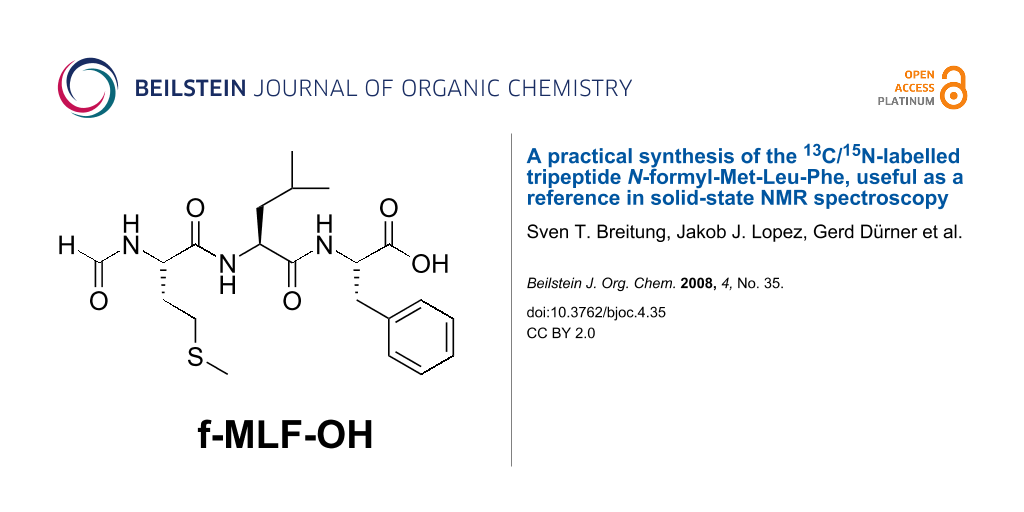
Abstract
A mild synthetic method for N-formyl-Met-Leu-Phe-OH (1) is described. After Fmoc solid phase peptide synthesis, on-bead formylation and HPLC purification, more than 30 mg of the fully 13C/15N-labelled tripeptide 1 could be isolated in a typical batch. This peptide can be easily crystallised and is therefore well suited as a standard sample for setting up solid-state NMR experiments.

Graphical Abstract
Introduction
There appears to be a general lack of widely available and standardised samples for setting up new solid-state NMR experiments. Such a standard sample should show a small 13C and 15N linewidth and a short 1H T1 relaxation time. It should contain a number of molecular groups with different chemical shifts for setting up correlation spectra. The 13C/15N-labelled tripeptide N-formyl-Met-Leu-Phe-OH (f-MLF-OH) (1) has been shown in a number of solid-state NMR studies to fulfil these criteria. It has been used in great detail to examine spin dynamics in peptide and for distance measurements [1-9]. MLF is a chemotactic peptide which plays an important role in antibody research [10].
Although this tripeptide was briefly commercially available in the past, only one synthesis of f-MLF-OH (1) by solid phase methods has been published so far [11]. Rigorous reaction conditions (EtOH, reflux, 24–65 h) were required to couple the first Boc-protected amino acid to the solid support (chloromethyl resin) and long reaction times (18 h) were necessary to attach further building blocks to the growing peptide chain. The formylation of the N-terminus with formic acid/acetic anhydride was carried out after cleavage from the resin with liquid hydrogen fluoride [11,12]. No experimental procedures were given in all subsequent publications. In this work, we present for the first time in detail an improved practical synthesis for fully 13C/15N-labelled f-MLF-OH (1) based on the Fmoc-strategy.
Results and Discussion
Peptide Synthesis
The synthesis of the MLF tripeptide started with the immobilisation of 13C/15N-labelled Fmoc-Phe-OH to the solid support (Wang resin 2). This esterification step, leading to 3 quantitatively, was performed by activating the COOH group with MSNT under mild reaction conditions (Scheme 1) [13]. Full conversion of the resin bound hydroxy groups could be demonstrated by a colourimetric test with Dabcyl-COOH [14]. After removal of Fmoc with piperidine, the 13C/15N-labelled monomers Fmoc-Leu-OH and Fmoc-Met-OH were successively coupled to the solid support with DIC/HOBt [15]. Quantitative conversion in each reaction was monitored by the Kaiser test [16,17]. The resulting tripeptide 4 was formylated under mild conditions with 13C-labelled ethyl formate to obtain 5 [18]. The main advantage of using ethyl formate is the possibility of re-isolation of the expensive 13C-labelled reagent for further formylation reactions, due to the absence of activating agents. To the best of our knowledge, formylation reactions of N-termini with ethyl formate have never been reported before in solid phase peptide synthesis. Other methods of formylation were not tested, since they require elevated reaction temperatures or acidic conditions [19,20], which might cause some premature removal of the peptide from the resin. The final step was the cleavage of f-MLF-OH (1) from the solid support with TFA [21,22]. To prevent by-product formation from electrophilic intermediates present in the cleavage process, EDT and TIS were added as scavengers [23,24].
