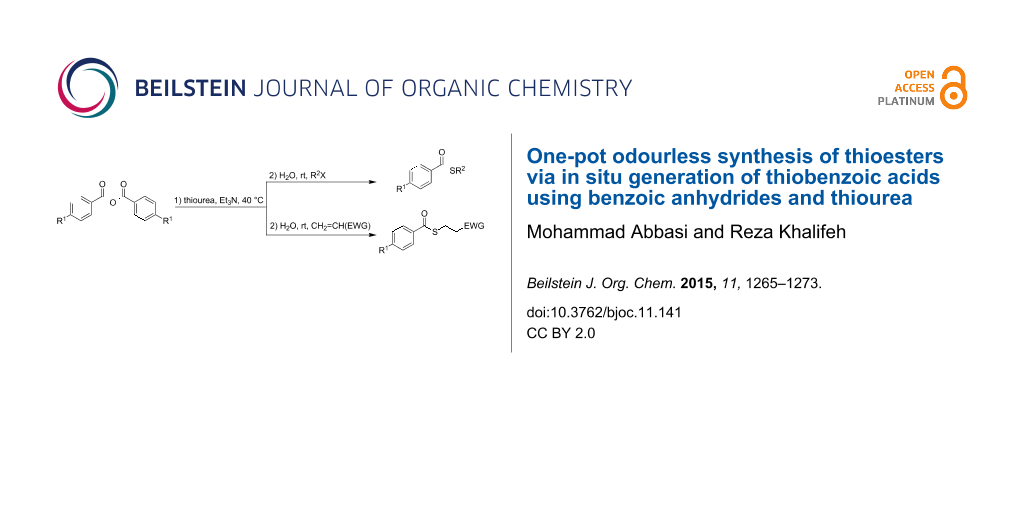
Abstract
An efficient and odourless procedure for a one-pot synthesis of thioesters by the reaction of benzoic anhydrides, thiourea and various organic halides (primary, allylic, and benzylic) or structurally diverse, electron-deficient alkenes (ketones, esters, and nitriles) in the presence of Et3N has been developed. In this method, thiobenzoic acids were in situ generated from the reaction of thiourea with benzoic anhydrides, which were subjected to conjugate addition with electron-deficient alkenes or a nucleophilic displacement reaction with alkyl halides.

Graphical Abstract
Introduction
Thioesters have many uses in organic synthesis as intermediates, mild acyl transfer agents and thiol sources [1-19]. Thioesters can be synthesized in the laboratory by means of different methods using diverse reagents and substrates [20-26]. The reaction of active carboxylic acid derivatives with thiols [27-41] and the coupling of thiols with carboxylic acids using activating agents [42-45] have been mainly used for the synthesis of thioesters in organic synthesis. Thiol-based reactions have a foul smell, making them unpleasant. Thioesters can also be prepared through the conjugate addition and nucleophilic displacement reactions using thioacid nucleophiles. Nevertheless, the reactions of thioacids have not been widely studied because they are not sufficiently available. On the other hand, the usual methods for preparing thioacids involve the action of toxic and unpleasant smelling, gaseous hydrogen sulfide on carboxylic acid derivatives [46,47]. Also, thioacids as thiols have a strong and repulsive smell. The in situ generation of thioacids using odourless, easy to handle and inoffensive substrates is an appropriate solution to these problems. In this regard, the synthesis of S-functionalized thioesters using thioaroylate ions in situ generated from acyloxyphosphonium salts and tetrathiomolybdate has been reported [48-50].
Results and Discussion
The preparation of alkane thiols using thiourea and alkyl halides is a well-known reaction in organic synthesis. In addition, two methods for the synthesis of thioacids from acyl chlorides using N,N-dimethylthioformamide [51] or thioacetamide [52] have been reported. As far as we know, a similar reaction using thiourea and carboxylic acid derivatives to prepare thioacids has not yet been introduced. Hence, this work is focused on the development of new reaction sequences for the synthesis of thioacid derivatives via in situ generation of thioacids using thiourea as an inexpensive and easy to handle sulfur surrogate.
Recently, a one-pot procedure for the preparation of thioesters via the reaction of in situ generated thiols (as suggested by other authors) with benzoyl chlorides was reported (Scheme 1, path a) [53]. In this protocol, an alkyl halide was treated with thiourea and a benzoyl chloride in an aqueous Triton X-100 micelle [53]. Alternatively, the thioester could be synthesized from the reaction of the alkyl halide with the thioacid in situ generated from the reaction of thiourea and benzoyl chlorides (Scheme 1, path b).
![[1860-5397-11-141-i1]](/bjoc/content/inline/1860-5397-11-141-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-pot procedure for the preparation of thioesters.
Scheme 1: One-pot procedure for the preparation of thioesters.
In order to explore this possibility, the alkyl halide was removed from the procedure and butyl acrylate was treated with benzoyl chloride and thiourea (Scheme 2). Thiobenzoic acid or its corresponding thia-Michael adduct were not formed after 24 h. The butyl acrylate was intact and the starting benzoyl chloride was mainly converted to potassium benzoate.
![[1860-5397-11-141-i2]](/bjoc/content/inline/1860-5397-11-141-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of benzoyl chloride, thiourea and butyl acrylate.
Scheme 2: Reaction of benzoyl chloride, thiourea and butyl acrylate.
