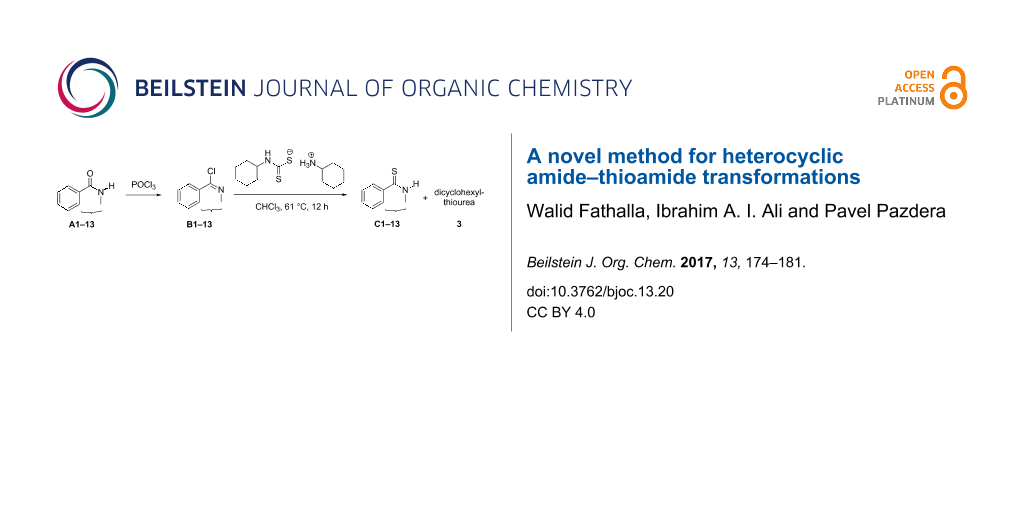
Abstract
In this paper, we introduce a novel and convenient method for the transformation of heterocyclic amides into heteocyclic thioamides. A two-step approach was applied for this transformation: Firstly, we applied a chlorination of the heterocyclic amides to afford the corresponding chloroheterocycles. Secondly, the chloroherocycles and N-cyclohexyl dithiocarbamate cyclohexylammonium salt were heated in chloroform for 12 h at 61 °C to afford heteocyclic thioamides in excellent yields.

Graphical Abstract
Introduction
Transforming heterocyclic amides into thioamides is an important task in organic synthesis. Earlier reports for this type of O/S conversions were achieved by several thiating reagents; for instance, Lawesson's reagent (2,4-bis(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetane 2,4-disulfide) [1-3], Berzelius reagent [4-6] (P4S10), and phosphorus pentasulfide [7] in dry toluene, xylene or pyridine under reflux conditions. A two-step approach for the purpose of thiation of heterocyclic amides attracted our attention: as a first step, we applied a chlorination of heterocyclic amides, followed by thiation via reaction with thiourea on the basis of reagent-promoted desulfurylation of isothiourea under strong basic conditions [8,9]. Aiming to continue our reseach work on the structure modification of functionalized heterocyclic amides and thioamides [10-17], we found it interesting to design a new convenient and simple method for the thiation of heterocyclic amides.
Results and Discussion
Many synthetic methods related to thiation of heterocyclic amides have been reported to date. Most methods suffer from the employment of expensive specific reagents, high temperature, use of strong basic conditions, ultra-dry solvents, bad smell, low yield, difficulties in work-up procedures or from a narrow substrate scope. Therefore, the development of a more efficient method for the transformation of heterocyclic amides to heterocyclic thioamides gained great attention.
The reaction of three molar equivalents of cyclohexylamine (1) with one molar equivalent of carbon disulfide in water typically afforded N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) as an excellent new thiating reagent in high yield, Scheme 1.
![[1860-5397-13-20-i1]](/bjoc/content/inline/1860-5397-13-20-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2).
Scheme 1: Synthesis of N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2).
The structure assignment of the prepared N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) is based on 1H and 13C NMR spectral and physicochemical analysis. The 1H NMR spectrum displays a broad singlet signal at 8.01 ppm associated with three NH protons. The 1H NMR spectrum also shows three multiplet signals at 4.15–3.95 and 3.05–2.96 and 1.98–0.96 ppm corresponding to two CH and 10 CH2 groups, respectively. The 13C NMR spectrum of 2 displays signals at δ 212.4, 55.3 and 50.0 ppm associated with (C=S) and two CH groups, respectively. The 13C NMR spectrum of 2 also shows signals at 32.3, 30.9, 25.8, 25.5, 25.1, and 24.3 ppm due to cyclohexyl CH2 groups.
Heterocyclic amides A1–13 used in this context were prepared as described in literature expanding simple one-step procedures to multi-step sequential reactions. Quinazoline-4-one (A1) [18] was prepared by Niementowski reaction by fusion of anthranilic acid with formamide at 120 °C for 5 h. A number of quinazoline derivatives A2–A6 [19-21] were prepared via sequential steps starting from easily available carboxylic acid chlorides. The acid chlorides reacted with anthranilic acid to afford benzoxazines, followed by sequential reaction with ammonia to afford the benzanilide derivatives and finally, benzanilides were cyclized by heating in sodium hydroxide solution and gave quinazolines A2–A6. Methyl 1,2-dihydro-2-oxoquinoline-4-carboxylate (A9) [22,23] was prepared by heating isatine with malonic acid followed by esterification of the produced quinoline carboxylic acid with methanol in the presence of sulfuric acid at 80 °C for 6 h. 4-Arylphthalazin-1(2H)-ones A7 and A8 [24,25] were prepared by Friedel–Crafts acylation reaction of N-aminophthalimide with either benzene or toluene in the presence of AlCl3, respectively. A number of quinoxalin-2-one derivatives A10–13 [26-29] were prepared by the reaction of o-phenylenediamine with oxoacids or oxoesters either in HCl solution or in ethanol.
