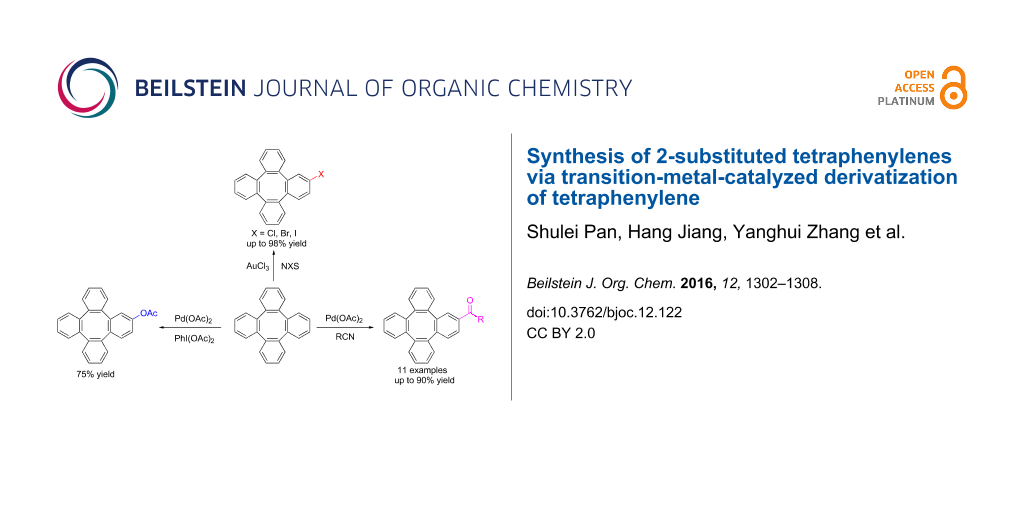
Abstract
A new strategy for the synthesis of 2-substituted tetraphenylenes through a transition-metal-catalyzed derivatization has been developed. Three types of functionalities, including OAc, X (Cl, Br, I) and carbonyl, were introduced onto tetraphenylene, which allows the easy access to a variety of monosubstituted tetraphenylenes. These reactions could accelerate research on the properties and application of tetraphenylene derivatives.

Graphical Abstract
Introduction
Tetraphenylene (1) is one of the simplest motifs in the eight-membered ring aromatic compounds (Figure 1) [1,2]. Based on its nonplanar distinct saddle-shaped structure [3,4], tetraphenylene and its derivatives have found broad applications in materials science [5-11], supramolecular chemistry [12-18], and asymmetric catalysis [19-21].
![[1860-5397-12-122-1]](/bjoc/content/figures/1860-5397-12-122-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Tetraphenylene and its saddle-shaped structure.
Figure 1: Tetraphenylene and its saddle-shaped structure.
Since Rapson and co-workers reported the first synthesis of tetraphenylene in 1943 [22], in which 2,2’-dibromobiphenyl was converted to its corresponding Grignard reagent and subsequent addition of copper(II) chloride provided 1 in 16% yield, a variety of methods for constructing the tetraphenylene skeleton have been developed [23-42]. While most of these traditional approaches suffer from harsh conditions or complicated procedures, a novel strategy via transition-metal-catalyzed C–H activation has attracted great attention and emerged as a powerful methodology for the synthesis of tetraphenylenes [43,44]. However, the methods of this strategy have a relatively limited substrate scope and are primarily applicable to the synthesis of symmetrically substituted tetraphenylenes. Among the various reactions that have been developed for the construction of the tetraphenylene skeleton, the methods for the synthesis of tetraphenylene derivatives via the direct derivatization of tetraphenylene are rare. More importantly, the direct derivatization of tetraphenylene would provide an efficient method for the synthesis of tetraphenylene derivatives, in particular for unsymmetrically substituted ones. Although direct bromination [22], nitration [22], and acetylation [45] of tetraphenylene via electrophilic aromatic substitution have been reported, it is still desirable to develop new methods for the derivatization of tetraphenylenes. Herein we report several synthetic protocols for the transition-metal-catalyzed derivatization of tetraphenylene, which provide a new method for the synthesis of 2-substituted tetraphenylenes.
Results and Discussion
The acetoxy group is an important functional group because it can be transformed into a variety of other functionalities [46,47], thus making the acetoxylation a highly interesting reaction. The Sanford and Wang group, respectively, developed a highly efficient palladium and gold-catalyzed direct acetoxylation of arenes with iodobenzene diacetate [48,49]. Based on these excellent works, we surveyed the reaction conditions for the acetoxylation of tetraphenylene (1). For this, 1 was allowed to react with PhI(OAc)2 (2a) in the presence of Pd(OAc)2/pyridine as catalysis system in a mixture of AcOH and Ac2O at 100 °C. Gratefully, the desired acetylated product 3a was formed in 52% yield (Table 1, entry 1). Prolonging the reaction time or carrying out the reaction at 120 °C led to lower yields (Table 1, entries 2 and 3). However, the yields increased when increasing amounts of PhI(OAc)2 were used (58% for 3.0 equiv and 70% for 4.0 equiv, respectively). Further increase of PhI(OAc)2 beyond 4.0 equiv failed to further improve the yield. On the other hand, the yield increased to 75% by using higher concentrations of the reactants.