This result confirms that the thioester product cannot be produced through generation of the thioacid from thiourea and benzoyl chloride.
To establish a protocol for the one-pot synthesis of thioacid derivatives via in situ generation of thioacids, considerable preliminary tests were accomplished using thiourea and carboxylic acid derivatives such as esters, acyl halides and anhydrides under various conditions. First, acetyl and benzoyl chlorides were separately treated with an equivalent amount of thiourea by using various solvents and temperatures. The results of these experiments were not satisfactory and after work-up with basic aqueous solutions, a complex mixture of unidentified products was obtained in all experiments. The similar experiments with ethyl acetate and ethyl benzoate did not lead to the formation of any product and the substrates were intact under all experimental conditions tested. Next, a possible reaction between thiourea and carboxylic anhydrides was investigated. As a model reaction, a mixture of well-powdered benzoic anhydride (1 mmol) and thiourea (1.1 mmol) in Et3N (0.5 mL) was stirred at 40 °C. The starting benzoic anhydride was gradually consumed within 0.5 h and a two-phase system containing Et3N and a thick brick-red liquid was formed. Next, H2O (1 mL) and H2O2 (1.2 mmol) were added to the reaction mixture and the resulting solution was stirred for 1 h at room temperature. Extractive work-up with EtOAc followed by silica gel column chromatography afforded benzoyl disulfide in 91% yield.
We then focused our attention on developing a practical method to prepare thia-Michael adducts of thioacids. In this regard, a mixture of benzoic anhydride and thiourea in Et3N was stirred at 40 °C for 0.5 h. Then, water and n-butyl acrylate were added to the mixture and stirring was continued for another 1 h at room temperature. The best results were obtained using 1.3 equiv of thiourea and 1.2 equiv of benzoic anhydride per 1 equiv of butyl acrylate. The desired thia-Michael product was obtained in 86% yield (based on the starting butyl acrylate) and 72% (based on the starting benzoic anhydride) after extractive work-up with EtOAc followed by silica gel column chromatography.
The optimized reaction conditions were then applied to further reactions using structurally diverse benzoic anhydrides and different electron-deficient alkenes. The results are summarized in Table 1.
Table 1: One-pot synthesis of thioesters using benzoic anhydrides, thiourea and electron-deficient alkenesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-141-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R1 | EWG | t (h) | Product | Isolated yield (%)b |
|---|---|---|---|---|---|
| 1 | H | COCH3 | 0.5 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-141-i5.svg?max-width=637&scale=1.0)
1 [48] |
80 |
| 2 | H | CN | 0.5 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-141-i6.svg?max-width=637&scale=1.0)
2 [20] |
88 |
| 3 | H | CO2Et | 0.5 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-141-i7.svg?max-width=637&scale=1.0)
3 |
90 |
| 4 | H | CO2n-Bu | 0.5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-141-i8.svg?max-width=637&scale=1.0)
4 |
86 |
| 5 | CH3 | COCH3 | 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-141-i9.svg?max-width=637&scale=1.0)
5 |
80 |
| 6 | CH3 | CN | 5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-141-i10.svg?max-width=637&scale=1.0)
6 |
84 |
| 7 | CH3 | CO2Et | 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-141-i11.svg?max-width=637&scale=1.0)
7 |
88 |
| 8 | CH3 | CO2n-Bu | 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-141-i12.svg?max-width=637&scale=1.0)
8 |
90 |
| 9 | Cl | COCH3 | 2 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-141-i13.svg?max-width=637&scale=1.0)
9 |
83 |
| 10 | Cl | CN | 2 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-141-i14.svg?max-width=637&scale=1.0)
10 |
85 |
| 11 | Cl | CO2Et | 2 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-141-i15.svg?max-width=637&scale=1.0)
11 |
88 |
| 12 | Cl | CO2n-Bu | 2 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-141-i16.svg?max-width=637&scale=1.0)
12 |
88 |
| 13c | CH3O | COCH3 | 2 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-141-i17.svg?max-width=637&scale=1.0)
13 |
83 |
| 14c | CH3O | CN | 2 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-141-i18.svg?max-width=637&scale=1.0)
14 |
86 |
| 15c | CH3O | CO2Et | 2 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-141-i19.svg?max-width=637&scale=1.0)
15 |
88 |
| 16c | CH3O | CO2n-Bu | 2 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-141-i20.svg?max-width=637&scale=1.0)
16 |
90 |
aFirst-step reaction conditions: anhydride (1.2 mmol), thiourea (1.3 mmol), Et3N (0.5 mL), 40 °C; Second-step reaction conditions: Micheal acceptor (1 mmol), H2O (1 mL), rt. bThe yields were calculated based on Michael acceptors as limiting reagents. To achieve yields based on the starting anhydrides or thiourea, they should be divided by 1.2 and 1.3, respectively. cThe first step reaction was conducted at 80 °C.
It can be seen that benzoic anhydrides have readily produced the corresponding thioesters through the in situ generation of thioacids. The reaction of aliphatic anhydrides such as acetic, phenylacetic and hexanoic anhydrides with thiourea under the reaction conditions did not lead to the desired thioacids after 24 h. In another project, the optimized reaction conditions were applied to further reactions using structurally diverse benzoic anhydrides and different alkyl halides. The results are shown in Table 2.
Table 2: One-pot synthesis of thioesters using benzoic anhydrides, thiourea and alkyl halidesa.