Heterocyclic amides A1–9 were heated with POCl3 for 2–5 h as reported in literature to afford the respective chloroheterocycles [30-37] B1–9 and 13 and were purified using flash column chromatography; petroleum ether (60–80)/ethyl acetate (9:1) as an eluent. Best results for the preparation of chloroquinoxalinones B11 and B12 [38,39] were achieved by dropwise addition of N,N-dimethylaniline to a stirred cold solution of quinoxalinones A11 and A12 and POCl3, the reaction mixture was refluxed for 15 minutes.
Thus, N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) was added to 4-chloro-2-phenylquinazoline (B2) solution in CHCl3, the reaction mixture was heated at 61 °C for 12 h. The reaction mixture was evaporated and poured in ethanol to give bright yellow crystals as only isolated product, identified as 2-phenylquinazoline-4(3H)-thione (C2). The filtrate was once again evaporated and crystalized from ethanol/water to give dicyclohexylthiourea (3, Scheme 2). We have extended the scope of this process to involve the transformation of a number of heterocyclic amides; quinazolin-4(3H)-one (A1), 2-substituted quinazolin-4(3H)-one A3–A6 and 4-subsituted phthalazin-1(2H)-ones A7 and A8 into the corresponding heterocyclic thioamides C1 and C3–C8, respectively (Scheme 2, Table 1 and Table 2).
![[1860-5397-13-20-i2]](/bjoc/content/inline/1860-5397-13-20-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The two-step thiation of quinazolin-4-one A1–6 and phthalazin-1-ones A7 and A8.
Scheme 2: The two-step thiation of quinazolin-4-one A1–6 and phthalazin-1-ones A7 and A8.
Table 1: Synthesis of quinazolin-4-thionesa.
| No. |
heterocyclic
amide A |
chloro-
heterocycles B |
heterocyclic
thioamide C |
Yieldb % |
|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-20-i5.svg?max-width=637&scale=1.0)
A1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-20-i6.svg?max-width=637&scale=1.0)
B1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-20-i7.svg?max-width=637&scale=1.0)
C1 |
76% |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-20-i8.svg?max-width=637&scale=1.0)
A2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-20-i9.svg?max-width=637&scale=1.0)
B2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-20-i10.svg?max-width=637&scale=1.0)
C2 |
92% |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-20-i11.svg?max-width=637&scale=1.0)
A3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-20-i12.svg?max-width=637&scale=1.0)
B3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-20-i13.svg?max-width=637&scale=1.0)
C3 |
84% |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-20-i14.svg?max-width=637&scale=1.0)
A4 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-20-i15.svg?max-width=637&scale=1.0)
B4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-20-i16.svg?max-width=637&scale=1.0)
C4 |
89% |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-20-i17.svg?max-width=637&scale=1.0)
A5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-20-i18.svg?max-width=637&scale=1.0)
B5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-20-i19.svg?max-width=637&scale=1.0)
C5 |
95% |
| 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-20-i20.svg?max-width=637&scale=1.0)
A6 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-20-i21.svg?max-width=637&scale=1.0)
B6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-20-i22.svg?max-width=637&scale=1.0)
C6 |
81% |
aReaction conditions: chloroheterocycles (20 mmol) and N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2, 20 mmol) were heated in CHCl3 (25 mL) at 61 °C for 12 h. bYields refer to isolated pure product of the reaction from B to C.
Table 2: Synthesis of phthalizin-1-thiones C7 and C8a.
| No. |
heterocyclic
amide A |
chloro-
heterocycles B |
heterocyclic
thioamide C |
Yieldb % |
|---|---|---|---|---|
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-20-i23.svg?max-width=637&scale=1.0)
A7 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-20-i24.svg?max-width=637&scale=1.0)
B7 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-20-i25.svg?max-width=637&scale=1.0)
C7 |
91% |
| 8 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-20-i26.svg?max-width=637&scale=1.0)
A8 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-20-i27.svg?max-width=637&scale=1.0)
B8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-13-20-i28.svg?max-width=637&scale=1.0)
C8 |
78% |
aReaction conditions as described before. bYields refer to isolated pure product of the reaction from B to C.
The N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) has been found to be an excellent reagent for thiation of heterocyclic amides into thioamides at position 4, Scheme 2, Table 1 and Table 2. We have extended the scope of this thiation process to involve heterocyclic amides at positions 2 and 3. Thus, methyl 1,2-dihydro-2-oxoquinoline-4-carboxylate (A9) and 3-substituted quinoxalin-2(1H)-ones A10–13 reacted similarly with phosphorous oxychloride to afford the chloro derivatives B9–13 which were subsequently converted into the corresponding thioamides C9–13 by the reaction with N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) in CHCl3 under reflux conditions for 12 h (Scheme 3, Table 3).
![[1860-5397-13-20-i3]](/bjoc/content/inline/1860-5397-13-20-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Thiation of quinoline A9 and quinoxalinone A10–13.
Scheme 3: Thiation of quinoline A9 and quinoxalinone A10–13.
Table 3: Synthesis of quinolin-2-thiones C9 and quinoxalin-2-thiones C10–C13a.
| No. |
heterocyclic
amide A |
chloro-
heterocycles B |
heterocyclic
thioamide C |
Yieldb % |
|---|---|---|---|---|
| 9 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-13-20-i29.svg?max-width=637&scale=1.0)
A9 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-13-20-i30.svg?max-width=637&scale=1.0)
B9 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-13-20-i31.svg?max-width=637&scale=1.0)
C9 |
76% |
| 10 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-13-20-i32.svg?max-width=637&scale=1.0)
A10 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-13-20-i33.svg?max-width=637&scale=1.0)
B10 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-13-20-i34.svg?max-width=637&scale=1.0)
C10 |
69% |
| 11 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-13-20-i35.svg?max-width=637&scale=1.0)
A11 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-13-20-i36.svg?max-width=637&scale=1.0)
B11 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-13-20-i37.svg?max-width=637&scale=1.0)
C11 |
83% |
| 12 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-13-20-i38.svg?max-width=637&scale=1.0)
A12 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-13-20-i39.svg?max-width=637&scale=1.0)
B12 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-13-20-i40.svg?max-width=637&scale=1.0)
C12 |
72% |
| 13 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-13-20-i41.svg?max-width=637&scale=1.0)
A13 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-13-20-i42.svg?max-width=637&scale=1.0)
B13 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-13-20-i43.svg?max-width=637&scale=1.0)
C13 |
91% |
aReaction conditions as described before. bYields refers to isolated pure product of the reaction from B to C.
