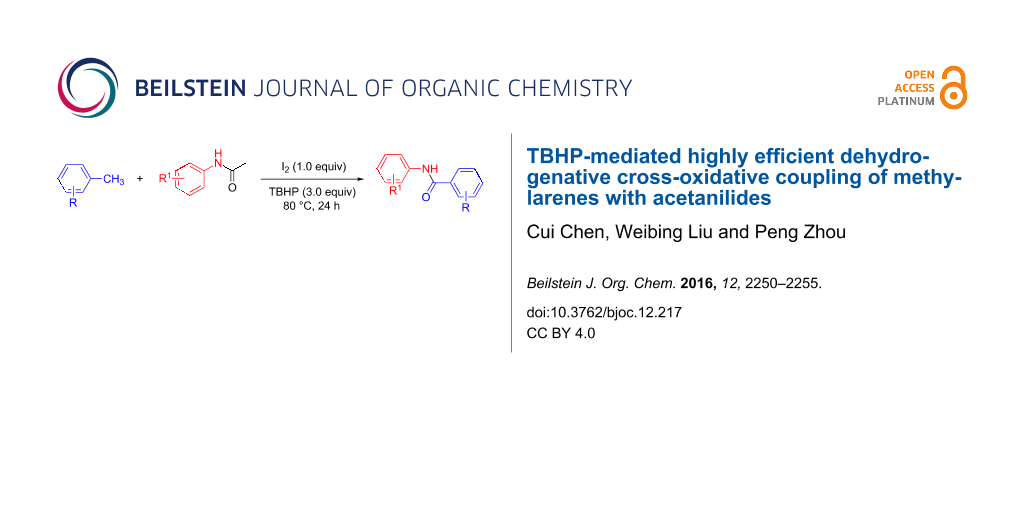
Abstract
A TBHP-mediated dehydrogenative cross-oxidative-coupling approach has been developed for the synthesis of N-arylbenzamides from methylarenes and acetanilides. This cross-coupling method is free of transition metal catalysts and ligands, and no extra organic solvents are required, which make it an useful and attractive strategy for the straightforward construction of C–N bonds. Besides, this conversion is an important complement to the conventional C–N forming strategies.

Graphical Abstract
Introduction
Recently, amides have attracted more and more attention due to their extensive utilization in pharmaceutical and agrochemical applications [1-3], as well as for precursors in organic synthesis for the construction of natural products, polymers and organic materials [4-6]. The database of medicinal chemistry indicates that around 25% of synthetic drugs contain the amide moiety [7]. What is more, the amide motif has also served as pivotal intermediate to generate several other organic functionalities [8,9]. To date, a large number of amidation reactions have been established [10,11], such as the condensation of carboxylic acid derivatives with amines [12], the rearrangement of ketoximes [13], the C–N cross coupling between aromatic amides or amines and aryl(pseudo)halides [14,15] or aldehydes [16-20]. However, to the best of our knowledge, the studies of dehydrogenative cross-oxidative-coupling reactions between methylarenes and amines for the formation of amides are rather limited, and which would be an important complement to the conventional C–N forming strategies. Herein, we disclose a dehydrogenative C–N cross-oxidative-coupling reaction of methylarenes with acetanilides, using TBHP as an oxidant to afford N-arylamides in moderate to good yields (Scheme 1).
![[1860-5397-12-217-i1]](/bjoc/content/inline/1860-5397-12-217-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
We began by studying the reaction of toluene (1a) and acetanilide (2a) as model substrates to identify the optimal conditions (Table 1). In the presence of I2 (0.1 equiv) and tert-butyl hydroperoxide (TBHP, 2.0 equiv), the study commenced to optimize the reaction time (Table 1, entries 1–3). The results show that the reaction was completed after 24 h and led to the desired N-phenylbenzamide 3aa in 62% GC yield (Table 1, entry 2). Disappointingly, other peroxides like di-tert-butylperoxide (DTBP), benzoyl peroxide, dicumyl peroxide (DCP), methyl ethyl ketone peroxide (MEKP), tert-butylperoxy benzoate (TBPB) and cumene hydroperoxide (CHP) proved wholly ineffective for this conversion and no product was observed (Table 1, entries 4–9). Next, the effect of other iodine sources on the reaction was monitored. Pleasingly, ICl and NIS afforded the desired product 3aa successfully, but led to a marked decrease in yield (Table 1, entries 10 and 11). When the loading of I2 was increased to 1.0 equivalent, a pronounced improvement in the reaction yield was observed (Table 1, entry 12). Further increasing the loading of I2 did not show any beneficial effect (Table 1, entry 13). However, the reaction does not occur in the absence of molecular iodine (Table 1, entry 14), which indicates that molecular iodine is requisite for this conversion. We were pleased to find that an excellent product yield of 86% was obtained (Table 1, entry 15) when increasing the loading of TBHP to 3.0 equivalents. However, decreasing the loading of TBHP to 1.0 equivalent led to a drastic drop in the yield (Table 1, entry 16). It is worth mentioning that this conversion did not show any beneficial effect with nitrogen protection.
