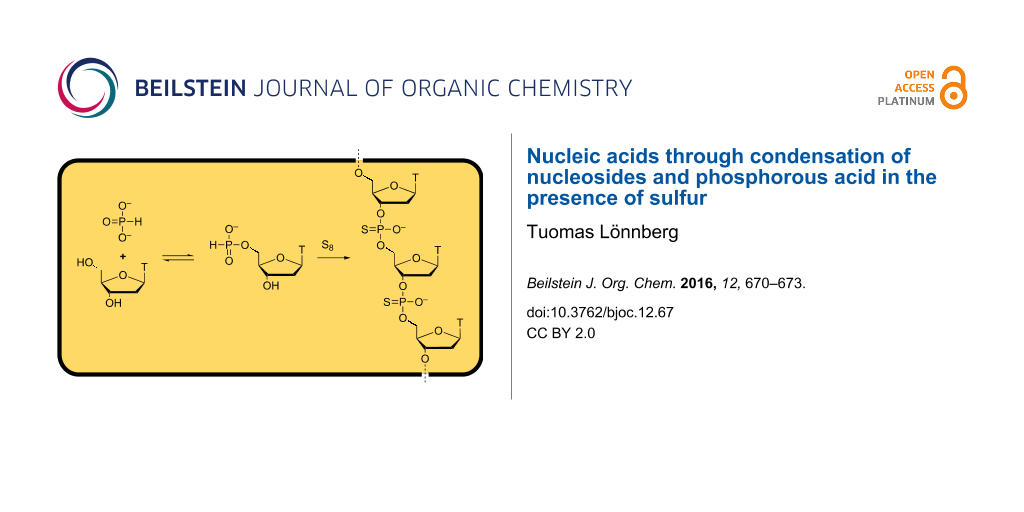
Abstract
Short phosphorothioate oligonucleotides have been prepared by refluxing an equimolar mixture of thymidine and triethylammonium phosphite in toluene in the presence of elemental sulfur. Desulfurization and subsequent digestion of the products by P1 nuclease revealed that nearly 80% of the internucleosidic linkages thus formed were of the canonical 3´,5´-type.

Graphical Abstract
Introduction
Arguably the most crucial step in the origin of life was the prebiotic formation of information carrying polymers. For the polymers playing this role in contemporary biochemistry, i.e., DNA and RNA, such a process appears difficult owing to the low reactivity and solubility of phosphate. Indeed, all of the enzyme-free nucleic acid polymerizations described so far have utilized activated starting materials, such as cyclic phosphates [1,2] or phosphoroimidazolides [3,4].
The trivalent phosphorus atom of phosphorous acid is much more susceptible to a nucleophilic attack than the pentavalent phosphorus atom of phosphoric acid [5]. Furthermore, phosphite salts are up to 1000-fold more soluble in water than phosphate salts [6]. For these reasons, compounds of reduced phosphorus (i.e., phosphorus at oxidation state lower than +5) were first proposed to have played a role in prebiotic phosphorylation reactions as early as 1955 [7]. Since then, both terrestrial [8] and extraterrestrial [9] sources of reduced phosphorus have been identified. Recent studies suggest the presence of significant amounts of phosphite in the Archean ocean, lending support to the idea of prebiotic phosphite chemistry [10,11].
Monoesters of phosphorous acid are hydrolytically stable compounds that are readily formed upon concentration of aqueous solutions of alcohols and phosphite salts [12]. The monoesters may react further to H-phosphonate diesters but this is an equilibrium process that under aqueous conditions favors the starting materials [13]. Oxidation of the H-phosphonate diester products, however, converts them to the stable phosphodiester counterparts. It is interesting to note that this reaction is faster for H-phosphonate diesters than for the respective monoesters or phosphorous acid itself [12], providing the driving force for polymerization. Proposed oxidants for prebiotic phosphite chemistry include H2O2 and Fe(III).
The present paper describes the polymerization of thymidine and triethylammonium phosphite, with elemental sulfur acting as the oxidant. Up to pentameric oligonucleotides with internucleosidic phosphorothioate linkages of predominantly 3´,5´-regiochemistry were formed by this method. Elemental sulfur may have been abundant on primitive Earth but its availability is limited by its poor solubility. In the present study this problem was addressed by carrying out the model reaction in toluene. On primitive Earth, solubilization by micelles [14,15] or a prebiotic oil slick [16] appears a more plausible scenario [17].