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-141-i21.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R1 | R2X | t (h) | Product | Isolated yield (%)b |
|---|---|---|---|---|---|
| 1 | H | n-C10H21I | 0.5 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-141-i22.svg?max-width=637&scale=1.0)
17 [53] |
85 |
| 2 | H |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-141-i23.svg?max-width=637&scale=1.0)
|
0.5 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-141-i24.svg?max-width=637&scale=1.0)
18 |
90 |
| 3 | H |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-141-i25.svg?max-width=637&scale=1.0)
|
0.5 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-141-i26.svg?max-width=637&scale=1.0)
19 [54] |
81 |
| 4 | H | CH3I | 0.5 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-141-i27.svg?max-width=637&scale=1.0)
20 |
92 |
| 5 | H | n-C8H17Br | 0.5 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-141-i28.svg?max-width=637&scale=1.0)
21 [53] |
87 |
| 6 | H |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-141-i29.svg?max-width=637&scale=1.0)
|
0.5 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-141-i30.svg?max-width=637&scale=1.0)
22 [55] |
90 |
| 7 | CH3 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-141-i31.svg?max-width=637&scale=1.0)
|
5 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-141-i32.svg?max-width=637&scale=1.0)
23 [56] |
91 |
| 8 | CH3 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-141-i33.svg?max-width=637&scale=1.0)
|
5 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-141-i34.svg?max-width=637&scale=1.0)
24 |
80 |
| 9 | CH3 | n-C8H17Br | 5 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-141-i35.svg?max-width=637&scale=1.0)
25 |
84 |
| 10 | CH3 | CH3I | 5 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-141-i36.svg?max-width=637&scale=1.0)
26 [20] |
90 |
| 11 | CH3 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-11-141-i37.svg?max-width=637&scale=1.0)
|
5 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-11-141-i38.svg?max-width=637&scale=1.0)
27 |
90 |
| 12 | Cl | n-C10H21I | 2 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-11-141-i39.svg?max-width=637&scale=1.0)
28 [57] |
86 |
| 13 | Cl | CH3I | 2 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-11-141-i40.svg?max-width=637&scale=1.0)
29 [20] |
91 |
| 14 | Cl |
![[Graphic 38]](/bjoc/content/inline/1860-5397-11-141-i41.svg?max-width=637&scale=1.0)
|
2 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-11-141-i42.svg?max-width=637&scale=1.0)
30 |
87 |
| 15 | Cl |
![[Graphic 40]](/bjoc/content/inline/1860-5397-11-141-i43.svg?max-width=637&scale=1.0)
|
2 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-11-141-i44.svg?max-width=637&scale=1.0)
31 |
84 |
| 16c | CH3O |
![[Graphic 42]](/bjoc/content/inline/1860-5397-11-141-i45.svg?max-width=637&scale=1.0)
|
2 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-11-141-i46.svg?max-width=637&scale=1.0)
32 |
88 |
| 17c | CH3O |
![[Graphic 44]](/bjoc/content/inline/1860-5397-11-141-i47.svg?max-width=637&scale=1.0)
|
2 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-11-141-i48.svg?max-width=637&scale=1.0)
33 |
81 |
| 18c | CH3O |
![[Graphic 46]](/bjoc/content/inline/1860-5397-11-141-i49.svg?max-width=637&scale=1.0)
|
2 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-11-141-i50.svg?max-width=637&scale=1.0)
34 |
87 |
| 19c | CH3O | n-C8H17Br | 2 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-11-141-i51.svg?max-width=637&scale=1.0)
35 [58] |
88 |
| 20c | CH3O | CH3I | 2 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-11-141-i52.svg?max-width=637&scale=1.0)
36 |
91 |
aFirst-step reaction conditions: anhydride (1.2 mmol), thiourea (1.3 mmol), Et3N (0.5 mL), 40 °C; Second-step reaction conditions: R2X (1 mmol), H2O (1 mL), rt. bThe yields have been calculated based on alkyl halides as limiting reagents. To achieve yields based on the starting anhydrides or thiourea, they should be divided by 1.2 and 1.3, respectively. cThe first step reaction was conducted at 80 °C.
As evident from the results presented in Table 2, by this method, primary, benzylic, allylic and propargylic halides have been easily converted to the related thioesters within appropriate reaction times in good to excellent yields (Table 2, entries 1–20).
A general pathway for the reaction has been proposed as presented in Scheme 3. Similar to reactions of acyl chlorides with N,N-dimethylthioformamide [51] or thioacetamide [52], a benzoic anhydride undergoes a nucleophilic acyl substitution reaction with thiourea to produce the corresponding S-benzoylisothiouronium salt as an intermediate. After addition of H2O to the reaction mixture, the salt is hydrolyzed by the hydroxide anion to generate the thiobenzoate anion. Finally, the thiobenzoate reacts with a Michael acceptor or an alkyl halide to give the thioester product.
![[1860-5397-11-141-i3]](/bjoc/content/inline/1860-5397-11-141-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, an efficient, versatile, and odourless protocol for a one-pot preparation of thioesters from non-thiolic precursors under mild conditions has been developed. In this protocol, a mixture of a benzoic anhydride, thiourea and an alkyl halide (primary, allylic or benzylic), or a conjugated olefin (ketones, esters, nitriles), Et3N and H2O produced the related thioesters in good to excellent yields.