Table 1: Optimization studiesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-217-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst (0.1 equiv) | Oxidant (2.0 equiv) | Time (h) | Yield (%)b |
|---|---|---|---|---|
| 1 | I2 | TBHP | 12 | 47 |
| 2 | I2 | TBHP | 24 | 62 |
| 3 | I2 | TBHP | 36 | 62 |
| 4 | I2 | DTBP | 24 | 0 |
| 5 | I2 | benzoyl peroxide | 24 | 0 |
| 6 | I2 | DCP | 24 | 0 |
| 7 | I2 | MEKP | 24 | 0 |
| 8 | I2 | TBPB | 24 | 0 |
| 9 | I2 | CHP | 24 | 0 |
| 10 | ICl | TBHP | 24 | 31 |
| 11 | NIS | TBHP | 24 | 39 |
| 12c | I2 | TBHP | 24 | 71 |
| 13d | I2 | TBHP | 24 | 71 |
| 14 | – | TBHP | 24 | – |
| 15c,e | I2 | TBHP | 24 | 86 |
| 16c,f | I2 | TBHP | 24 | 37 |
aUnless otherwise specified, all the reactions were carried out on 2a 0.25 mmol scale, catalyst 0.1 equivalents, oxidant 2.0 equivalents, toluene 2.0 mL; bGC yield; ciodine 1.0 equivalents; diodine 1.5 equivalents; eTBHP 3.0 equivalents; fTBHP 1.0 equivalent.
After identifying the optimized conditions, we next explored the substrate scope of this transformation. As detailed in Table 2, a wide variety of acetanilides having substituent groups such as methyl, methoxy, ethoxyl, chloro and cyano at different positions were employed to react under the standard conditions. We were pleased to find that all these tested substrates were successfully converted into the desired N-arylbenzamides 3. Notably, this conversion appears quite sensitive with respect to the nature (electron-donating or electron-withdrawing) and positions of the substituent groups under the stipulated conditions. para-Methoxy- and para-ethoxy-substituted acetanilides led to a drastic drop in yield as compared to para-cyano- and para-chloro-substituted acetanilides (Table 2, entries 5–8). As well as ortho-methyl- and ortho-chloro-substituted substrates also led to a marked decrease in yield as compared to their corresponding para-substituted substrates (Table 2, entries 2, 3, 5, and 9). To further extend the adaptability of this transformation, other methyl arenes were also checked under the standard conditions. Pleasingly, m-xylene and p-xylene afforded the desired product 3 successfully, but in lower yields (Table 2, entries 10 and 11). In the same manner, we next investigated the reactions of p-xylene with N-phenylacetamide, N-p-tolylacetamide, N-(4-methoxyphenyl)acetamide, N-(2-chlorophenyl)acetamide and N-(3-chlorophenyl)acetamide under the standard conditions. Gratifyingly, all the tested acetamides reacted with p-xylene successfully, offering the desired N-arylbenzamides in moderate yields, from 51% to 69% (Table 2, entries 12–14). It is noteworthy that the reactions did not result in the desired product when using aniline and diethylamine as the partners of acetanilide.