The synthetic procedure for the formation of C1–13 reported herein have the advantage of operational simplicity and availability of both the substrate and the reagents giving a series of very interesting compounds. This method also was adjusted to involve a one-pot strategy starting from heterocyclic amides A1–13 to directly afford the heterocyclic thioamides C1–13. Thus, 2-phenylquinazolin-4(3H)-one (A2) was heated with phosphorous oxychloride for 2 h. The reaction mixture was evaporated and poured in ice-cold ammonia solution, then extracted with chloroform and dried over sodium sulfate. N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) was added to the chloroform solution of chloroquinazoline B2 and heated at 61 °C for 12 h. The reaction mixture was evapourated and ethanol was added successively to give the desired product C2.
The structure assignment of the prepared heterocyclic thioamides C1–13 is based on 1H and 13C NMR spectral and physicochemical analyses. The 1H NMR spectrum of 2-(4-methoxyphenyl)quinazoline-4(3H)-thione (C5) gave a broad singlet and a singlet signal at δ 13.71 and 3.87 ppm, associated with NH and OCH3 groups, respectively. The significant downfield shift of the NH proton is probably due to intermolecular hydrogen bond interactions of the type NH···S=C. All the isolated thioureas C1–13 exhibited similar 1H NMR spectral patterns with the NH protons at similar chemical shifts and they adopt paired thioamide structures (vide infra). The 1H NMR spectrum also shows four doublet and two triplet signals at δ 8.60, 8.19, 7.75, 7.11, 7.88, 7.56, respectively due to eight aromatic protons. The 13C NMR spectrum of C5 displays signals at δ 187.9 and 56.0 ppm due to C=S and OCH3, respectively.
A mechanistic rationalization for this interesting rearrangement is given in Scheme 4. The reaction of 4-chloro-2-phenylquinazoline (B2) with N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2) in CHCl3 at 61 °C for 12 h was principally expected to give 2-phenylquinazolin-4-yl cyclohexylcarbamodithioate (I) and cyclohexylamine hydrochloride. Cyclohexylamine hydrochloride under heating conditions will eliminate an HCl molecule forming the free cyclohexylamine base.
![[1860-5397-13-20-i4]](/bjoc/content/inline/1860-5397-13-20-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Rational mechanism of the reaction of 4-chloro-2-phenylquinazoline (B2) to 2-phenylquinazolin-4(3H)-thione.
Scheme 4: Rational mechanism of the reaction of 4-chloro-2-phenylquinazoline (B2) to 2-phenylquinazolin-4(3H)...
Cyclohexylamine will further abstract a proton from I followed by electron delocalization and the overall formation of cyclohexyl isothiocyanate (4) via C–S bond cleavage and the formation of quinazoline thiol anion II having a negative charge concerted on the nitrogen atom. The protonated cyclohexylamine in the previous step will transfer this extra proton to II to afford the quinazoline thione C2. On the other hand the free cyclohexylamine will add to cyclohexyl isothiocyanate (4) to form the thiourea 3. Similar results were obtained by Furumoto [40], and Sun [41] reported the application of cyanuric chloride (2,4,6-trichloro-1,3,5-triazine, TCT) as a desulfurylation reagent in the synthesis of carbodiimides or alkyl isothiocyanates from thioureas under mild conditions.
Conclusion
Several synthetic procedures related to thiation of heterocyclic amides have been reported to date. The drawback of the existing methods is the use of expensive specific reagents, high temperature, use of strong basic conditions, ultra-dry solvents, bad smell, low yield, difficulties in work-up procedures or from a narrow substrate scope. In this work, we successfully developed a facile and convenient general method for the transformation of heterocyclic amides into heterocyclic thioamides. Generally, in the proposed technique we transformed heterocyclic amides to chloroheterocyclic compounds by the action of phosphorous oxychloride. Subsequently, chloroheterocyclic derivatives reacted with N-cyclohexyl dithiocarbamate cyclohexylammonium salt in chloroform at 61 °C for 12 h to finally afford the heterocyclic thioamides in excellent yields. Furthermore, this method is advantageous over existing methods in the matter of simplicity of the work-up procedure, higher yield, odorless, lower reaction temperature and finally the availability of both precursors and reagent.
Experimental
General procedures
Solvents were purified and dried by standard procedures. The boiling range of the petroleum ether used was 40–60 °C. Thin-layer chromatography (TLC): silica gel 60 F254 plastic plates (E. Merck, layer thickness 0.2 mm) detected by UV absorption. Elemental analyses were performed on a Flash EA-1112 instrument at the Microanalytical laboratory, Faculty of Science, Suez Canal University, Ismailia, Egypt. Melting points were determined on a Büchi 510 melting-point apparatus and the values are uncorrected. 1H and 13C NMR spectra were recorded at 300 MHz and 75.5 MHz, respectively (Bruker AC 300) in CDCl3 and DMSO solution with tetramethylsilane as an internal standard. The NMR analyses were performed at the Organic Chemistry Department Masaryk University, Brno, Czech Republic. Compounds A1–13 and B1–13 were obtained by published methods [18-39], and their melting points and 1H and 13C NMR spectra corresponded to those given in the literature.