Experimental
S-3-Oxobutyl benzothioate (1)
Colorless oil; 1H NMR (250 MHz, CDCl3) δ 7.95–7.91 (m, 2H), 7.58–7.52 (m, 1H), 7.45–7.39 (m, 2H), 3.24 (t, J = 6.7 Hz, 2H), 2.86 (t, J = 6.7 Hz, 2H), 2.16 (s, 3H); 13C NMR (62.5 MHz, CDCl3) δ 206.5, 191.9, 136.8, 133.5, 128.6, 127.2, 43.4, 29.9, 22.7; IR (neat) ν (cm−1): 1717 (C=O ketone), 1655 (C=O thioester); anal. calcd for (C11H12O2S): C, 63.43; H, 5.81; S, 15.40; found: C, 63.51; H, 5.77; S, 15.42.
Butyl 3-(benzoylthio)propanoate (4)
Colorless oil; 1H NMR (250 MHz, CDCl3) δ 7.95–7.91 (m, 2H), 7.54–7.51 (m, 1H), 7.44–7.38 (m, 2H), 4.09 (t, J = 6.6 Hz, 2H), 3.30 (t, J = 7.0 Hz, 2H), 2.71 (t, J = 7.0 Hz, 2H), 1.65–1.54 (m, 2H), 1.43–1.31 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (62.5 MHz, CDCl3) δ 191.5, 171.8, 136.8, 133.5, 128.6, 127.2, 64.7, 34.5, 30.6, 24.1, 19.1, 13.7; IR (neat) ν (cm−1): 1732 (C=O ester), 1666 (C=O thioester); anal. calcd for (C14H18O3S): C, 63.13; H, 6.81; S, 12.04; found: C, 63.19; H, 6.85; S, 11.98.
S-Decyl benzothioate (17)
1H NMR (250 MHz, CDCl3) δ 7.91–7.86 (m, 2H), 7.51–7.33 (m, 3H), 2.99 (t, J = 7.3 Hz, 2H), 1.65–1.54 (m, 2H), 1.38–1.19 (m, 14H), 0.83–0.78 (m, 3H); 13C NMR (62.5 MHz, CDCl3) δ 192.0, 137.3, 133.1, 128.9, 127.4, 31.9, 29.6, 29.6, 29.5, 29.3, 29.2, 29.0, 29.0, 22.7, 14.1; IR (neat) ν (cm−1): 1666 (C=O thioester); anal. calcd for (C17H26OS): C, 73.33; H, 9.41; S, 11.52; found: C, 73.31; H, 9.48; S, 11.46.
S-Prop-2-yn-1-yl benzothioate (19)
1H NMR (250 MHz, CDCl3) δ 7.88–7.84 (m, 2H), 7.53–7.46 (m, 1H), 7.40–7.33 (m, 2H), 3.75 (d, J = 2.7 Hz, 2H), 2.15 (t, J = 2.7 Hz, 1H); 13C NMR (62.5 MHz, CDCl3) δ 190.1, 136.2, 133.8, 128.8, 127.3, 78.9, 71.1, 17.5; IR (neat) ν (cm−1): 3294, 2125, 1670; anal. calcd for (C10H8OS): C, 68.15; H, 4.58; S, 18.19; found: C, 68.20; H, 4.55; S, 18.26.
S-(2-Methylallyl) 4-methylbenzothioate (27)
1H NMR (250 MHz, CDCl3) δ 7.79 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H), 4.95 (s, 1H), 4.80–4.79 (m, 1H), 3.62 (s, 2H), 2.31 (s, 3H), 1.73 (s, 3H); 13C NMR (62.5 MHz, CDCl3) δ 191.0, 144.2, 140.9, 134.4, 129.3, 127.3, 114.2, 35.7, 21.7, 21.3; IR (neat) ν (cm−1): 1659; anal. calcd for (C12H14OS): C, 69.86; H, 6.84; S, 15.54; found: C, 69.80; H, 6.88; S, 15.61.
References
-
Cremlyn, R. J. An Introduction to Organosulfur Chemistry; John Wiley & Sons: Chichester, 1996.
Return to citation in text: [1] -
Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; John Wiley & Sons: New York, 1999.