Table 2: Scope of the N-arylamidesa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-217-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | 2 | 3 | Yield(%)b |
|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-217-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-217-i7.svg?max-width=637&scale=1.0)
|
80 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-217-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-217-i9.svg?max-width=637&scale=1.0)
|
75 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-217-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-217-i11.svg?max-width=637&scale=1.0)
|
72 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-217-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-217-i13.svg?max-width=637&scale=1.0)
|
81 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-217-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-217-i15.svg?max-width=637&scale=1.0)
|
83 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-217-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-217-i17.svg?max-width=637&scale=1.0)
|
63 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-217-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-217-i19.svg?max-width=637&scale=1.0)
|
62 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-217-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-217-i21.svg?max-width=637&scale=1.0)
|
85 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-217-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-217-i23.svg?max-width=637&scale=1.0)
|
71 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-217-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-217-i25.svg?max-width=637&scale=1.0)
|
59 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-217-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-217-i27.svg?max-width=637&scale=1.0)
|
66 |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-217-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-12-217-i29.svg?max-width=637&scale=1.0)
|
65 |
| 13 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-12-217-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-12-217-i31.svg?max-width=637&scale=1.0)
|
57 |
| 14 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-12-217-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-12-217-i33.svg?max-width=637&scale=1.0)
|
69 |
| 15 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-12-217-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-12-217-i35.svg?max-width=637&scale=1.0)
|
51 |
aUnless otherwise specified, all the reactions were carried out on 2 1.0 mmol scale, 1 2.0 mL; bisolated yield.
In order to gain insight into the nature of this conversion, two experiments were conducted (Scheme 2). With the addition of Na2CO3 (sodium carbonate) into the reaction between 1a and 2a, the yield of 3aa decreased dramatically to 37%, which confirmed that Na2CO3 has an inhibitory effect for this transformation. At the same time, the radical scavenger TEMPO (2,2,6,6-tetramethylpiperidinoxyl) completely inhibited the reaction and almost no product was obtained. The result indicated that the mechanism may involve a radical pathway. To our delight, reactions using benzaldehyde and 1-(iodomethyl)benzene as the surrogates of toluene afforded 3aa in excellent yields. These results evidence that either 1-(iodomethyl)benzene or benzaldehyde may be the reaction intermediates derived in situ from toluene.