Table 1: The Pd(OA)2-catalyzed acetoxylation of tetraphenylene (1).
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-122-i2.svg?max-width=637&scale=1.0)
|
||||
| Entry | PhI(OAc)2 (equiv) | Temperature, T (°C) | AcOH/Ac2O (mL) | Yielda (%) |
|---|---|---|---|---|
| 1 | 2.0 | 100 | 0.90:0.10 | 52 |
| 2 | 2.0 | 100 | 0.90:0.10 | 42b |
| 3 | 2.0 | 120 | 0.90:0.10 | 34 |
| 4 | 3.0 | 100 | 0.90:0.10 | 58 |
| 5 | 4.0 | 100 | 0.90:0.10 | 70 |
| 6 | 3.0 | 100 | 0.45:0.05 | 75 (72)c |
| 7 | 4.0 | 100 | 0.45:0.05 | 74 |
aThe yields were determined by 1H NMR analysis of the crude products using CH2Br2 as the internal standard. bReaction time 24 h. cIsolated yields based on tetraphenylene (1).
Halogen-substituted compounds are another important substance class in organic synthesis, since these substituents allow access to a variety of functionalities [50-55]. In this context especially the direct halogenation of tetraphenylene attracted our attention. The Wang group reported an efficient and mild protocol for a gold-catalyzed direct C–H halogenation of arenes with N-halosuccinimides [56,57]. Therefore, we initially investigated the chlorination of tetraphenylene by subjecting it to Wang’s conditions, and the reaction gave 3b in 28% (Table 2, entry 1). The yield decreased when the reaction was carried out at a lower or higher temperature (Table 2, entries 2 and 3). Gratefully, the yield was dramatically enhanced to 72% when 0.4 equiv of BF3·Et2O was added (Table 2, entry 4), and further to 90% when the reaction was run for 24 hours (Table 2, entry 5). Finally, the optimal 94% yield was achieved using 2.0 equiv NCS (Table 2, entry 6).
Table 2: The AuCl3-catalyzed chlorination of tetraphenylene (1).
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-122-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | NCS (equiv) | Additive (equiv) | T (°C) | t (h) | Yielda (%) |
|---|---|---|---|---|---|
| 1 | 1.0 | – | 80 | 12 | 28 |
| 2 | 1.0 | – | 100 | 12 | 26 |
| 3 | 1.0 | – | 60 | 24 | 24 |
| 4 | 1.0 | BF3·Et2O (0.4) | 80 | 12 | 72 |
| 5 | 1.0 | BF3·Et2O (0.4) | 80 | 24 | 90 |
| 6 | 2.0 | BF3·Et2O (0.4) | 80 | 24 | 94 (91)b |
aThe yields were determined by 1H NMR analysis of the crude products using CH2Br2 as the internal standard. bIsolated yields based on tetraphenylene (1).
Subsequently, the bromination of tetraphenylene (1) was examined. The reaction yielded the desired brominated product 3c under Wang’s conditions in 64% yield (Table 3, entry 1). Increasing or lowering the temperature again failed to improve the yield (Table 3, entries 2 and 3). The yield increased to 86% when 1.5 equiv NBS was used (Table 3, entry 4), and was further optimized to 98% when the reaction time was prolonged to 24 hours (Table 3, entry 5).
Table 3: The AuCl3-catalyzed bromination of tetraphenylene (1).
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-122-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | NBS (equiv) | T (°C) | t (h) | Yielda (%) |
|---|---|---|---|---|
| 1 | 1.0 | 80 | 12 | 64 |
| 2 | 1.0 | 50 | 12 | 54 |
| 3 | 1.0 | 100 | 12 | 64 |
| 4 | 1.5 | 80 | 12 | 86 |
| 5 | 1.5 | 80 | 24 | 98 (95)b |
aThe yields were determined by 1H NMR analysis of the crude products using CH2Br2 as the internal standard. bIsolated yields based on tetraphenylene (1).
Next, we surveyed the reaction conditions for the iodination of tetraphenylene with NIS (2d). Under the reaction conditions developed by the Wang group, the desired iodinated tetraphenylene 3d was obtained in 18% yield as shown in Table 4 (entry 1). When the reaction was performed at 60 °C, the yield of 3d increased to 32% (Table 4, entry 2). However, further enhancing the temperature failed to give a higher yield (Table 4, entry 3). The addition of 2.0 equiv 2d improved the yield remarkably (Table 4, entry 4). The yields decreased when the reaction time was shortened or prolonged (Table 4, entries 5 and 6).
Table 4: The AuCl3-catalyzed iodination of tetraphenylene (1).