Return to citation in text: [1] -
Abbasi, M. Tetrahedron Lett. 2012, 53, 2608–2610. doi:10.1016/j.tetlet.2012.03.045
Return to citation in text: [1] -
Abbasi, M. Tetrahedron Lett. 2012, 53, 3683–3685. doi:10.1016/j.tetlet.2012.05.042
Return to citation in text: [1] -
Villalobos, J. M.; Srogl, J.; Liebeskind, L. S. J. Am. Chem. Soc. 2007, 129, 15734–15735. doi:10.1021/ja074931n
Return to citation in text: [1] -
Prokopcová, H.; Pisani, L.; Kappe, C. O. Synlett 2007, 43–46. doi:10.1055/s-2006-958443
Return to citation in text: [1] -
Morita, A.; Kuwahara, S. Org. Lett. 2006, 8, 1613–1616. doi:10.1021/ol053122a
Return to citation in text: [1] -
Lengar, A.; Kappe, C. O. Org. Lett. 2004, 6, 771–774. doi:10.1021/ol036496h
Return to citation in text: [1] -
Camarero, J. A.; Hackel, B. J.; de Yoreo, J. J.; Mitchell, A. R. J. Org. Chem. 2004, 69, 4145–4151. doi:10.1021/jo040140h
Return to citation in text: [1] -
Lalic, G.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc. 2003, 125, 2852–2853. doi:10.1021/ja029452x
Return to citation in text: [1] -
Agapiou, K.; Krische, M. J. Org. Lett. 2003, 5, 1737–1740. doi:10.1021/ol030035e
Return to citation in text: [1] -
Ingenito, R.; Wenschuh, H. Org. Lett. 2003, 5, 4587–4590. doi:10.1021/ol035742m
Return to citation in text: [1] -
Aggarwal, V. K.; Esquivel-Zamora, B. N. J. Org. Chem. 2002, 67, 8618–8621. doi:10.1021/jo026410i
Return to citation in text: [1] -
Boeckman, R. K., Jr.; Clark, T. J.; Shook, B. C. Org. Lett. 2002, 4, 2109–2112. doi:10.1021/ol026101e
Return to citation in text: [1] -
Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. J. Am. Chem. Soc. 2002, 124, 6552–6554. doi:10.1021/ja026216d
Return to citation in text: [1] -
Alphonse, F.-A.; Suzenet, F.; Keromnes, A.; Lebert, B.; Guillaumet, G. Synlett 2002, 447–450. doi:10.1055/s-2002-20462
Return to citation in text: [1] -
Longbottom, D. A.; Morrison, A. J.; Dixon, D. J.; Ley, S. V. Angew. Chem., Int. Ed. 2002, 41, 2786–2790. doi:10.1002/1521-3773(20020802)41:15<2786::AID-ANIE2786>3.0.CO;2-Z
Return to citation in text: [1] -
Bu, X.; Wu, X.; Xie, G.; Guo, Z. Org. Lett. 2002, 4, 2893–2895. doi:10.1021/ol0263191
Return to citation in text: [1] -
Evans, D. A.; Rajapakse, H. A.; Chiu, A.; Stenkamp, D. Angew. Chem., Int. Ed. 2002, 41, 4573–4576. doi:10.1002/1521-3773(20021202)41:23<4573::AID-ANIE4573>3.0.CO;2-S
Return to citation in text: [1] -
Huang, Y.-T.; Lu, S.-Y.; Yi, C.-L.; Lee, C.-F. J. Org. Chem. 2014, 79, 4561–4568. doi:10.1021/jo500574p
Return to citation in text: [1] [2] [3] [4] -
van Zijl, A. W.; Minnaard, A. J.; Feringa, B. L. J. Org. Chem. 2008, 73, 5651–5653. doi:10.1021/jo800879e
Return to citation in text: [1] -
Wang, L.; He, W.; Yu, Z. Chem. Soc. Rev. 2013, 42, 599–621. doi:10.1039/C2CS35323G
Return to citation in text: [1] -
Nambu, H.; Hata, K.; Matsugi, M.; Kita, Y. Chem. Commun. 2002, 1082–1083. doi:10.1039/b202129c
Return to citation in text: [1] -
Zeng, J.-W.; Liu, Y.-C.; Hsieh, P.-A.; Huang, Y.-T.; Yi, C.-L.; Badsara, S. S.; Lee, C.-F. Green Chem. 2014, 16, 2644–2652. doi:10.1039/c4gc00025k
Return to citation in text: [1] -
Yi, C.-L.; Huang, Y.-T.; Lee, C.-F. Green Chem. 2013, 15, 2476–2484. doi:10.1039/c3gc40946e
Return to citation in text: [1] -
Abbasi, M. Synth. Commun. 2013, 43, 1759–1765. doi:10.1080/00397911.2012.667183
Return to citation in text: [1] -
Basu, B.; Paul, S.; Nanda, A. K. Green Chem. 2010, 12, 767–771. doi:10.1039/b925620b
Return to citation in text: [1] -
Zieba, A.; Suwinska, K. Heterocycles 2008, 75, 2649–2657. doi:10.3987/COM-08-11421
Return to citation in text: [1] -
Petersson, M. J.; Marchal, C.; Loughlin, W. A.; Jenkins, I. D.; Healy, P. C.; Almesaker, A. Tetrahedron 2007, 63, 1395–1401. doi:10.1016/j.tet.2006.11.090
Return to citation in text: [1] -
Chakraborti, A. K.; Gulhane, R. Tetrahedron Lett. 2003, 44, 6749–6753. doi:10.1016/S0040-4039(03)01641-1
