Asymmetry 1998, 9, 3431–3436. doi:10.1016/S0957-4166(98)00358-9 |
| 64. | Kokubo, M.; Naito, T.; Kobayashi, S. Tetrahedron 2010, 66, 1111–1118. doi:10.1016/j.tet.2009.11.018 |
| 65. | Gao, B.; Xie, M.; Sun, A.; Hu, X.; Ding, X.; Liu, X.; Lin, L.; Feng, X. Adv. Synth. Catal. 2012, 354, 1509–1518. doi:10.1002/adsc.201200142 |
| 66. | Boudou, M.; Ogawa, C.; Kobayashi, S. Adv. Synth. Catal. 2006, 348, 2585–2589. doi:10.1002/adsc.200600290 |
| 67. | Yang, M.; Zhu, C.; Yuan, F.; Huang, Y.; Pan, Y. Org. Lett. 2005, 7, 1927–1930. doi:10.1021/ol0503034 |
| 68. | Matsunaga, S.; Das, J.; Roels, J.; Vogl, E. M.; Yamamoto, N.; Lida, T.; Yamaguchi, K.; Shibasaki, M. J. Am. Chem. Soc. 2000, 122, 2252–2260. doi:10.1021/ja993650f |
| 69. | Arai, K.; Salter, M. M.; Yamashita, Y.; Kobayashi, S. Angew. Chem., Int. Ed. 2007, 46, 955–957. doi:10.1002/anie.200603787 |
| 70. | Chen, Y.-J.; Chen, C. Tetrahedron: Asymmetry 2007, 18, 1313–1319. doi:10.1016/j.tetasy.2007.04.030 |
| 71. | Gansäuer, A.; Fan, C.-A.; Keller, F.; Karbaum, P. Chem.–Eur. J. 2007, 13, 8084–8090. doi:10.1002/chem.200701021 |
| 72. | Belokon, Y. N.; Chusov, D.; Peregudov, A. S.; Yashkina, L. V.; Timofeeva, G. I.; Maleev, V. I.; North, M.; Kagan, H. B. Adv. Synth. Catal. 2009, 351, 3157–3167. doi:10.1002/adsc.200900523 |
| 51. | Zhang, Y.; Kee, C. W.; Lee, R.; Fu, X.; Soh, J. Y.-T.; Loh, E. M. F.; Huang, K.-W.; Tan, C.-H. Chem. Commun. 2011, 47, 3897–3899. doi:10.1039/C0CC05840H |
| 52. | Ohmatsu, K.; Hamajima, Y.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 8794–8797. doi:10.1021/ja3028668 |
| 49. | Lattanzi, A.; Della Sala, G. Eur. J. Org. Chem. 2009, 1845–1848. doi:10.1002/ejoc.200900095 |
| 50. | Mita, T.; Jacobsen, E. N. Synlett 2009, 1680–1684. doi:10.1055/s-0029-1217344 |
| 47. | Senatore, M.; Lattanzi, A.; Santoro, S.; Santi, C.; Della Sala, G. Org. Biomol. Chem. 2011, 9, 6205–6207. doi:10.1039/C1OB05837A |
| 91. | Malkov, A. V.; Gordon, M. R.; Stončius, S.; Hussain, J.; Kočovský, P. Org. Lett. 2009, 11, 5390–5393. doi:10.1021/ol902148s |
| 92. | Hayashi, M.; Shiomi, N.; Funahashi, Y.; Nakamura, S. J. Am. Chem. Soc. 2012, 134, 19366–19369. doi:10.1021/ja309963w |
| 93. | Wang, Z.; Chen, Z.; Sun, J. Angew. Chem., Int. Ed. 2013, 52, 6685–6688. doi:10.1002/anie.201300188 |
| 94. | Chawla, R.; Singh, A. K.; Yadav, L. D. S. RSC Adv. 2013, 3, 11385–11403. doi:10.1039/C3RA00175J |
| 95. | Cao, Y.-M.; Zhang, F.-T.; Shen, F.-F.; Wang, R. Chem.–Eur. J. 2013, 19, 9476–9480. doi:10.1002/chem.201300297 |
© 2013 Wang; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)