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-122-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | NIS (equiv) | T (°C) | t (h) | Yielda (%) |
|---|---|---|---|---|
| 1 | 1.0 | rt | 12 | 18 |
| 2 | 1.0 | 60 | 12 | 32 |
| 3 | 1.0 | 80 | 12 | 30 |
| 4 | 2.0 | 60 | 12 | 80 (78)b |
| 5 | 2.0 | 60 | 6 | 72 |
| 6 | 2.0 | 60 | 16 | 52 |
aThe yields were determined by 1H NMR analysis of the crude products using CH2Br2 as the internal standard. bIsolated yields based on tetraphenylene (1).
Having successfully developed protocols for introducing OAc and X (Cl, Br, I) onto tetraphenylene (1), we next turned to investigate the carbonylation of tetraphenylene (1). The carbonyl group is a common structural element present in both natural products and functional materials and can be transformed into other functionalities [58,59]. The Larock group reported a novel Pd-catalyzed addition of nitriles to an arene C–H bond for the synthesis of aryl ketones [60,61]. Following the Larock’s conditions, we investigated the carbonylation of tetraphenylene (1) and the carbonylated product 5a was obtained in 20% yield (Table 5, entry 1). While the yield was improved to 42% in the presence of 2.0 equiv DMSO (Table 5, entry 2), it decreased when 4.0 equiv DMSO were used (Table 5, entry 3). Since the solubility of 1 in trifluoroacetic acid is low, we envisaged that the addition of co-solvents would promote the reaction. Therefore, we screened the effect of different co-solvents on the reaction and dichloromethane was found to be the best choice (Table 5, entries 4 and 5). The yield was remarkably improved by increasing the amount of Pd(OAc)2 (Table 5, entry 6). Raising or lowering the temperature led to lower yields (Table 5, entries 7 and 8). The variation of the amount of PhCN lead to an optimal yield of 83% when 2.5 equiv PhCN were employed (Table 5, entry 9). However, using 3.0 equiv PhCN failed to enhance the yield (Table 5, entry 10).
Table 5: The Pd(OAc)2-catalyzed carbonylation of tetraphenylene (1).
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-122-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | PhCN (equiv) | Pd(OAc)2 (equiv) | DMSO (equiv) | Additive (0.2 mL) | Yielda (%) |
|---|---|---|---|---|---|
| 1 | 2.0 | 10% | 1.0 | / | 20 |
| 2 | 2.0 | 10% | 2.0 | / | 42 |
| 3 | 2.0 | 10% | 4.0 | / | 28 |
| 4 | 2.0 | 10% | 2.0 | DCM | 52 |
| 5 | 2.0 | 10% | 2.0 | DCE | 36 |
| 6 | 2.0 | 20% | 2.0 | DCM | 71 |
| 7 | 2.0 | 20% | 2.0 | DCM | 60b |
| 8 | 2.0 | 20% | 2.0 | DCM | 64c |
| 9 | 2.5 | 20% | 2.0 | DCM | 83 (80)d |
| 10 | 3.0 | 20% | 2.0 | DCM | 60 |
aThe yields were determined by 1H NMR analysis of the crude products using CH2Br2 as the internal standard. bReaction temperature 90 °C. cReaction temperature 110 °C. dIsolated yields based on tetraphenylene (1).
Under the optimal reaction conditions, various nitriles 4b–k including aromatic and aliphatic ones, were reacted with tetraphenylene (1) to give the corresponding carbonyl products 5b–k (Scheme 1). Both substrates containing either an electron-donating methyl group or electron-withdrawing trifluoromethyl group were suitable for the reaction. In addition, halogen-substituted nitriles, including F, Cl, and Br substituents, were well-tolerated under the standard reaction conditions. Also, methyl 4-cyanobenzoate and 1-naphthonitrile were successfully reacted with tetraphenylene to form the corresponding carbonylated products. It is worth mentioning that aliphatic nitriles 4j,k were also found to be reactive under the conditions.
![[1860-5397-12-122-i1]](/bjoc/content/inline/1860-5397-12-122-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The Pd(OAc)2-catalyzed reaction of nitriles with tetraphenylene (1).
Scheme 1: The Pd(OAc)2-catalyzed reaction of nitriles with tetraphenylene (1).
Conclusion
In conclusion, three reactions for halogenation, acetoxylation, and carbonylation of tetraphenylene (1) have been developed via a transition-metal-catalyzed direct derivatization. The reactions provide new methods for the synthesis of a variety of 2-substituted tetraphenylenes, which could accelerate the research on the properties and application of tetraphenylene derivatives.
Supporting Information
| Supporting Information File 1: Experimental section and characterization of the synthesized compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21372176), Tongji University 985 Phase III funds, the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, and Shanghai Science and Technology Commission (14DZ2261100).