![[1860-5397-4-35-i1]](/bjoc/content/inline/1860-5397-4-35-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of f-MLF-OH (1). a) Fmoc-Phe-OH, MSNT, MeIm, over night. b) 1. piperidine, 30 min; 2. Fmoc-Leu-OH, DIC, HOBt*H2O, 3 h. c) 1. piperidine, 30 min; 2. Fmoc-Met-OH, DIC, HOBt*H2O, 3 h; 3. piperidine, 30 min. d) HCOOEt, over night. e) TFA, EDT, H2O, TIS (94 : 2.5 : 2.5 : 1), 4.5 h. All reactions were carried out at room temperature. Abbreviations: MSNT = 1-(mesitylene-2-sulphonyl)-3-nitro-1,2,4-triazole, MeIm = N-methylimidazole, DIC = diisopropylcarbodiimide, HOBt = 1-hydroxybenzotriazole, TFA = trifluoroacetic acid, EDT = ethanedithiole, TIS = triisopropylsilane.
Scheme 1: Synthesis of f-MLF-OH (1). a) Fmoc-Phe-OH, MSNT, MeIm, over night. b) 1. piperidine, 30 min; 2. Fmo...
Conclusion
Compared to the solution phase synthesis of peptides, the solid phase synthesis offers a simplified purification of the intermediates. With the improved synthetic protocol based on Fmoc building blocks, where all reaction steps were carried out at room temperature, we were able to obtain the per-13C/15N-labelled formylated Met-Leu-Phe tripeptide as carboxylic acid in acceptable yields [32.8 mg (23%) after HPLC purification]. Due to the selected reagents, i.e. MSNT for the esterification, DIC/HOBt for the peptide coupling and ethyl formate for the formylation, shorter reaction time and quantitative conversion could be accomplished when compared to the previous protocol from 1976 [11], where yields are not given. In contrast to the Boc-strategy, the Fmoc-method for solid phase peptide synthesis has the advantage of orthogonal conditions for the removal of N-protective groups and the cleavage from the Wang resin [25]. Unintentional release of the growing peptide chain thus could be excluded. In addition, the use of liquid hydrogen fluoride is no longer required.
Experimental
General: All reagents were obtained from commercial suppliers and were used without further purification. HPLC gradient: 0–5 min (0.1% TFA/MeCN 99:1), 5–20 min (0.1% TFA/MeCN 99:1 to 30:70), 20–25 min (0.1% TFA/MeCN 30:70), 25–30 min (0.1% TFA/MeCN 30:70 to 99:1), 30–40 min (0.1% TFA/MeCN 99:1). ESI-MS: Fisons VG Plattform II. NMR: Bruker AM 300 (1H: 300 MHz; 13C: 75.5 MHz).
Immobilisation of the first monomer: Wang resin (485 mg, 0.315 mmol, capacity: 0.65 mmol/g) was swelled under argon with dry CH2Cl2 (1.5 mL) in an oven dried reaction vessel for 15 min. In a second dry vessel, 13C/15N-labelled Fmoc-Phe-OH (250 mg, 0.63 mmol) was dissolved in dry CH2Cl2 (3 mL) and MeIm (94 µL, 1.18 mmol). Afterwards MSNT (467 mg, 1.576 mmol) was added. The reaction mixture was shaken under argon until MSNT had completely dissolved. The amino acid solution was transferred via syringe to the resin and the suspension was flushed with argon. The sealed reaction vessel was gently agitated over night.
The beads were transferred to a syringe with a filter and washed with dry CH2Cl2 (5 ×). Some beads were dried in vacuo for the following colourimetric test with Dabcyl-COOH for the detection of polymer-supported OH groups [14]. (See Supporting Information File 1.)