General method for the preparation of thiating reagent N-cyclohexyl dithiocarbamate cyclohexylammonium salt (2). To a mixture of freshly distilled cyclohexylamine (60 mmol) and water (50 mL) was added carbon disulfide (21 mmol) dropwise. The reaction mixture was stirred at room temperature for 2 h. The white solid obtained was filtered, washed with water, dried and crystalized from ethanol to provide the pure product. Yield 98% (ethanol 95%) white crystals, mp 188–189 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.01 (bs, 3H, 3NH), 4.15–3.95 (m, 1H, CH), 3.05–2.96 (m, 1H, CH), 1.98–0.96 (20H, m, 10CH2); 13C NMR (75.0 MHz, DMSO-d6) δ 212.4 (C=S), 55.3 (CH), 50.0 (CH), 32.3 (2CH2), 30.9 (2CH2), 25.8 (CH2), 25.5 (2CH2), 25.1 (CH2), 24.3 (2CH2); anal. calcd for C13H26N2S2 (274.2): C, 56.56; H, 9.43; N, 10.09; found: C, 56.88; H, 9.55; N, 10.21.
General method for the preparation of heterocyclic thioamides
Method A. To a solution of chloroheterocycles (2.5 mmol) in CHCl3 (25 mL) was added (0.69 g, 2.5 mmol) of N-cyclohexyl dithiocarbamate cyclohexylammonium salt. The reaction mixture was refluxed at 61 °C for 12 h. The reaction mixture was evaporated under reduced pressure and 25 mL of ethanol was added to the solid residue. The yellowish–orange precipitate was filtered to give the desired product. The crude compounds were pure enough for analytical purposes. Purification of products for analysis was achieved by crystallization from the appropriate solvent; chromatographed with the appropriate eluent or by repeated dissolution in KOH and reprecipitation by acetic acid. The filtrate was evaporated once again and the solid obtained was crystalized from ethanol water to give symmetrical dicyclohexylthiourea (3).
Method B. To a cold solution of heterocyclic amide (2.5 mmol) in POCl3 (25 mL) was added dimethylaniline (2.5 mmol). The reaction mixture was stirred under reflux (100–105 °C) for 1.5–2 h. The excess POCl3 was removed under reduced pressure. The residue was poured into a mixture of chloroform (50 mL), ice water (80 mL) and ammonia (5 mL). The chloroform layer was separated, dried over Na2SO4 and filtered. To this chloroform solution of the in situ generated chloroheterocycles was added (0.69 g, 2.5 mmol) of N-cyclohexyl dithiocarbamate cyclohexylammonium salt. The reaction mixture was refluxed at 61 °C for 12 h. The reaction mixture was evaporated under reduced pressure and 25 mL of ethanol was added to the solid residue. The yellowish–orange precipitate was filtered to give the desired product. The crude compounds were pure enough for analytical purposes. Purification of products for analysis was achieved by crystallization from the appropriate solvent; chromatographed with the appropriate eluent or by repeated dissolution in KOH and reprecipitation by acetic acid.
Dicyclohexylthiourea (3) [42]: Yield 65% (ethanol 95%–H2O) white crystals, mp 180–181 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.05 (bs, 2H, NH), 4.05–3.89 (m, 2H, 2CH), 1.87–1.52 (m, 10H, 5CH2) 1.29–1.12 (m, 10H, 5CH2); 13C NMR (75.0 MHz, DMSO-d6) δ 180.5 (C=S), 51.9 (CH), 32.8 (2CH2), 25.7 (2CH2), 25.0 (CH2); anal. calcd for C13H24N2S (240.2): C, 64.95; H, 10.06; N, 11.65; found: C, 64.82; H, 10.01; N, 11.46.
Quinazoline-4(3H)-thione (C1) [43]: Yield 76% (H2O) yellow crystals, mp 320–321 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.83 (bs, 1H, NH), 8.55–7.28 (m, 5H, ArH); 13C NMR (75.0 MHz, DMSO-d6) δ 186.2 (C=S), 144.8 (C Ar), 144.2 (CHAr), 135.7 (CHAr), 129.7 (CHAr), 129.4 (C Ar), 128.7 (CHAr), 128.5 (CHAr); anal. calcd for C8H6N2S (162.0): C, 59.23; H, 3.73; N, 17.27; found: C, 59.17; H, 3.69; N, 17.15.
2-Phenylquinazoline-4(3H)-thione (C2) [44]: Yield 92% (ethanol 95%–DMF) yellow crystals, mp 222–223 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.75 (bs, 1H, NH), 8.63 (d, J = 8.0 Hz, 1H, ArH), 8.17 (d, J = 8.0 Hz, 2H, ArH), 7.93–7.89 (m, 3H, ArH), 7.82–7.57 (m, 3H, ArH); 13C NMR (75.0 MHz, DMSO-d6) δ 188.5 (C=S), 152.1 (C Ar), 144.8 (C Ar), 135.8 (CHAr), 132.8 (C Ar), 131.9 (CHAr), 129.8 (CHAr), 128.9 (CHAr), 128.6 (CHAr), 128.4 (CHAr), 128.1 (C Ar); anal. calcd for C14H10N2S (238.1): C, 70.56; H, 4.23; N, 11.76; found: C, 70.48; H, 4.16; N, 11.49.
2-o-Tolylquinazoline-4(3H)-thione (C3) [45]: Yield 84% (ethanol 95%–DMF) yellow crystals, mp 183–184 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.97 (bs, 1H, NH), 8.65 (d, J = 8.0 Hz, 1H, ArH), 7.96 (t, J = 8.0 Hz, 1H, ArH), 7.75 (d, J = 8.0 Hz, 1H, ArH), 7.66–7.35 (m, 5H, ArH), 2.39 (s, 3H, CH3); 13C NMR (75.0 MHz, DMSO-d6) δ 187.7 (C=S), 155.5 (C Ar), 144.7 (C Ar), 136.8 (C Ar), 135.8 (CHAr), 134.0 (CHAr), 130.9 (C Ar), 130.6 (CHAr), 130.0 (CHAr), 129.7 (CHAr), 128.6 (CHAr), 128.0 (CHAr), 126.1 (C Ar), 19.9 (CH3); anal. calcd for C15H12N2S (252.1): C, 71.40; H, 4.79; N, 11.10; found: C, 71.21; H, 4.65; N, 10.94.