References
-
Karle, I. L.; Brockway, L. O. J. Am. Chem. Soc. 1944, 66, 1974–1979. doi:10.1021/ja01239a057
Return to citation in text: [1] -
Irngartinger, H.; Reibel, W. R. K. Acta Crystallogr., Sect. B 1981, 37, 1724–1728. doi:10.1107/S0567740881006985
Return to citation in text: [1] -
Huang, H.; Hau, C.-K.; Law, C. C. M.; Wong, H. N. C. Org. Biomol. Chem. 2009, 7, 1249–1257. doi:10.1039/B818029F
Return to citation in text: [1] -
Han, J.-W.; Chen, J.-X.; Li, X.; Peng, X.-S.; Wong, H. N. C. Synlett 2013, 24, 2188–2198. doi:10.1055/s-0033-1339859
Return to citation in text: [1] -
Rajca, A.; Safronov, A.; Rajca, S.; Ross, C. R., II; Stezowski, J. J. J. Am. Chem. Soc. 1996, 118, 7272–7279. doi:10.1021/ja961065y
Return to citation in text: [1] -
Rajca, A.; Safronov, A.; Rajca, S.; Shoemaker, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 488–491. doi:10.1002/anie.199704881
Return to citation in text: [1] -
Elliott, E. L.; Orita, A.; Hasegawa, D.; Gantzel, P.; Otera, J.; Siegel, J. S. Org. Biomol. Chem. 2005, 3, 581–583. doi:10.1039/B416820H
Return to citation in text: [1] -
Hisaki, I.; Sonoda, M.; Tobe, Y. Eur. J. Org. Chem. 2006, 833–847. doi:10.1002/ejoc.200500705
Return to citation in text: [1] -
Rajca, A.; Rajca, S.; Pink, M.; Miyasaka, M. Synlett 2007, 1799–1822. doi:10.1055/s-2007-984538
Return to citation in text: [1] -
Hau, C.-K.; Chui, S. S.-Y.; Lu, W.; Che, C.-M.; Cheng, P.-S.; Mak, T. C. W.; Miao, Q.; Wong, H. N. C. Chem. Sci. 2011, 2, 1068–1075. doi:10.1039/C1SC00053E
Return to citation in text: [1] -
Xiong, X.-D.; Deng, C.-L.; Peng, X.-S.; Miao, Q.; Wong, H. N. C. Org. Lett. 2014, 16, 3252–3255. doi:10.1021/ol501267z
Return to citation in text: [1] -
Mak, T. C. W.; Wong, H. N. C. Tetraphenylene and Related Hosts. In Comprehensive Supramolecular Chemistry; MacNicol, D. D.; Toda, F.; Bishop, P., Eds.; Pergamon Press: Oxford, 1996; Vol. 6, pp 351–369.
Return to citation in text: [1] -
Mak, T. C. W.; Wong, H. N. C. Top. Curr. Chem. 1987, 140, 141–164. doi:10.1007/BFb0003839
Return to citation in text: [1] -
Man, Y.-M.; Mak, T. C. W.; Wong, H. N. C. J. Org. Chem. 1990, 55, 3214–3221. doi:10.1021/jo00297a043
Return to citation in text: [1] -
Yang, X.-P.; Du, D.-M.; Li, Q.; Mak, T. C. W.; Wong, H. N. C. Chem. Commun. 1999, 1607–1608. doi:10.1039/A904144C
Return to citation in text: [1] -
Lai, C. W.; Lam, C. K.; Lee, H. K.; Mak, T. C. W.; Wong, H. N. C. Org. Lett. 2003, 5, 823–826. doi:10.1021/ol020253s
Return to citation in text: [1] -
Wen, J.-F.; Hong, W.; Yuan, K.; Mak, T. C. W.; Wong, H. N. C. J. Org. Chem. 2003, 68, 8918–8931. doi:10.1021/jo0302408
Return to citation in text: [1] -
Lin, F.; Peng, H.-Y.; Chen, J.-X.; Chik, D. T. W.; Cai, Z.; Wong, K. M. C.; Yam, V. W. W.; Wong, H. N. C. J. Am. Chem. Soc. 2010, 132, 16383–16392. doi:10.1021/ja106599j
Return to citation in text: [1] -
Peng, H.-Y.; Lam, C.-K.; Mak, T. C. W.; Cai, Z.; Ma, W.-T.; Li, Y.-X.; Wong, H. N. C. J. Am. Chem. Soc. 2005, 127, 9603–9611. doi:10.1021/ja051013l
Return to citation in text: [1] -
Wu, A.-H.; Hau, C.-K.; Wong, H. N. C. Adv. Synth. Catal. 2007, 349, 601–608. doi:10.1002/adsc.200600499
Return to citation in text: [1] -
Rajca, A.; Rajca, S. Angew. Chem., Int. Ed. 2010, 49, 672–674. doi:10.1002/anie.200905421
Return to citation in text: [1] -
Rapson, W. S.; Shuttleworth, R. G.; van Niekerk, J. N. J. Chem. Soc. 1943, 326–327. doi:10.1039/JR9430000326
Return to citation in text: [1] [2] [3] -
Wang, C.; Xi, Z. Chem. Commun. 2007, 5119–5133. doi:10.1039/B709839A
Return to citation in text: [1] -
Wittig, G.; Lehmann, G. Chem. Ber. 1957, 90, 875–892. doi:10.1002/cber.19570900606
Return to citation in text: [1] -
Wittig, G.; Klar, G. Justus Liebigs Ann. Chem. 1967, 704, 91–108. doi:10.1002/jlac.19677040108
Return to citation in text: [1] -
Hellwinkel, D.; Reiff, G.; Nykodym, V. Justus Liebigs Ann. Chem. 1977, 1013–1025. doi:10.1002/jlac.197719770615