Return to citation in text: [1] -
Kumar, P.; Pandey, R. K.; Bodas, M. S.; Dagade, S. P.; Dongare, M. K.; Ramaswamy, A. V. J. Mol. Catal. A: Chem. 2002, 181, 207–213. doi:10.1016/S1381-1169(01)00365-X
Return to citation in text: [1] -
Nakae, Y.; Kusaki, I.; Sato, T. Synlett 2001, 1584–1586. doi:10.1055/s-2001-17483
Return to citation in text: [1] -
Orita, A.; Tanahashi, C.; Kakuda, A.; Otera, J. J. Org. Chem. 2001, 66, 8926–8934. doi:10.1021/jo0107453
Return to citation in text: [1] -
Vedejs, E.; MacKay, J. A. Org. Lett. 2001, 3, 535–536. doi:10.1021/ol006923g
Return to citation in text: [1] -
Sarvanan, P.; Singh, V. K. Tetrahedron Lett. 1999, 40, 2611–2614. doi:10.1016/S0040-4039(99)00229-4
Return to citation in text: [1] -
Procopiou, P. A.; Baugh, S. P. D.; Flack, S. S.; Inglis, G. G. A. J. Org. Chem. 1998, 63, 2342–2347. doi:10.1021/jo980011z
Return to citation in text: [1] -
Ishihara, K.; Kubota, M.; Kurihara, H.; Yamamoto, H. J. Org. Chem. 1996, 61, 4560–4567. doi:10.1021/jo952237x
Return to citation in text: [1] -
Ishihara, K.; Kobuta, M.; Yamamoto, H. Synlett 1996, 265–266. doi:10.1055/s-1996-5376
Return to citation in text: [1] -
Vedejs, E.; Bennet, N. S.; Conn, L. M.; Diver, S. T.; Gingras, M.; Lin, S.; Oliver, P. M.; Peterson, M. J. J. Org. Chem. 1993, 58, 7286–7288. doi:10.1021/jo00077a064
Return to citation in text: [1] -
Iqbal, J.; Srivastava, R. R. J. Org. Chem. 1992, 57, 2001–2007. doi:10.1021/jo00033a020
Return to citation in text: [1] -
McGarvey, G. J.; Williams, J. M.; Hiner, R. N.; Matsubara, Y.; Oh, T. J. Am. Chem. Soc. 1986, 108, 4943–4952. doi:10.1021/ja00276a040
Return to citation in text: [1] -
Iranpoor, N.; Firouzabadi, H.; Khalili, D.; Motevalli, S. J. Org. Chem. 2008, 73, 4882–4887. doi:10.1021/jo8000782
Return to citation in text: [1] -
Katritzky, A. R.; Shestopalov, A. A.; Suzuki, K. Synthesis 2004, 1806–1813. doi:10.1055/s-2004-829126
Return to citation in text: [1] -
Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. doi:10.1002/anie.197805221
Return to citation in text: [1] -
Liu, H.-J.; Sabesan, S. I. Can. J. Chem. 1980, 58, 2645–2648. doi:10.1139/v80-423
Return to citation in text: [1] -
Loeliger, P.; Fliickiger, E. Org. Synth. 1976, 55, 127–133. doi:10.15227/orgsyn.055.0127
Return to citation in text: [1] -
Shin, H.-C.; Quinn, D. M. Lipids 1993, 28, 73–74. doi:10.1007/BF02536365
Return to citation in text: [1] -
Gopinath, P.; Debasree, C.; Vidyarini, R. S.; Chandrasekaran, S. Tetrahedron 2010, 66, 7001–7011. doi:10.1016/j.tet.2010.06.028
Return to citation in text: [1] [2] -
Gopinath, P.; Vidyarini, R. S.; Chandrasekaran, S. Eur. J. Org. Chem. 2009, 6043–6047. doi:10.1002/ejoc.200900956
Return to citation in text: [1] -
Gopinath, P.; Vidyarini, R. S.; Chandrasekaran, S. J. Org. Chem. 2009, 74, 6291–6294. doi:10.1021/jo9009694
Return to citation in text: [1] -
Kobayashi, Y.; Itabashi, K. Synthesis 1985, 671–672. doi:10.1055/s-1985-31303
Return to citation in text: [1] [2] -
Toriyama, M.; Kamijo, H.; Motohashi, S.; Takido, T.; Itabashi, K. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 1661–1665. doi:10.1080/10426500307837
Return to citation in text: [1] [2] -
Lu, G.-P.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059
Return to citation in text: [1] [2] [3] [4] -
Cao, H.; McNamee, L.; Alper, H. J. Org. Chem. 2008, 73, 3530–3534. doi:10.1021/jo800287s
Return to citation in text: [1] -
Uno, T.; Inokuma, T.; Takemoto, Y. Chem. Commun. 2012, 48, 1901–1903. doi:10.1039/c2cc17183j
Return to citation in text: [1] -
Olah, G. A.; Bruce, M. R.; Clouet, F. L. J. Org. Chem. 1981, 46, 438–442. doi:10.1021/jo00315a040
Return to citation in text: [1] -
Burhardt, M. N.; Taaning, R. H.; Skrydstrup, T. Org. Lett. 2013, 15, 948–951. doi:10.1021/ol400138m
Return to citation in text: [1] -