2-p-Tolylquinazoline-4(3H)-thione (C4) [46]: Yield 89% (ethanol 95%–DMF) yellow crystals, mp 218–219 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.78 (bs, 1H, NH), 8.62 (d, J = 8.0 Hz, 1H, ArH), 8.10 (d, J = 8.0 Hz, 2H, ArH), 7.93–7.76 (m, 2H, ArH), 7.58 (t, J = 8.0 Hz, 1H, ArH), 7.37 (d, J = 8.0 Hz, 2H, ArH), 2.41 (s, 3H, CH3); 13C NMR (75.0 MHz, DMSO-d6) δ 187.9 (C=S), 151.9 (C Ar), 149.3 (C Ar), 142.1 (C Ar), 135.9 (CHAr), 130.4 (C Ar), 129.8 (CHAr), 129.7 (CHAr), 129.6 (CHAr), 128.8 (CHAr), 126.3 (CHAr), 128.0 (CHAr), 126.4 (C Ar), 21.5 (CH3); anal. calcd for C15H12N2S (252.1): C, 71.40; H, 4.79; N, 11.10; found: C, 71.28; H, 4.61; N, 11.84.
The yield, 1H, 13C NMR spectral data and physicochemical analysis of other prepared thioamides (C5–C13) are presented in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Additional experimental and analytical data. | ||
| Format: PDF | Size: 234.9 KB | Download |
References
-
Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2007, 107, 5210. doi:10.1021/cr040650b
Return to citation in text: [1] -
Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2010, 110, 3419. doi:10.1021/cr900243d
Return to citation in text: [1] -
Curphey, T. J. J. Org. Chem. 2002, 67, 6461. doi:10.1021/jo0256742
Return to citation in text: [1] -
Libermann, R. Bull. Soc. Chim. Fr. 1959, 1793.
Return to citation in text: [1] -
Leonard, N. J.; Curtin, D. Y. J. Org. Chem. 1946, 11, 349. doi:10.1021/jo01174a007
Return to citation in text: [1] -
Armstrong, R. T. Vulcanization accelerators. U.S. Patent US2382769, Aug 14, 1945.
Return to citation in text: [1] -
Morrison, D. C.; Furst, A. J. Org. Chem. 1956, 21, 470. doi:10.1021/jo01110a026
Return to citation in text: [1] -
Asano, K. Yakugaku Zasshi 1958, 78, 729.
Return to citation in text: [1] -
El-Hawash, S. A. M.; Abdel Wahab, A. E. Arch. Pharm. 2006, 339, 437. doi:10.1002/ardp.200600012
Return to citation in text: [1] -
Fathalla, W.; Čajan, M.; Pazdera, P. Molecules 2000, 5, 1210. doi:10.3390/51201210
Return to citation in text: [1] -
Fathalla, W.; Čajan, M.; Pazdera, P. Molecules 2001, 6, 557. doi:10.3390/60600557
Return to citation in text: [1] -
Fathalla, W.; Pazdera, P.; Marek, J. J. Heterocycl. Chem. 2002, 39, 1139. doi:10.1002/jhet.5570390605
Return to citation in text: [1] -
Fathalla, W.; Marek, J.; Pazdera, P. J. Sulfur Chem. 2008, 29, 31. doi:10.1080/17415990701759685
Return to citation in text: [1] -
Fathalla, W. ARKIVOC 2008, xii, 245.
Return to citation in text: [1] -
Ali, I. A. I.; Fathalla, W. Heteroat. Chem. 2006, 17, 280. doi:10.1002/hc.20203
Return to citation in text: [1] -
Fathalla, W.; El Rayes, S.; Ali, I. A. I. ARKIVOC 2008, xiii, 179.
Return to citation in text: [1] -
Fathalla, W. Chem. Heterocycl. Compd. 2015, 51, 67. doi:10.1007/s10593-015-1661-1
Return to citation in text: [1] -
Yamamoto, Y. Methods of molecular hetarenes and related ring systems; Science of Synthesis: Houben-Weyl, Vol. 16; 2003; pp 726 ff.
Return to citation in text: [1] [2] -
Sundaram, R.; Yuvaraj, E.; Babu, G. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1999, 38, 905.
Return to citation in text: [1] [2] -
Lee, E. S.; Son, J. K.; Na, Y. H.; Jahng, Y. Heterocycl. Commun. 2004, 10, 325. doi:10.1515/HC.2004.10.4-5.325
Return to citation in text: [1] [2] -
Okada, K.; Sakuma, H.; Inoue, S. Chem. Lett. 1979, 8, 131. doi:10.1246/cl.1979.131
Return to citation in text: [1] [2] -
Chawla, H. M.; Gupta, T. J. Inclusion Phenom. Macrocyclic Chem. 2015, 81, 49. doi:10.1007/s10847-014-0432-4
Return to citation in text: [1] [2] -
Derbala, H. A. Y. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1994, 33, 779.
Return to citation in text: [1] [2] -
Ismail, M. F.; Kandile, N. G. Acta Chim. Hung. 1991, 128, 251.