Return to citation in text: [1] -
Rajca, A.; Safronov, A.; Rajca, S.; Wongsriratanakul, J. J. Am. Chem. Soc. 2000, 122, 3351–3357. doi:10.1021/ja993286k
Return to citation in text: [1] -
Kabir, S. M. H.; Iyoda, M. Synthesis 2000, 1839–1843. doi:10.1055/s-2000-8239
Return to citation in text: [1] -
Kabir, S. M. H.; Hasegawa, M.; Kuwatani, Y.; Yoshida, M.; Matsuyama, H.; Iyoda, M. J. Chem. Soc., Perkin Trans. 1 2001, 159–165. doi:10.1039/B006375O
Return to citation in text: [1] -
Rajca, A.; Wang, H.; Bolshov, P.; Rajca, S. Tetrahedron 2001, 57, 3725–3735. doi:10.1016/S0040-4020(01)00241-1
Return to citation in text: [1] -
Li, X.; Han, J.-W.; Wong, H. N. C. Asian J. Org. Chem. 2016, 5, 74–81. doi:10.1002/ajoc.201500327
Return to citation in text: [1] -
Perthuisot, C.; Edelbach, B. L.; Zubris, D. L.; Simhai, N.; Iverson, C. N.; Müller, C.; Satoh, T.; Jones, W. D. J. Mol. Catal. A 2002, 189, 157–168. doi:10.1016/S1381-1169(02)00203-0
Return to citation in text: [1] -
Eisch, J. J.; Piotrowski, A. M.; Han, K. I.; Krüger, C.; Tsay, Y. H. Organometallics 1985, 4, 224–231. doi:10.1021/om00121a003
Return to citation in text: [1] -
Schwager, H.; Spyroudis, S.; Vollhardt, K. P. C. J. Organomet. Chem. 1990, 382, 191–200. doi:10.1016/0022-328X(90)85227-P
Return to citation in text: [1] -
Edelbach, B. L.; Lachicotte, R. J.; Jones, W. D. J. Am. Chem. Soc. 1998, 120, 2843–2853. doi:10.1021/ja973368d
Return to citation in text: [1] -
Simhai, N.; Iverson, C. N.; Edelbach, B. L.; Jones, W. D. Organometallics 2001, 20, 2759–2766. doi:10.1021/om010111m
Return to citation in text: [1] -
Beck, R.; Johnson, S. A. Chem. Commun. 2011, 47, 9233–9235. doi:10.1039/C1CC12657A
Return to citation in text: [1] -
Lindow, D. F.; Friedman, L. J. Am. Chem. Soc. 1967, 89, 1271–1272. doi:10.1021/ja00981a046
Return to citation in text: [1] -
Friedman, L.; Lindow, D. F. J. Am. Chem. Soc. 1968, 90, 2324–2328. doi:10.1021/ja01011a021
Return to citation in text: [1] -
Xing, Y.-D.; Huang, N. Z. J. Org. Chem. 1982, 47, 140–142. doi:10.1021/jo00340a030
Return to citation in text: [1] -
Wang, X.-M.; Hou, X.; Zhou, Z.; Mak, T. C. W.; Wong, H. N. C. J. Org. Chem. 1993, 58, 7498–7506. doi:10.1021/jo00078a031
Return to citation in text: [1] -
Song, Q.; Lebeis, C. W.; Shen, X.; Ho, D. M.; Pascal, R. A., Jr. J. Am. Chem. Soc. 2005, 127, 13732–13737. doi:10.1021/ja0541985
Return to citation in text: [1] -
Masselot, D.; Charmant, J. P. H.; Gallagher, T. J. Am. Chem. Soc. 2006, 128, 694–695. doi:10.1021/ja056964d
Return to citation in text: [1] -
Jiang, H.; Zhang, Y.; Chen, D.; Zhou, B.; Zhang, Y. Org. Lett. 2016, 18, 2032–2035. doi:10.1021/acs.orglett.6b00641
Return to citation in text: [1] -
Figeys, H. P.; Dralants, A. Tetrahedron Lett. 1971, 3901–3904. doi:10.1016/S0040-4039(01)97319-8
Return to citation in text: [1] -
Hageman, H. J. Chem. Commun. 1968, 401–402. doi:10.1039/C19680000401
Return to citation in text: [1] -
Mori, T.; Takamoto, M.; Wada, T.; Inoue, Y. Photochem. Photobiol. Sci. 2003, 2, 1187–1199. doi:10.1039/B305898K
Return to citation in text: [1] -
Emmert, M. H.; Gary, J. B.; Villalobos, J. M.; Sanford, M. S. Angew. Chem., Int. Ed. 2010, 49, 5884–5886. doi:10.1002/anie.201002351
Return to citation in text: [1] -
Qiu, D.; Zheng, Z.; Mo, F.; Xiao, Q.; Tian, Y.; Zhang, Y.; Wang, J. Org. Lett. 2011, 13, 4988–4991. doi:10.1021/ol202075x
Return to citation in text: [1] -
Miyaura, N.; Yamada, K.; Suzuki, A. Tetrahedron Lett. 1979, 20, 3437–3440. doi:10.1016/S0040-4039(01)95429-2
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/C39790000866
Return to citation in text: [1] -
Miyaura, N. Top. Curr. Chem. 2002, 219, 11–59. doi:10.1007/3-540-45313-X_2
Return to citation in text: [1] -
Suzuki, A.; Brown, H. C. Organic Synthesis via Boranes; Aldrich: Milwaukee, 2003; Vol. 3.