![[1860-5397-12-217-i2]](/bjoc/content/inline/1860-5397-12-217-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The mechanism for this conversion is unclear. Based on literature reports and our present experimental results, a plausible reaction mechanism has been proposed in Scheme 3 and exemplified by the formation of 3aa. Initially, toluene (1a) reacted with molecular iodine and TBHP to produce the 1-(iodomethyl)benzene (4) and benzaldehyde (5) [21,22]. Intermediate 6 was generated from the coupling of 2a with intermediate 4 [22], by eliminating a molecule of HI. According to the results of the control experiments, intermediate 6 also could be obtained from the reaction of benzaldehyde with 2a under the standard conditions. Then, intermediate 6 underwent a sequence of steps including fast oxidation and dehydration to give N-acetyl-N-phenylbenzamide (8) under the stipulated conditions, similar to the report of An [23]. Finally, the acidic hydrolysis of N-acetyl-N-phenylbenzamide (8) furnished the final product N-phenylbenzamide (3aa).
![[1860-5397-12-217-i3]](/bjoc/content/inline/1860-5397-12-217-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, a dehydrogenative C–N cross oxidative coupling approach has been developed for the synthesis of N-arylbenzamides from methylarenes and acetanilides. In this protocol, the C–N cross oxidative coupling is free of transition metal catalysts, which makes the present method a useful and attractive strategy for the straightforward construction of C–N bonds. Besides, this conversion is an important complement to the conventional C–N forming strategies. More applications of this novel protocol and the study of the detailed mechanism are currently underway.
Supporting Information
| Supporting Information File 1: Full experimental details and copies of NMR spectral data. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Ganesh, T.; Jiang, J. X.; Yang, M. S.; Dingledine, R. J. Med. Chem. 2014, 57, 4173. doi:10.1021/jm5000672
Return to citation in text: [1] -
Valverde, I. E.; Vomstein, S.; Fischer, C. A.; Mascarin, A.; Mindt, T. L. J. Med. Chem. 2015, 58, 7475. doi:10.1021/acs.jmedchem.5b00994
Return to citation in text: [1] -
Good, J. A. D.; Silver, J.; Núñez-Otero, C.; Bahnan, W.; Krishnan, K. S.; Salin, O.; Engström, P.; Svensson, R.; Artursson, P.; Bergström, S.; Almqvist, F. J. Med. Chem. 2016, 59, 2094. doi:10.1021/acs.jmedchem.5b01759
Return to citation in text: [1] -
Otrubova, K.; Brown, M.; McCormick, M. S.; Han, G. W.; Cravatt, B. F.; Stevens, R. C.; Lichtman, A. H.; Boger, D. L. J. Am. Chem. Soc. 2013, 135, 6289. doi:10.1021/ja4014997
Return to citation in text: [1] -
Guo, X. G.; Facchetti, A.; Marks, T. J. Chem. Rev. 2014, 114, 8943. doi:10.1021/cr500225d
Return to citation in text: [1] -
Martí-Centelles, V.; Burguete, M. I.; Luis, S. V. J. Org. Chem. 2016, 81, 2143. doi:10.1021/acs.joc.5b02676
Return to citation in text: [1] -
Racine, E.; Monnier, F.; Vorsb, J. P.; Taillefer, M. Chem. Commun. 2013, 49, 7412. doi:10.1039/c3cc42575d
Return to citation in text: [1] -
Tobisu, M.; Nakamura, K.; Chatani, N. J. Am. Chem. Soc. 2014, 136, 5587. doi:10.1021/ja501649a
Return to citation in text: [1] -
Panahi, F.; Jamedi, F.; Iranpoor, N. Eur. J. Org. Chem. 2016, 780. doi:10.1002/ejoc.201501349
Return to citation in text: [1] -
Zhang, Z. M.; Wu, L. H.; Liao, J. H.; Wu, W. Q.; Jiang, H. F.; Li, J. X.; Li, J. W. J. Org. Chem. 2015, 80, 7594. doi:10.1021/acs.joc.5b01178
Return to citation in text: [1] -
Zhang, F. L.; Zhu, X.; Chiba, S. Org. Lett. 2015, 17, 3138. doi:10.1021/acs.orglett.5b01458
Return to citation in text: [1] -
Chen, P. J.; Wang, H. Y.; Peng, A. Y. RSC Adv. 2015, 5, 94328. doi:10.1039/C5RA18459B
Return to citation in text: [1] -
Babak, K.; Hesam, B. Synlett 2010, 2019. doi:10.1055/s-0030-1258484
Return to citation in text: [1] -
Dai, H. M.; Yu, C.; Lu, C. S.; Yan, H. Eur. J. Org. Chem. 2016, 1255. doi:10.1002/ejoc.201501551
Return to citation in text: [1] -
Iranpoor, N.; Panahi, F.; Roozbin, F.; Erfan, S.; Rahimi, S. Eur. J. Org. Chem. 2016, 1781. doi:10.1002/ejoc.201501607
Return to citation in text: [1] -
Ding, Y. Z.; Zhang, X.; Zhang, D. Y.; Chen, Y. T.; Wu, Z. B.; Wang, P. Y.; Xue, W.; Song, B. A.; Yang, S. Tetrahedron Lett. 2015, 56, 831. doi:10.1016/j.tetlet.2014.12.113
Return to citation in text: [1] -