Return to citation in text: [1] [2] -
Ajani, O. O.; Obafemi, C. A.; Ikpo, C. O.; Ogunniran, K. O.; Nwinyi, O. C. Chem. Heterocycl. Compd. 2009, 45, 1370. doi:10.1007/s10593-010-0435-z
Return to citation in text: [1] [2] -
Noolvi, M. N.; Patel, H. M.; Bhardwaj, V.; Chauhan, A. Eur. J. Med. Chem. 2011, 46, 2327. doi:10.1016/j.ejmech.2011.03.015
Return to citation in text: [1] [2] -
Mahesh, R.; Dhar, A. K.; Sasank, T. V. N. V. T.; Thirunavukkarasu, S.; Devadoss, T. Chin. Chem. Lett. 2011, 22, 389. doi:10.1016/j.cclet.2010.11.002
Return to citation in text: [1] [2] -
Elhelby, A. A.; Ayyad, R. R.; Zayed, M. F. Arzneim. Forsch. 2011, 61, 379. doi:10.1055/s-0031-1296214
Return to citation in text: [1] [2] -
Shi, L.; Wu, T.-T.; Wang, Z.; Xu, J.-Y.; Xu, Y.-G. Bioorg. Med. Chem. 2014, 22, 4735. doi:10.1016/j.bmc.2014.07.008
Return to citation in text: [1] [2] -
Marvania, B.; Lee, P.-C.; Chaniyara, R.; Dong, H.; Suman, S.; Kakadiya, R.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Bioorg. Med. Chem. 2011, 19, 1987. doi:10.1016/j.bmc.2011.01.055
Return to citation in text: [1] [2] -
Aly, A. A. J. Chin. Chem. Soc. 2007, 54, 437. doi:10.1002/jccs.200700061
Return to citation in text: [1] [2] -
Browner, M.; Clark, D.; Cushing, T.; Hao, X.; Hawley, R.; He, X.; Jaen, J.; Lebadie, S.; Smith, M.-L.; Talmas, F.; Walker, N.; Labelle, M. Antiinflammation agents. U.S. Pat. Appl. US 20020161004 A1, Oct 31, 2002.
Return to citation in text: [1] [2] -
Rival, Y.; Hoffmann, R.; Didier, B.; Rybaltchenko, V.; Bourguignon, J.; Wermuth, C. G. J. Med. Chem. 1998, 41, 311. doi:10.1021/jm9705418
Return to citation in text: [1] [2] -
Hemdan, M. M.; Taha, S. M.; Gabr, A. M.; Elkady, M. Y. J. Chem. Res. 2010, 34, 102. doi:10.3184/030823410X12658886079090
Return to citation in text: [1] [2] -
Galal, S. A.; Abdelsamie, A. S.; Tokuda, H.; Suzuki, N.; Lida, A.; ElHefnawi, M. M.; Ramadan, R. A.; Atta, M. H. E.; El Diwani, H. I. Eur. J. Med. Chem. 2011, 46, 327. doi:10.1016/j.ejmech.2010.11.022
Return to citation in text: [1] [2] -
Da Silva Miranda, F.; Signori, A. M.; Vicente, J.; De Souza, B.; Priebe, J. P.; Szpoganicz, B.; Gonçalves, N. S.; Neves, A. Tetrahedron 2008, 64, 5410. doi:10.1016/j.tet.2008.02.097
Return to citation in text: [1] [2] -
Aggarwal, R.; Masan, E.; Sumran, G. Synth. Commun. 2013, 43, 1842. doi:10.1080/00397911.2012.674168
Return to citation in text: [1] [2] -
Wagle, S.; Adhikari, A. V.; Kumari, N. S. Eur. J. Med. Chem. 2009, 44, 1135. doi:10.1016/j.ejmech.2008.06.006
Return to citation in text: [1] [2] -
Koshel, N. G.; Kovalev, E. G.; Postovskii, I. Ya. Chem. Heterocycl. Compd. 1970, 6, 791. doi:10.1007/BF00470545
Return to citation in text: [1] [2] -
Furumoto, S. Nippon Kagaku Zasshi 1971, 92, 1005. doi:10.1246/nikkashi1948.92.1005
Return to citation in text: [1] -
Sun, N.; Li, B.; Shao, J.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. Beilstein J. Org. Chem. 2012, 8, 61. doi:10.3762/bjoc.8.6
Return to citation in text: [1] -
Ramadas, K.; Janarthanan, N.; Velmathi, S. Synth. Commun. 1997, 27, 2255. doi:10.1080/00397919708003379
Return to citation in text: [1] -
Sánchez, A. I.; Martinez-Barrasa, V.; Burgos, C.; Vaquero, J. J.; Alvarez-Builla, J.; Terricabras, E.; Segarra, V. Bioorg. Med. Chem. 2013, 21, 2370. doi:10.1016/j.bmc.2013.01.067
Return to citation in text: [1] -
Lee, N. K.; Lee, J. W.; Lee, S.; Im, G.-J.; Han, H. Y.; Kim, T. K.; Kwak, W.-j.; Kim, S. W.; Ha, J.; Kim, E. K.; Lee, J. K.; Yoo, C. Y.; Lee, D. Y. Quinazoline derivatives for the treatment and prevention of diabetes and obesity. U.S. Pat. Appl. US20080207614 A1, Aug 28, 2008.
Return to citation in text: [1] -
Legrand, L. Bull. Soc. Chim. Fr. 1961, 620.