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Felpin, F.-X.; Ayad, T.; Mitra, S. Eur. J. Org. Chem. 2006, 2679–2690. doi:10.1002/ejoc.200501004
Return to citation in text: [1] -
Mo, F.; Yan, J. M.; Qiu, D.; Li, F.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 2028–2032. doi:10.1002/anie.200906699
Return to citation in text: [1] -
Qiu, D.; Mo, F.; Zheng, Z.; Zhang, Y.; Wang, J. Org. Lett. 2010, 12, 5474–5477. doi:10.1021/ol102350v
Return to citation in text: [1] -
Boger, D. L.; Hong, J.; Hikota, M.; Ishida, M. J. Am. Chem. Soc. 1999, 121, 2471–2477. doi:10.1021/ja983631q
Return to citation in text: [1] -
Romines, K. R.; Freeman, G. A.; Schaller, L. T.; Cowan, J. R.; Gonzales, S. S.; Tidwell, J. H.; Andrews, C. W., III; Stammers, D. K.; Hazen, R. J.; Ferris, R. G.; Short, S. A.; Chan, J. H.; Boone, L. R. J. Med. Chem. 2006, 49, 727–739. doi:10.1021/jm050670l
Return to citation in text: [1] -
Zhou, C.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 2302–2303. doi:10.1021/ja038687l
Return to citation in text: [1] -
Zhou, C.; Larock, R. C. J. Org. Chem. 2006, 71, 3551–3558. doi:10.1021/jo060220g
Return to citation in text: [1]
| 60. | Zhou, C.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 2302–2303. doi:10.1021/ja038687l |
| 61. | Zhou, C.; Larock, R. C. J. Org. Chem. 2006, 71, 3551–3558. doi:10.1021/jo060220g |
| 1. | Karle, I. L.; Brockway, L. O. J. Am. Chem. Soc. 1944, 66, 1974–1979. doi:10.1021/ja01239a057 |
| 2. | Irngartinger, H.; Reibel, W. R. K. Acta Crystallogr., Sect. B 1981, 37, 1724–1728. doi:10.1107/S0567740881006985 |
| 19. | Peng, H.-Y.; Lam, C.-K.; Mak, T. C. W.; Cai, Z.; Ma, W.-T.; Li, Y.-X.; Wong, H. N. C. J. Am. Chem. Soc. 2005, 127, 9603–9611. doi:10.1021/ja051013l |
| 20. | Wu, A.-H.; Hau, C.-K.; Wong, H. N. C. Adv. Synth. Catal. 2007, 349, 601–608. doi:10.1002/adsc.200600499 |
| 21. | Rajca, A.; Rajca, S. Angew. Chem., Int. Ed. 2010, 49, 672–674. doi:10.1002/anie.200905421 |
| 56. | Mo, F.; Yan, J. M.; Qiu, D.; Li, F.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 2028–2032. doi:10.1002/anie.200906699 |
| 57. | Qiu, D.; Mo, F.; Zheng, Z.; Zhang, Y.; Wang, J. Org. Lett. 2010, 12, 5474–5477. doi:10.1021/ol102350v |
| 12. | Mak, T. C. W.; Wong, H. N. C. Tetraphenylene and Related Hosts. In Comprehensive Supramolecular Chemistry; MacNicol, D. D.; Toda, F.; Bishop, P., Eds.; Pergamon Press: Oxford, 1996; Vol. 6, pp 351–369. |
| 13. | Mak, T. C. W.; Wong, H. N. C. Top. Curr. Chem. 1987, 140, 141–164. doi:10.1007/BFb0003839 |
| 14. | Man, Y.-M.; Mak, T. C. W.; Wong, H. N. C. J. Org. Chem. 1990, 55, 3214–3221. doi:10.1021/jo00297a043 |
| 15. | Yang, X.-P.; Du, D.-M.; Li, Q.; Mak, T. C. W.; Wong, H. N. C. Chem. Commun. 1999, 1607–1608. doi:10.1039/A904144C |
| 16. | Lai, C. W.; Lam, C. K.; Lee, H. K.; Mak, T. C. W.; Wong, H. N. C. Org. Lett. 2003, 5, 823–826. doi:10.1021/ol020253s |
| 17. | Wen, J.-F.; Hong, W.; Yuan, K.; Mak, T. C. W.; Wong, H. N. C. J. Org. Chem. 2003, 68, 8918–8931. doi:10.1021/jo0302408 |