Okuyama, T.; Kitano, M.; Fueno, T. J. Org. Chem. 1987, 52, 2657–2661. doi:10.1021/jo00389a005
Return to citation in text: [1]
| 57. | Burhardt, M. N.; Taaning, R. H.; Skrydstrup, T. Org. Lett. 2013, 15, 948–951. doi:10.1021/ol400138m |
| 56. | Olah, G. A.; Bruce, M. R.; Clouet, F. L. J. Org. Chem. 1981, 46, 438–442. doi:10.1021/jo00315a040 |
| 20. | Huang, Y.-T.; Lu, S.-Y.; Yi, C.-L.; Lee, C.-F. J. Org. Chem. 2014, 79, 4561–4568. doi:10.1021/jo500574p |
| 1. | Cremlyn, R. J. An Introduction to Organosulfur Chemistry; John Wiley & Sons: Chichester, 1996. |
| 2. | Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; John Wiley & Sons: New York, 1999. |
| 3. | Abbasi, M. Tetrahedron Lett. 2012, 53, 2608–2610. doi:10.1016/j.tetlet.2012.03.045 |
| 4. | Abbasi, M. Tetrahedron Lett. 2012, 53, 3683–3685. doi:10.1016/j.tetlet.2012.05.042 |
| 5. | Villalobos, J. M.; Srogl, J.; Liebeskind, L. S. J. Am. Chem. Soc. 2007, 129, 15734–15735. doi:10.1021/ja074931n |
| 6. | Prokopcová, H.; Pisani, L.; Kappe, C. O. Synlett 2007, 43–46. doi:10.1055/s-2006-958443 |
| 7. | Morita, A.; Kuwahara, S. Org. Lett. 2006, 8, 1613–1616. doi:10.1021/ol053122a |
| 8. | Lengar, A.; Kappe, C. O. Org. Lett. 2004, 6, 771–774. doi:10.1021/ol036496h |
| 9. | Camarero, J. A.; Hackel, B. J.; de Yoreo, J. J.; Mitchell, A. R. J. Org. Chem. 2004, 69, 4145–4151. doi:10.1021/jo040140h |
| 10. | Lalic, G.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc. 2003, 125, 2852–2853. doi:10.1021/ja029452x |
| 11. | Agapiou, K.; Krische, M. J. Org. Lett. 2003, 5, 1737–1740. doi:10.1021/ol030035e |
| 12. | Ingenito, R.; Wenschuh, H. Org. Lett. 2003, 5, 4587–4590. doi:10.1021/ol035742m |
| 13. | Aggarwal, V. K.; Esquivel-Zamora, B. N. J. Org. Chem. 2002, 67, 8618–8621. doi:10.1021/jo026410i |
| 14. | Boeckman, R. K., Jr.; Clark, T. J.; Shook, B. C. Org. Lett. 2002, 4, 2109–2112. doi:10.1021/ol026101e |
| 15. | Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. J. Am. Chem. Soc. 2002, 124, 6552–6554. doi:10.1021/ja026216d |
| 16. | Alphonse, F.-A.; Suzenet, F.; Keromnes, A.; Lebert, B.; Guillaumet, G. Synlett 2002, 447–450. doi:10.1055/s-2002-20462 |
| 17. | Longbottom, D. A.; Morrison, A. J.; Dixon, D. J.; Ley, S. V. Angew. Chem., Int. Ed. 2002, 41, 2786–2790. doi:10.1002/1521-3773(20020802)41:15<2786::AID-ANIE2786>3.0.CO;2-Z |
| 18. | Bu, X.; Wu, X.; Xie, G.; Guo, Z. Org. Lett. 2002, 4, 2893–2895. doi:10.1021/ol0263191 |
| 19. | Evans, D. A.; Rajapakse, H. A.; Chiu, A.; Stenkamp, D. Angew. Chem., Int. Ed. 2002, 41, 4573–4576. doi:10.1002/1521-3773(20021202)41:23<4573::AID-ANIE4573>3.0.CO;2-S |
| 46. | Loeliger, P.; Fliickiger, E. Org. Synth. 1976, 55, 127–133. doi:10.15227/orgsyn.055.0127 |
| 47. | Shin, H.-C.; Quinn, D. M. Lipids 1993, 28, 73–74. doi:10.1007/BF02536365 |
| 53. | Lu, G.-P.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059 |
| 42. | Iranpoor, N.; Firouzabadi, H.; Khalili, D.; Motevalli, S. J. Org. Chem. 2008, 73, 4882–4887. doi:10.1021/jo8000782 |
| 43. | Katritzky, A. R.; Shestopalov, A. A.; Suzuki, K. Synthesis 2004, 1806–1813. doi:10.1055/s-2004-829126 |
| 44. | Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. doi:10.1002/anie.197805221 |
| 45. | Liu, H.-J.; Sabesan, S. I. Can. J. Chem. 1980, 58, 2645–2648. doi:10.1139/v80-423 |
| 55. | Uno, T.; Inokuma, T.; Takemoto, Y. Chem. Commun. 2012, 48, 1901–1903. doi:10.1039/c2cc17183j |
| 27. | Basu, B.; Paul, S.; Nanda, A. K. Green Chem. 2010, 12, 767–771. doi:10.1039/b925620b |
| 28. | Zieba, A.; Suwinska, K. Heterocycles 2008, 75, 2649–2657. doi:10.3987/COM-08-11421 |
| 29. | Petersson, M. J.; Marchal, C.; Loughlin, W. A.; Jenkins, I. D.; Healy, P. C.; Almesaker, A. Tetrahedron 2007, 63, 1395–1401. doi:10.1016/j.tet.2006.11.090 |
| 30. | Chakraborti, A. K.; Gulhane, R. Tetrahedron Lett. 2003, 44, 6749–6753. doi:10.1016/S0040-4039(03)01641-1 |