Return to citation in text: [1] -
Hanusek, J.; Hejtmánková, L.; Kubicová, L.; Sedlák, M. Molecules 2001, 6, 323. doi:10.3390/60400323
Return to citation in text: [1]
| 43. | Sánchez, A. I.; Martinez-Barrasa, V.; Burgos, C.; Vaquero, J. J.; Alvarez-Builla, J.; Terricabras, E.; Segarra, V. Bioorg. Med. Chem. 2013, 21, 2370. doi:10.1016/j.bmc.2013.01.067 |
| 44. | Lee, N. K.; Lee, J. W.; Lee, S.; Im, G.-J.; Han, H. Y.; Kim, T. K.; Kwak, W.-j.; Kim, S. W.; Ha, J.; Kim, E. K.; Lee, J. K.; Yoo, C. Y.; Lee, D. Y. Quinazoline derivatives for the treatment and prevention of diabetes and obesity. U.S. Pat. Appl. US20080207614 A1, Aug 28, 2008. |
| 1. | Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2007, 107, 5210. doi:10.1021/cr040650b |
| 2. | Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2010, 110, 3419. doi:10.1021/cr900243d |
| 3. | Curphey, T. J. J. Org. Chem. 2002, 67, 6461. doi:10.1021/jo0256742 |
| 10. | Fathalla, W.; Čajan, M.; Pazdera, P. Molecules 2000, 5, 1210. doi:10.3390/51201210 |
| 11. | Fathalla, W.; Čajan, M.; Pazdera, P. Molecules 2001, 6, 557. doi:10.3390/60600557 |
| 12. | Fathalla, W.; Pazdera, P.; Marek, J. J. Heterocycl. Chem. 2002, 39, 1139. doi:10.1002/jhet.5570390605 |
| 13. | Fathalla, W.; Marek, J.; Pazdera, P. J. Sulfur Chem. 2008, 29, 31. doi:10.1080/17415990701759685 |
| 14. | Fathalla, W. ARKIVOC 2008, xii, 245. |
| 15. | Ali, I. A. I.; Fathalla, W. Heteroat. Chem. 2006, 17, 280. doi:10.1002/hc.20203 |
| 16. | Fathalla, W.; El Rayes, S.; Ali, I. A. I. ARKIVOC 2008, xiii, 179. |
| 17. | Fathalla, W. Chem. Heterocycl. Compd. 2015, 51, 67. doi:10.1007/s10593-015-1661-1 |
| 18. | Yamamoto, Y. Methods of molecular hetarenes and related ring systems; Science of Synthesis: Houben-Weyl, Vol. 16; 2003; pp 726 ff. |
| 19. | Sundaram, R.; Yuvaraj, E.; Babu, G. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1999, 38, 905. |
| 20. | Lee, E. S.; Son, J. K.; Na, Y. H.; Jahng, Y. Heterocycl. Commun. 2004, 10, 325. doi:10.1515/HC.2004.10.4-5.325 |
| 21. | Okada, K.; Sakuma, H.; Inoue, S. Chem. Lett. 1979, 8, 131. doi:10.1246/cl.1979.131 |
| 22. | Chawla, H. M.; Gupta, T. J. Inclusion Phenom. Macrocyclic Chem. 2015, 81, 49. doi:10.1007/s10847-014-0432-4 |
| 23. | Derbala, H. A. Y. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1994, 33, 779. |
| 24. | Ismail, M. F.; Kandile, N. G. Acta Chim. Hung. 1991, 128, 251. |
| 25. | Ajani, O. O.; Obafemi, C. A.; Ikpo, C. O.; Ogunniran, K. O.; Nwinyi, O. C. Chem. Heterocycl. Compd. 2009, 45, 1370. doi:10.1007/s10593-010-0435-z |
| 26. | Noolvi, M. N.; Patel, H. M.; Bhardwaj, V.; Chauhan, A. Eur. J. Med. Chem. 2011, 46, 2327. doi:10.1016/j.ejmech.2011.03.015 |
| 27. | Mahesh, R.; Dhar, A. K.; Sasank, T. V. N. V. T.; Thirunavukkarasu, S.; Devadoss, T. Chin. Chem. Lett. 2011, 22, 389. doi:10.1016/j.cclet.2010.11.002 |
| 28. | Elhelby, A. A.; Ayyad, R. R.; Zayed, M. F. Arzneim. Forsch. 2011, 61, 379. doi:10.1055/s-0031-1296214 |
| 29. | Shi, L.; Wu, T.-T.; Wang, Z.; Xu, J.-Y.; Xu, Y.-G. Bioorg. Med. Chem. 2014, 22, 4735. doi:10.1016/j.bmc.2014.07.008 |
| 30. | Marvania, B.; Lee, P.-C.; Chaniyara, R.; Dong, H.; Suman, S.; Kakadiya, R.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Bioorg. Med. Chem. 2011, 19, 1987. doi:10.1016/j.bmc.2011.01.055 |
| 31. | Aly, A. A. J. Chin. Chem. Soc. 2007, 54, 437. doi:10.1002/jccs.200700061 |
| 32. | Browner, M.; Clark, D.; Cushing, T.; Hao, X.; Hawley, R.; He, X.; Jaen, J.; Lebadie, S.; Smith, M.-L.; Talmas, F.; Walker, N.; Labelle, M. Antiinflammation agents. U.S. Pat. Appl. US 20020161004 A1, Oct 31, 2002. |
| 33. | Rival, Y.; Hoffmann, R.; Didier, B.; Rybaltchenko, V.; Bourguignon, J.; Wermuth, C. G. J. Med. Chem. 1998, 41, 311. doi:10.1021/jm9705418 |
| 34. | Hemdan, M. M.; Taha, S. M.; Gabr, A. M.; Elkady, M. Y. J. Chem. Res. 2010, 34, 102. doi:10.3184/030823410X12658886079090 |
| 35. | Galal, S. A.; Abdelsamie, A. S.; Tokuda, H.; Suzuki, N.; Lida, A.; ElHefnawi, M. M.; Ramadan, R. A.; Atta, M. H. E.; El Diwani, H. I. Eur. J. Med. Chem. 2011, 46, 327. doi:10.1016/j.ejmech.2010.11.022 |
| 36. | Da Silva Miranda, F.; Signori, A. M.; Vicente, J.; De Souza, B.; Priebe, J. P.; Szpoganicz, B.; Gonçalves, N. S.; Neves, A. Tetrahedron 2008, 64, 5410. doi:10.1016/j.tet.2008.02.097 |