Results and Discussion
Preparation of phosphorothioate oligonucleotides
Equimolar amounts of thymidine and aq triethylammonium phosphite and a fourfold excess of S8 were suspended in toluene. The mixture was refluxed at 130 °C in a Dean–Stark apparatus for 90 h, after which it was evaporated to dryness. The brown glassy residue was suspended in water and the resulting mixture filtered. A sample of the filtrate was evaporated to dryness and the residue analyzed by 31P NMR (Figure 1). According to the 31P NMR spectrum, approximately 18% of the phosphite starting material had been converted to diverse phosphorothioate products.
![[1860-5397-12-67-1]](/bjoc/content/figures/1860-5397-12-67-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 31P NMR spectrum (162 MHz, D2O) of the crude product mixture after refluxing equimolar amounts of thymidine and triethylammonium phosphite and 4 equiv of S8 in toluene for 90 h.
Figure 1: 31P NMR spectrum (162 MHz, D2O) of the crude product mixture after refluxing equimolar amounts of t...
The product mixture was fractioned first by ion-exchange (IE) and then by reversed-phase (RP) HPLC (Figure 2, RP chromatograms presented in Supporting Information File 1). Electrospray ionization mass spectrometric (ESIMS) analysis of the collected fractions suggested the main products to be short oligonucleotides with internucleosidic phosphorothioate linkages. As could be expected, retention times in the IE HPLC correlated with the number of the negatively charged phosphorus-containing groups (Figure 2). While the shortest oligonucleotides (dimers and trimers) predominated in the mixture, up to pentameric products could be identified.
![[1860-5397-12-67-2]](/bjoc/content/figures/1860-5397-12-67-2.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 2: IE HPLC trace of the crude product mixture after refluxing equimolar amounts of thymidine and triethylammonium phosphite and 4 equiv of S8 in toluene for 90 h; Dionex DNASwiftTM column (150 × 5 mm, monolithic); flow rate = 1.5 mL min−1; linear gradient (0 to 50% over 25 min) of 330 mM NaClO4 in 20 mM TRIS buffer (pH 7.0).
Figure 2: IE HPLC trace of the crude product mixture after refluxing equimolar amounts of thymidine and triet...
All of the expected phosphorothioate oligonucleotides were accompanied by slower eluting (in IE HPLC) products exhibiting molecular weights 80 or 160 Da higher than the parent oligonucleotide. Similar derivatives of monomeric thymidine were also detected in the fastest eluting fractions. The 80 Da difference in molecular weight could arise from capping of a free hydroxy function by a phosphate or an H-phosphonothioate group and distinguishing between these two alternatives on the basis of MS alone is challenging. To verify the structure of the capping groups, a sample of the most abundant product, comprising two thymidines and two phosphorus atoms, was analyzed by 31P NMR. Phosphorothioate and H-phosphonothioate signals accounted for approximately 90% of the overall intensity (data presented in Supporting Information File 1), suggesting the product to be a bis(thymidinyl)phosphorothioate diester with one of the free hydroxy functions capped by an H-phosphonothioate group (Figure 3). It seems likely that also the other capped oligonucleotides had H-phosphonothioate, rather than phosphate termini. Evidently sulfurization and esterification of H-phosphonate monoesters compete under the experimental conditions.
![[1860-5397-12-67-3]](/bjoc/content/figures/1860-5397-12-67-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Possible structures of the most abundant product.
Figure 3: Possible structures of the most abundant product.
Determination of the regiochemistry of the phosphorothioate linkages
To establish the regiochemistry of the phosphorothioate linkages formed, a purified sample consisting of a mixture of tetrameric products (Figure 4A) was first desulfurized by treatment with iodine in aq pyridine (Figure 4B). According to a previous report, phosphodiester linkages are stable under these conditions [18]. The resulting phosphate-linked oligonucleotides were then subjected to digestion by P1 nuclease (Figure 4C). Thymidine and thymidine-5´-monophosphate accounted for approximately 78% of the final product mixture. As cleavage by P1 nuclease is limited to 3´,5´-phosphodiester linkages of single-stranded DNA or RNA, the results of the digestion experiment indicate that nearly 80% of the internucleosidic linkages formed upon condensation of thymidine and triethylammonium phosphite in the presence of sulfur had the natural 3´,5´-regiochemistry. The most likely explanation for this regioselectivity is the faster phosphitylation of the primary 5´-hydroxy function [12] – most of the thymidine starting material is first converted to thymidine-5´-H-phosphonate that subsequently polymerizes (Scheme 1).