| 31. | Kumar, P.; Pandey, R. K.; Bodas, M. S.; Dagade, S. P.; Dongare, M. K.; Ramaswamy, A. V. J. Mol. Catal. A: Chem. 2002, 181, 207–213. doi:10.1016/S1381-1169(01)00365-X |
| 32. | Nakae, Y.; Kusaki, I.; Sato, T. Synlett 2001, 1584–1586. doi:10.1055/s-2001-17483 |
| 33. | Orita, A.; Tanahashi, C.; Kakuda, A.; Otera, J. J. Org. Chem. 2001, 66, 8926–8934. doi:10.1021/jo0107453 |
| 34. | Vedejs, E.; MacKay, J. A. Org. Lett. 2001, 3, 535–536. doi:10.1021/ol006923g |
| 35. | Sarvanan, P.; Singh, V. K. Tetrahedron Lett. 1999, 40, 2611–2614. doi:10.1016/S0040-4039(99)00229-4 |
| 36. | Procopiou, P. A.; Baugh, S. P. D.; Flack, S. S.; Inglis, G. G. A. J. Org. Chem. 1998, 63, 2342–2347. doi:10.1021/jo980011z |
| 37. | Ishihara, K.; Kubota, M.; Kurihara, H.; Yamamoto, H. J. Org. Chem. 1996, 61, 4560–4567. doi:10.1021/jo952237x |
| 38. | Ishihara, K.; Kobuta, M.; Yamamoto, H. Synlett 1996, 265–266. doi:10.1055/s-1996-5376 |
| 39. | Vedejs, E.; Bennet, N. S.; Conn, L. M.; Diver, S. T.; Gingras, M.; Lin, S.; Oliver, P. M.; Peterson, M. J. J. Org. Chem. 1993, 58, 7286–7288. doi:10.1021/jo00077a064 |
| 40. | Iqbal, J.; Srivastava, R. R. J. Org. Chem. 1992, 57, 2001–2007. doi:10.1021/jo00033a020 |
| 41. | McGarvey, G. J.; Williams, J. M.; Hiner, R. N.; Matsubara, Y.; Oh, T. J. Am. Chem. Soc. 1986, 108, 4943–4952. doi:10.1021/ja00276a040 |
| 53. | Lu, G.-P.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059 |
| 20. | Huang, Y.-T.; Lu, S.-Y.; Yi, C.-L.; Lee, C.-F. J. Org. Chem. 2014, 79, 4561–4568. doi:10.1021/jo500574p |
| 21. | van Zijl, A. W.; Minnaard, A. J.; Feringa, B. L. J. Org. Chem. 2008, 73, 5651–5653. doi:10.1021/jo800879e |
| 22. | Wang, L.; He, W.; Yu, Z. Chem. Soc. Rev. 2013, 42, 599–621. doi:10.1039/C2CS35323G |
| 23. | Nambu, H.; Hata, K.; Matsugi, M.; Kita, Y. Chem. Commun. 2002, 1082–1083. doi:10.1039/b202129c |
| 24. | Zeng, J.-W.; Liu, Y.-C.; Hsieh, P.-A.; Huang, Y.-T.; Yi, C.-L.; Badsara, S. S.; Lee, C.-F. Green Chem. 2014, 16, 2644–2652. doi:10.1039/c4gc00025k |
| 25. | Yi, C.-L.; Huang, Y.-T.; Lee, C.-F. Green Chem. 2013, 15, 2476–2484. doi:10.1039/c3gc40946e |
| 26. | Abbasi, M. Synth. Commun. 2013, 43, 1759–1765. doi:10.1080/00397911.2012.667183 |
| 54. | Cao, H.; McNamee, L.; Alper, H. J. Org. Chem. 2008, 73, 3530–3534. doi:10.1021/jo800287s |
| 53. | Lu, G.-P.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059 |
| 48. | Gopinath, P.; Debasree, C.; Vidyarini, R. S.; Chandrasekaran, S. Tetrahedron 2010, 66, 7001–7011. doi:10.1016/j.tet.2010.06.028 |
| 51. | Kobayashi, Y.; Itabashi, K. Synthesis 1985, 671–672. doi:10.1055/s-1985-31303 |
| 52. | Toriyama, M.; Kamijo, H.; Motohashi, S.; Takido, T.; Itabashi, K. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 1661–1665. doi:10.1080/10426500307837 |
| 20. | Huang, Y.-T.; Lu, S.-Y.; Yi, C.-L.; Lee, C.-F. J. Org. Chem. 2014, 79, 4561–4568. doi:10.1021/jo500574p |
| 52. | Toriyama, M.; Kamijo, H.; Motohashi, S.; Takido, T.; Itabashi, K. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 1661–1665. doi:10.1080/10426500307837 |
| 51. | Kobayashi, Y.; Itabashi, K. Synthesis 1985, 671–672. doi:10.1055/s-1985-31303 |
| 20. | Huang, Y.-T.; Lu, S.-Y.; Yi, C.-L.; Lee, C.-F. J. Org. Chem. 2014, 79, 4561–4568. doi:10.1021/jo500574p |
| 48. | Gopinath, P.; Debasree, C.; Vidyarini, R. S.; Chandrasekaran, S. Tetrahedron 2010, 66, 7001–7011. doi:10.1016/j.tet.2010.06.028 |
| 49. | Gopinath, P.; Vidyarini, R. S.; Chandrasekaran, S. Eur. J. Org. Chem. 2009, 6043–6047. doi:10.1002/ejoc.200900956 |
| 50. | Gopinath, P.; Vidyarini, R. S.; Chandrasekaran, S. J. Org. Chem. 2009, 74, 6291–6294. doi:10.1021/jo9009694 |
| 53. | Lu, G.-P.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059 |
| 58. | Okuyama, T.; Kitano, M.; Fueno, T. J. Org. Chem. 1987, 52, 2657–2661. doi:10.1021/jo00389a005 |
© 2015 Abbasi and Khalifeh; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)