| 37. | Aggarwal, R.; Masan, E.; Sumran, G. Synth. Commun. 2013, 43, 1842. doi:10.1080/00397911.2012.674168 |
| 38. | Wagle, S.; Adhikari, A. V.; Kumari, N. S. Eur. J. Med. Chem. 2009, 44, 1135. doi:10.1016/j.ejmech.2008.06.006 |
| 39. | Koshel, N. G.; Kovalev, E. G.; Postovskii, I. Ya. Chem. Heterocycl. Compd. 1970, 6, 791. doi:10.1007/BF00470545 |
| 8. | Asano, K. Yakugaku Zasshi 1958, 78, 729. |
| 9. | El-Hawash, S. A. M.; Abdel Wahab, A. E. Arch. Pharm. 2006, 339, 437. doi:10.1002/ardp.200600012 |
| 42. | Ramadas, K.; Janarthanan, N.; Velmathi, S. Synth. Commun. 1997, 27, 2255. doi:10.1080/00397919708003379 |
| 7. | Morrison, D. C.; Furst, A. J. Org. Chem. 1956, 21, 470. doi:10.1021/jo01110a026 |
| 40. | Furumoto, S. Nippon Kagaku Zasshi 1971, 92, 1005. doi:10.1246/nikkashi1948.92.1005 |
| 4. | Libermann, R. Bull. Soc. Chim. Fr. 1959, 1793. |
| 5. | Leonard, N. J.; Curtin, D. Y. J. Org. Chem. 1946, 11, 349. doi:10.1021/jo01174a007 |
| 6. | Armstrong, R. T. Vulcanization accelerators. U.S. Patent US2382769, Aug 14, 1945. |
| 41. | Sun, N.; Li, B.; Shao, J.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. Beilstein J. Org. Chem. 2012, 8, 61. doi:10.3762/bjoc.8.6 |
| 24. | Ismail, M. F.; Kandile, N. G. Acta Chim. Hung. 1991, 128, 251. |
| 25. | Ajani, O. O.; Obafemi, C. A.; Ikpo, C. O.; Ogunniran, K. O.; Nwinyi, O. C. Chem. Heterocycl. Compd. 2009, 45, 1370. doi:10.1007/s10593-010-0435-z |
| 30. | Marvania, B.; Lee, P.-C.; Chaniyara, R.; Dong, H.; Suman, S.; Kakadiya, R.; Chou, T.-C.; Lee, T.-C.; Shah, A.; Su, T.-L. Bioorg. Med. Chem. 2011, 19, 1987. doi:10.1016/j.bmc.2011.01.055 |
| 31. | Aly, A. A. J. Chin. Chem. Soc. 2007, 54, 437. doi:10.1002/jccs.200700061 |
| 32. | Browner, M.; Clark, D.; Cushing, T.; Hao, X.; Hawley, R.; He, X.; Jaen, J.; Lebadie, S.; Smith, M.-L.; Talmas, F.; Walker, N.; Labelle, M. Antiinflammation agents. U.S. Pat. Appl. US 20020161004 A1, Oct 31, 2002. |
| 33. | Rival, Y.; Hoffmann, R.; Didier, B.; Rybaltchenko, V.; Bourguignon, J.; Wermuth, C. G. J. Med. Chem. 1998, 41, 311. doi:10.1021/jm9705418 |
| 34. | Hemdan, M. M.; Taha, S. M.; Gabr, A. M.; Elkady, M. Y. J. Chem. Res. 2010, 34, 102. doi:10.3184/030823410X12658886079090 |
| 35. | Galal, S. A.; Abdelsamie, A. S.; Tokuda, H.; Suzuki, N.; Lida, A.; ElHefnawi, M. M.; Ramadan, R. A.; Atta, M. H. E.; El Diwani, H. I. Eur. J. Med. Chem. 2011, 46, 327. doi:10.1016/j.ejmech.2010.11.022 |
| 36. | Da Silva Miranda, F.; Signori, A. M.; Vicente, J.; De Souza, B.; Priebe, J. P.; Szpoganicz, B.; Gonçalves, N. S.; Neves, A. Tetrahedron 2008, 64, 5410. doi:10.1016/j.tet.2008.02.097 |
| 37. | Aggarwal, R.; Masan, E.; Sumran, G. Synth. Commun. 2013, 43, 1842. doi:10.1080/00397911.2012.674168 |
| 22. | Chawla, H. M.; Gupta, T. J. Inclusion Phenom. Macrocyclic Chem. 2015, 81, 49. doi:10.1007/s10847-014-0432-4 |
| 23. | Derbala, H. A. Y. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1994, 33, 779. |
| 38. | Wagle, S.; Adhikari, A. V.; Kumari, N. S. Eur. J. Med. Chem. 2009, 44, 1135. doi:10.1016/j.ejmech.2008.06.006 |
| 39. | Koshel, N. G.; Kovalev, E. G.; Postovskii, I. Ya. Chem. Heterocycl. Compd. 1970, 6, 791. doi:10.1007/BF00470545 |
| 19. | Sundaram, R.; Yuvaraj, E.; Babu, G. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1999, 38, 905. |
| 20. | Lee, E. S.; Son, J. K.; Na, Y. H.; Jahng, Y. Heterocycl. Commun. 2004, 10, 325. doi:10.1515/HC.2004.10.4-5.325 |
| 21. | Okada, K.; Sakuma, H.; Inoue, S. Chem. Lett. 1979, 8, 131. doi:10.1246/cl.1979.131 |
| 46. | Hanusek, J.; Hejtmánková, L.; Kubicová, L.; Sedlák, M. Molecules 2001, 6, 323. doi:10.3390/60400323 |
| 18. | Yamamoto, Y. Methods of molecular hetarenes and related ring systems; Science of Synthesis: Houben-Weyl, Vol. 16; 2003; pp 726 ff. |
| 26. | Noolvi, M. N.; Patel, H. M.; Bhardwaj, V.; Chauhan, A. Eur. J. Med. Chem. 2011, 46, 2327. doi:10.1016/j.ejmech.2011.03.015 |
| 27. | Mahesh, R.; Dhar, A. K.; Sasank, T. V. N. V. T.; Thirunavukkarasu, S.; Devadoss, T. Chin. Chem. Lett. 2011, 22, 389. doi:10.1016/j.cclet.2010.11.002 |
| 28. | Elhelby, A. A.; Ayyad, R. R.; Zayed, M. F. Arzneim. Forsch. 2011, 61, 379. doi:10.1055/s-0031-1296214 |
| 29. | Shi, L.; Wu, T.-T.; Wang, Z.; Xu, J.-Y.; Xu, Y.-G. Bioorg. Med. Chem. 2014, 22, 4735. doi:10.1016/j.bmc.2014.07.008 |
© 2017 Fathalla et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)