| 18. | Lin, F.; Peng, H.-Y.; Chen, J.-X.; Chik, D. T. W.; Cai, Z.; Wong, K. M. C.; Yam, V. W. W.; Wong, H. N. C. J. Am. Chem. Soc. 2010, 132, 16383–16392. doi:10.1021/ja106599j |
| 58. | Boger, D. L.; Hong, J.; Hikota, M.; Ishida, M. J. Am. Chem. Soc. 1999, 121, 2471–2477. doi:10.1021/ja983631q |
| 59. | Romines, K. R.; Freeman, G. A.; Schaller, L. T.; Cowan, J. R.; Gonzales, S. S.; Tidwell, J. H.; Andrews, C. W., III; Stammers, D. K.; Hazen, R. J.; Ferris, R. G.; Short, S. A.; Chan, J. H.; Boone, L. R. J. Med. Chem. 2006, 49, 727–739. doi:10.1021/jm050670l |
| 5. | Rajca, A.; Safronov, A.; Rajca, S.; Ross, C. R., II; Stezowski, J. J. J. Am. Chem. Soc. 1996, 118, 7272–7279. doi:10.1021/ja961065y |
| 6. | Rajca, A.; Safronov, A.; Rajca, S.; Shoemaker, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 488–491. doi:10.1002/anie.199704881 |
| 7. | Elliott, E. L.; Orita, A.; Hasegawa, D.; Gantzel, P.; Otera, J.; Siegel, J. S. Org. Biomol. Chem. 2005, 3, 581–583. doi:10.1039/B416820H |
| 8. | Hisaki, I.; Sonoda, M.; Tobe, Y. Eur. J. Org. Chem. 2006, 833–847. doi:10.1002/ejoc.200500705 |
| 9. | Rajca, A.; Rajca, S.; Pink, M.; Miyasaka, M. Synlett 2007, 1799–1822. doi:10.1055/s-2007-984538 |
| 10. | Hau, C.-K.; Chui, S. S.-Y.; Lu, W.; Che, C.-M.; Cheng, P.-S.; Mak, T. C. W.; Miao, Q.; Wong, H. N. C. Chem. Sci. 2011, 2, 1068–1075. doi:10.1039/C1SC00053E |
| 11. | Xiong, X.-D.; Deng, C.-L.; Peng, X.-S.; Miao, Q.; Wong, H. N. C. Org. Lett. 2014, 16, 3252–3255. doi:10.1021/ol501267z |
| 48. | Emmert, M. H.; Gary, J. B.; Villalobos, J. M.; Sanford, M. S. Angew. Chem., Int. Ed. 2010, 49, 5884–5886. doi:10.1002/anie.201002351 |
| 49. | Qiu, D.; Zheng, Z.; Mo, F.; Xiao, Q.; Tian, Y.; Zhang, Y.; Wang, J. Org. Lett. 2011, 13, 4988–4991. doi:10.1021/ol202075x |
| 3. | Huang, H.; Hau, C.-K.; Law, C. C. M.; Wong, H. N. C. Org. Biomol. Chem. 2009, 7, 1249–1257. doi:10.1039/B818029F |
| 4. | Han, J.-W.; Chen, J.-X.; Li, X.; Peng, X.-S.; Wong, H. N. C. Synlett 2013, 24, 2188–2198. doi:10.1055/s-0033-1339859 |
| 50. | Miyaura, N.; Yamada, K.; Suzuki, A. Tetrahedron Lett. 1979, 20, 3437–3440. doi:10.1016/S0040-4039(01)95429-2 |
| 51. | Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/C39790000866 |
| 52. | Miyaura, N. Top. Curr. Chem. 2002, 219, 11–59. doi:10.1007/3-540-45313-X_2 |
| 53. | Suzuki, A.; Brown, H. C. Organic Synthesis via Boranes; Aldrich: Milwaukee, 2003; Vol. 3. |
| 54. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 55. | Felpin, F.-X.; Ayad, T.; Mitra, S. Eur. J. Org. Chem. 2006, 2679–2690. doi:10.1002/ejoc.200501004 |
| 22. | Rapson, W. S.; Shuttleworth, R. G.; van Niekerk, J. N. J. Chem. Soc. 1943, 326–327. doi:10.1039/JR9430000326 |
| 45. | Figeys, H. P.; Dralants, A. Tetrahedron Lett. 1971, 3901–3904. doi:10.1016/S0040-4039(01)97319-8 |
| 43. | Masselot, D.; Charmant, J. P. H.; Gallagher, T. J. Am. Chem. Soc. 2006, 128, 694–695. doi:10.1021/ja056964d |