![[1860-5397-12-67-4]](/bjoc/content/figures/1860-5397-12-67-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: IE HPLC traces of (A) a mixture of tetrameric products, (B) the product mixture after desulfurization by iodine and (C) the product mixture after P1 nuclease digestion of the desulfurized material; Dionex DNASwiftTM column (150 × 5 mm, monolithic); flow rate = 1.5 mL min−1; linear gradient (2 to 35% over 20 min) of 330 mM NaClO4 in 20 mM TRIS buffer (pH 7.0). The tall peak eluting at 1.5 min in chromatogram C corresponds to either the enzyme itself or an impurity in the commercial preparation.
Figure 4: IE HPLC traces of (A) a mixture of tetrameric products, (B) the product mixture after desulfurizati...
![[1860-5397-12-67-i1]](/bjoc/content/inline/1860-5397-12-67-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Phosphitylation and subsequent dimerization of thymidine.
Scheme 1: Phosphitylation and subsequent dimerization of thymidine.
Conclusion
Under dehydrating conditions and in the presence of sulfur, thymidine and triethylammonium phosphite condense into oligonucleotides with internucleosidic phosphorothioate linkages. Nearly 80% of these linkages have the natural 3´,5´-regiochemistry. Together with the recent evidence of phosphite in the Archean ocean, these results lend support to the hypothesis that phosphorous acid and its salts may have played a key role in the prebiotic synthesis of nucleic acids.
Supporting Information
| Supporting Information File 1: Experimental methods, HPLC chromatograms and mass spectra of the oligomerization products. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Costanzo, G.; Pino, S.; Ciciriello, F.; Di Mauro, E. J. Biol. Chem. 2009, 284, 33206–33216. doi:10.1074/jbc.M109.041905
Return to citation in text: [1] -
Tohidi, M.; Orgel, L. E. J. Mol. Evol. 1990, 30, 97–103. doi:10.1007/BF02099935
Return to citation in text: [1] -
Blain, J. C.; Ricardo, A.; Szostak, J. W. J. Am. Chem. Soc. 2014, 136, 2033–2039. doi:10.1021/ja411950n
Return to citation in text: [1] -
Huang, W.; Ferris, J. P. Chem. Commun. 2003, 1458–1459. doi:10.1039/B303134A
Return to citation in text: [1] -
Stawinski, J.; Kraszewski, A. Acc. Chem. Res. 2002, 35, 952–960. doi:10.1021/ar010049p
Return to citation in text: [1] -
Glindemann, D.; de Graaf, R. M.; Schwartz, A. W. Origins Life Evol. Biosphere 1999, 29, 555–561. doi:10.1023/A:1006622900660
Return to citation in text: [1] -
Gulick, A. Am. Sci. 1955, 43, 479–489.