Jia, X. F.; Zhang, S. H.; Wang, W. H.; Luo, F.; Cheng, J. Org. Lett. 2009, 11, 3120. doi:10.1021/ol900934g
Return to citation in text: [1] -
Wu, Y. N.; Li, B. Z.; Mao, F.; Li, X. S.; Kwong, F. Y. Org. Lett. 2011, 13, 3258. doi:10.1021/ol201201a
Return to citation in text: [1] -
Wang, J.; Liu, C.; Yuan, J. W.; Lei, A. W. Chem. Commun. 2014, 50, 4736. doi:10.1039/c4cc01447b
Return to citation in text: [1] -
Kumar, S.; Vanjari, R.; Guntreddi, T.; Singh, K. N. RSC Adv. 2015, 5, 9920. doi:10.1039/C4RA15140B
Return to citation in text: [1] -
Rout, S. K.; Guin, S.; Ali, W.; Gogoi, A.; Patel, B. K. Org. Lett. 2014, 16, 3086. doi:10.1021/ol5011906
Return to citation in text: [1] -
Majji, G.; Guin, S.; Gogoi, A.; Rout, S. K.; Patel, B. K. Chem. Commun. 2013, 49, 3031. doi:10.1039/c3cc40832a
Return to citation in text: [1] [2] -
Zou, Y.; Xiao, J.; Peng, Z. Z.; Dong, W. R.; An, D. L. Chem. Commun. 2015, 51, 14889. doi:10.1039/C5CC05483D
Return to citation in text: [1]
| 1. | Ganesh, T.; Jiang, J. X.; Yang, M. S.; Dingledine, R. J. Med. Chem. 2014, 57, 4173. doi:10.1021/jm5000672 |
| 2. | Valverde, I. E.; Vomstein, S.; Fischer, C. A.; Mascarin, A.; Mindt, T. L. J. Med. Chem. 2015, 58, 7475. doi:10.1021/acs.jmedchem.5b00994 |
| 3. | Good, J. A. D.; Silver, J.; Núñez-Otero, C.; Bahnan, W.; Krishnan, K. S.; Salin, O.; Engström, P.; Svensson, R.; Artursson, P.; Bergström, S.; Almqvist, F. J. Med. Chem. 2016, 59, 2094. doi:10.1021/acs.jmedchem.5b01759 |
| 10. | Zhang, Z. M.; Wu, L. H.; Liao, J. H.; Wu, W. Q.; Jiang, H. F.; Li, J. X.; Li, J. W. J. Org. Chem. 2015, 80, 7594. doi:10.1021/acs.joc.5b01178 |
| 11. | Zhang, F. L.; Zhu, X.; Chiba, S. Org. Lett. 2015, 17, 3138. doi:10.1021/acs.orglett.5b01458 |
| 8. | Tobisu, M.; Nakamura, K.; Chatani, N. J. Am. Chem. Soc. 2014, 136, 5587. doi:10.1021/ja501649a |
| 9. | Panahi, F.; Jamedi, F.; Iranpoor, N. Eur. J. Org. Chem. 2016, 780. doi:10.1002/ejoc.201501349 |
| 7. | Racine, E.; Monnier, F.; Vorsb, J. P.; Taillefer, M. Chem. Commun. 2013, 49, 7412. doi:10.1039/c3cc42575d |
| 4. | Otrubova, K.; Brown, M.; McCormick, M. S.; Han, G. W.; Cravatt, B. F.; Stevens, R. C.; Lichtman, A. H.; Boger, D. L. J. Am. Chem. Soc. 2013, 135, 6289. doi:10.1021/ja4014997 |
| 5. | Guo, X. G.; Facchetti, A.; Marks, T. J. Chem. Rev. 2014, 114, 8943. doi:10.1021/cr500225d |
| 6. | Martí-Centelles, V.; Burguete, M. I.; Luis, S. V. J. Org. Chem. 2016, 81, 2143. doi:10.1021/acs.joc.5b02676 |
| 16. | Ding, Y. Z.; Zhang, X.; Zhang, D. Y.; Chen, Y. T.; Wu, Z. B.; Wang, P. Y.; Xue, W.; Song, B. A.; Yang, S. Tetrahedron Lett. 2015, 56, 831. doi:10.1016/j.tetlet.2014.12.113 |
| 17. | Jia, X. F.; Zhang, S. H.; Wang, W. H.; Luo, F.; Cheng, J. Org. Lett. 2009, 11, 3120. doi:10.1021/ol900934g |
| 18. | Wu, Y. N.; Li, B. Z.; Mao, F.; Li, X. S.; Kwong, F. Y. Org. Lett. 2011, 13, 3258. doi:10.1021/ol201201a |
| 19. | Wang, J.; Liu, C.; Yuan, J. W.; Lei, A. W. Chem. Commun. 2014, 50, 4736. doi:10.1039/c4cc01447b |
| 20. | Kumar, S.; Vanjari, R.; Guntreddi, T.; Singh, K. N. RSC Adv. 2015, 5, 9920. doi:10.1039/C4RA15140B |
| 22. | Majji, G.; Guin, S.; Gogoi, A.; Rout, S. K.; Patel, B. K. Chem. Commun. 2013, 49, 3031. doi:10.1039/c3cc40832a |
| 14. | Dai, H. M.; Yu, C.; Lu, C. S.; Yan, H. Eur. J. Org. Chem. 2016, 1255. doi:10.1002/ejoc.201501551 |
| 15. | Iranpoor, N.; Panahi, F.; Roozbin, F.; Erfan, S.; Rahimi, S. Eur. J. Org. Chem. 2016, 1781. doi:10.1002/ejoc.201501607 |
| 23. | Zou, Y.; Xiao, J.; Peng, Z. Z.; Dong, W. R.; An, D. L. Chem. Commun. 2015, 51, 14889. doi:10.1039/C5CC05483D |
| 12. | Chen, P. J.; Wang, H. Y.; Peng, A. Y. RSC Adv. 2015, 5, 94328. doi:10.1039/C5RA18459B |
| 21. | Rout, S. K.; Guin, S.; Ali, W.; Gogoi, A.; Patel, B. K. Org. Lett. 2014, 16, 3086. doi:10.1021/ol5011906 |
| 22. | Majji, G.; Guin, S.; Gogoi, A.; Rout, S. K.; Patel, B. K. Chem. Commun. 2013, 49, 3031. doi:10.1039/c3cc40832a |
© 2016 Chen et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)