| 44. | Jiang, H.; Zhang, Y.; Chen, D.; Zhou, B.; Zhang, Y. Org. Lett. 2016, 18, 2032–2035. doi:10.1021/acs.orglett.6b00641 |
| 46. | Hageman, H. J. Chem. Commun. 1968, 401–402. doi:10.1039/C19680000401 |
| 47. | Mori, T.; Takamoto, M.; Wada, T.; Inoue, Y. Photochem. Photobiol. Sci. 2003, 2, 1187–1199. doi:10.1039/B305898K |
| 23. | Wang, C.; Xi, Z. Chem. Commun. 2007, 5119–5133. doi:10.1039/B709839A |
| 24. | Wittig, G.; Lehmann, G. Chem. Ber. 1957, 90, 875–892. doi:10.1002/cber.19570900606 |
| 25. | Wittig, G.; Klar, G. Justus Liebigs Ann. Chem. 1967, 704, 91–108. doi:10.1002/jlac.19677040108 |
| 26. | Hellwinkel, D.; Reiff, G.; Nykodym, V. Justus Liebigs Ann. Chem. 1977, 1013–1025. doi:10.1002/jlac.197719770615 |
| 27. | Rajca, A.; Safronov, A.; Rajca, S.; Wongsriratanakul, J. J. Am. Chem. Soc. 2000, 122, 3351–3357. doi:10.1021/ja993286k |
| 28. | Kabir, S. M. H.; Iyoda, M. Synthesis 2000, 1839–1843. doi:10.1055/s-2000-8239 |
| 29. | Kabir, S. M. H.; Hasegawa, M.; Kuwatani, Y.; Yoshida, M.; Matsuyama, H.; Iyoda, M. J. Chem. Soc., Perkin Trans. 1 2001, 159–165. doi:10.1039/B006375O |
| 30. | Rajca, A.; Wang, H.; Bolshov, P.; Rajca, S. Tetrahedron 2001, 57, 3725–3735. doi:10.1016/S0040-4020(01)00241-1 |
| 31. | Li, X.; Han, J.-W.; Wong, H. N. C. Asian J. Org. Chem. 2016, 5, 74–81. doi:10.1002/ajoc.201500327 |
| 32. | Perthuisot, C.; Edelbach, B. L.; Zubris, D. L.; Simhai, N.; Iverson, C. N.; Müller, C.; Satoh, T.; Jones, W. D. J. Mol. Catal. A 2002, 189, 157–168. doi:10.1016/S1381-1169(02)00203-0 |
| 33. | Eisch, J. J.; Piotrowski, A. M.; Han, K. I.; Krüger, C.; Tsay, Y. H. Organometallics 1985, 4, 224–231. doi:10.1021/om00121a003 |
| 34. | Schwager, H.; Spyroudis, S.; Vollhardt, K. P. C. J. Organomet. Chem. 1990, 382, 191–200. doi:10.1016/0022-328X(90)85227-P |
| 35. | Edelbach, B. L.; Lachicotte, R. J.; Jones, W. D. J. Am. Chem. Soc. 1998, 120, 2843–2853. doi:10.1021/ja973368d |
| 36. | Simhai, N.; Iverson, C. N.; Edelbach, B. L.; Jones, W. D. Organometallics 2001, 20, 2759–2766. doi:10.1021/om010111m |
| 37. | Beck, R.; Johnson, S. A. Chem. Commun. 2011, 47, 9233–9235. doi:10.1039/C1CC12657A |
| 38. | Lindow, D. F.; Friedman, L. J. Am. Chem. Soc. 1967, 89, 1271–1272. doi:10.1021/ja00981a046 |
| 39. | Friedman, L.; Lindow, D. F. J. Am. Chem. Soc. 1968, 90, 2324–2328. doi:10.1021/ja01011a021 |
| 40. | Xing, Y.-D.; Huang, N. Z. J. Org. Chem. 1982, 47, 140–142. doi:10.1021/jo00340a030 |
| 41. | Wang, X.-M.; Hou, X.; Zhou, Z.; Mak, T. C. W.; Wong, H. N. C. J. Org. Chem. 1993, 58, 7498–7506. doi:10.1021/jo00078a031 |
| 42. | Song, Q.; Lebeis, C. W.; Shen, X.; Ho, D. M.; Pascal, R. A., Jr. J. Am. Chem. Soc. 2005, 127, 13732–13737. doi:10.1021/ja0541985 |
| 22. | Rapson, W. S.; Shuttleworth, R. G.; van Niekerk, J. N. J. Chem. Soc. 1943, 326–327. doi:10.1039/JR9430000326 |
| 22. | Rapson, W. S.; Shuttleworth, R. G.; van Niekerk, J. N. J. Chem. Soc. 1943, 326–327. doi:10.1039/JR9430000326 |
© 2016 Pan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)