Return to citation in text: [1] -
de Graaf, R. M.; Schwartz, A. W. Origins Life Evol. Biosphere 2000, 30, 405–410. doi:10.1023/A:1006700512902
Return to citation in text: [1] -
Pasek, M. A.; Lauretta, D. S. Astrobiology 2005, 5, 515–535. doi:10.1089/ast.2005.5.515
Return to citation in text: [1] -
Pasek, M. A.; Harnmeijer, J. P.; Buick, R.; Gull, M.; Atlas, Z. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10089–10094. doi:10.1073/pnas.1303904110
Return to citation in text: [1] -
Pasek, M.; Herschy, B.; Kee, T. P. Origins Life Evol. Biosphere 2015, 45, 207–218. doi:10.1007/s11084-015-9420-y
Return to citation in text: [1] -
Graaf, R. M.; Schwartz, A. Origins Life Evol. Biosphere 2005, 35, 1–10. doi:10.1007/s11084-005-0093-9
Return to citation in text: [1] [2] [3] -
Peyser, J. R.; Ferris, J. P. Origins Life Evol. Biosphere 2001, 31, 363–380. doi:10.1023/A:1011871726600
Return to citation in text: [1] -
Steudel, R.; Holdt, G. Angew. Chem., Int. Ed. Engl. 1988, 27, 1358–1359. doi:10.1002/anie.198813581
Return to citation in text: [1] -
Deamer, D. W.; Pashley, R. M. Origins Life Evol. Biosphere 1989, 19, 21–38. doi:10.1007/BF01808285
Return to citation in text: [1] -
Lasaga, A. C.; Holland, H. D.; Dwyer, M. J. Science 1971, 174, 53–55. doi:10.1126/science.174.4004.53
Return to citation in text: [1] -
Cleaves, H. J.; Miller, S. L. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7260–7263. doi:10.1073/pnas.95.13.7260
Return to citation in text: [1] -
Connolly, B. A.; Potter, B. V. L.; Eckstein, F.; Pingoud, A.; Grotjahn, L. Biochemistry 1984, 23, 3443–3453. doi:10.1021/bi00310a010
Return to citation in text: [1]
| 1. | Costanzo, G.; Pino, S.; Ciciriello, F.; Di Mauro, E. J. Biol. Chem. 2009, 284, 33206–33216. doi:10.1074/jbc.M109.041905 |
| 2. | Tohidi, M.; Orgel, L. E. J. Mol. Evol. 1990, 30, 97–103. doi:10.1007/BF02099935 |
| 18. | Connolly, B. A.; Potter, B. V. L.; Eckstein, F.; Pingoud, A.; Grotjahn, L. Biochemistry 1984, 23, 3443–3453. doi:10.1021/bi00310a010 |
| 6. | Glindemann, D.; de Graaf, R. M.; Schwartz, A. W. Origins Life Evol. Biosphere 1999, 29, 555–561. doi:10.1023/A:1006622900660 |
| 12. | Graaf, R. M.; Schwartz, A. Origins Life Evol. Biosphere 2005, 35, 1–10. doi:10.1007/s11084-005-0093-9 |
| 5. | Stawinski, J.; Kraszewski, A. Acc. Chem. Res. 2002, 35, 952–960. doi:10.1021/ar010049p |
| 16. | Lasaga, A. C.; Holland, H. D.; Dwyer, M. J. Science 1971, 174, 53–55. doi:10.1126/science.174.4004.53 |
| 3. | Blain, J. C.; Ricardo, A.; Szostak, J. W. J. Am. Chem. Soc. 2014, 136, 2033–2039. doi:10.1021/ja411950n |
| 4. | Huang, W.; Ferris, J. P. Chem. Commun. 2003, 1458–1459. doi:10.1039/B303134A |
| 17. | Cleaves, H. J.; Miller, S. L. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7260–7263. doi:10.1073/pnas.95.13.7260 |
| 12. | Graaf, R. M.; Schwartz, A. Origins Life Evol. Biosphere 2005, 35, 1–10. doi:10.1007/s11084-005-0093-9 |
| 12. | Graaf, R. M.; Schwartz, A. Origins Life Evol. Biosphere 2005, 35, 1–10. doi:10.1007/s11084-005-0093-9 |
| 10. | Pasek, M. A.; Harnmeijer, J. P.; Buick, R.; Gull, M.; Atlas, Z. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10089–10094. doi:10.1073/pnas.1303904110 |
| 11. | Pasek, M.; Herschy, B.; Kee, T. P. Origins Life Evol. Biosphere 2015, 45, 207–218. doi:10.1007/s11084-015-9420-y |
| 14. | Steudel, R.; Holdt, G. Angew. Chem., Int. Ed. Engl. 1988, 27, 1358–1359. doi:10.1002/anie.198813581 |
| 15. | Deamer, D. W.; Pashley, R. M. Origins Life Evol. Biosphere 1989, 19, 21–38. doi:10.1007/BF01808285 |
| 9. | Pasek, M. A.; Lauretta, D. S. Astrobiology 2005, 5, 515–535. doi:10.1089/ast.2005.5.515 |
| 8. | de Graaf, R. M.; Schwartz, A. W. Origins Life Evol. Biosphere 2000, 30, 405–410. doi:10.1023/A:1006700512902 |
| 13. | Peyser, J. R.; Ferris, J. P. Origins Life Evol. Biosphere 2001, 31, 363–380. doi:10.1023/A:1011871726600 |
© 2016 Lönnberg; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)