Coupling of further building blocks: In case of quantitative conversion, the resin was washed with N,N-dimethylformamide (DMF, 5 ×) and treated three times (15 min, 10 min, 5 min) with a piperidine solution (25% in DMF). Then the resin was washed with N-methylpyrrolidone (NMP, 5 ×) and a solution of 13C/15N-labelled Fmoc-Leu-OH (250 mg, 0.69 mmol), HOBt*H2O (145 mg, 0.945 mmol) and DIC (145 µL, 0.945 mmol) in NMP (2.5 mL) was aspirated into the syringe for the next coupling step. After 3 h the resin was washed with NMP (5 ×) and a few beads were tested for quantitative conversion by the Kaiser test [16,17]. (See Supporting Information File 1.)
In case of a successful peptide extension, the Fmoc-protecting group was removed with piperidine (25% in DMF). Afterwards, the last coupling step was started by adding a solution of 13C/15N-labelled Fmoc-Met-OH (250 mg, 0.66 mmol), HOBt*H2O (145 mg, 0.945 mmol) and DIC (145 µL, 0.945 mmol) in NMP (2.5 mL). In case of quantitative coupling (Kaiser test), the Fmoc-protecting group was removed and the resin was washed with DMF (5 ×) and CH2Cl2 (5 ×). The beads were dried in the syringe under reduced pressure.
Formylation of the N-terminus: For the following formylation, 13C-labelled ethyl formate (2 mL) was aspirated and the syringe was shaken over night at room temperature. The quantitative conversion was proved by the Kaiser test, after washing the resin with CH2Cl2 (5 ×).
Cleavage of the peptide from the solid support: For a successful cleavage, it is highly recommended to dry the beads in vacuo for at least 3 h. The cleavage solution [TFA (1.88 mL), H2O (50 µL), EDT (50 µL) and TIS (20 µL)] was added to the syringe. After shaking (3 × 90 min), the liberated peptide was filtered off into Millipore™ water. This solution was lyophilised and the residue was purified by HPLC. Yield: 32.8 mg (23%).
HPLC conditions: Preparative: Reprosil AQ, 250 × 20, 10 µm, 0.1% TFA/MeCN (100:60), 10 mL/min; analytical: gradient: Reprosil AQ, 125 × 4.6 mm, 5 µm, 0.8 mL/min, tR = 21.08 min; isocratic: Lichrospher RP8 (Merck), 125 × 4.0 mm, 5 µm, 0.1% TFA/MeCN (66:34), 0.8 mL/min, tR = 5.07 min.
13C NMR (δ[ppm] 75.5 MHz, MeCN-d3): 173.5 (m, 1C), 172.6 (m, 1C), 171.8 (m, 1C), 162.8 (d, J = 12.8 Hz, 1C), 138.0 (m, 1C), 131.1-126.8 (m, 5C), 55.1-51.3 (m, 3C), 41.3 (t, J = 34.4 Hz, 1C), 37.8 (m, 1C), 32.4 (t, J = 35.5 Hz, 1C), 30.4 (m, 1C), 25.4 (m, 1C), 23.2 (m, 1C), 21.7 (m, 1C), 15.3 (s, 1C).
MS (ESI): m/z (%) = 460.3 (100.0) [M–H]–, 13C21H3115N3O5S calcd. 461.26.
Supporting Information
Procedures for colourimetric resin tests (including synthesis and analytical data of Dabcyl-COOH), crystallisation protocol, ESI, 13C NMR and 13C/15N MAS-NMR spectra of f-MLF-OH (1) are available in the Supporting Information.
| Supporting Information File 1: A practical synthesis of the 13C/15N-labelled tripeptide N-Formyl-Met-Leu-Phe, useful as a reference in solid-state NMR spectroscopy. | ||
| Format: DOC | Size: 1.2 MB | Download |
References
-
Rienstra, C. M.; Hohwy, M.; Hong, M.; Griffin, R. G. J. Am. Chem. Soc. 2000, 122, 10979–10990. doi:10.1021/ja001092v
Return to citation in text: [1] -
Rienstra, C. M.; Hohwy, M.; Mueller, L. J.; Jaroniec, C. P.; Reif, B.; Griffin, R. G. J. Am. Chem. Soc. 2002, 124, 11908–11922. doi:10.1021/ja020802p
Return to citation in text: [1] -
Rienstra, C. M.; Tucker-Kellogg, L.; Jaroniec, C. P.; Hohwy, M.; Reif, B.; McMahon, M. T.; Tidor, B.; Lozano-Pérez, T.; Griffin, R. G. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10260–10265. doi:10.1073/pnas.152346599
Return to citation in text: [1] -
Reif, B.; Hohwy, M.; Jaroniec, C. P.; Rienstra, C. M.; Griffin, R. G. J. Magn. Reson. 2000, 145, 132–141. doi:10.1006/jmre.2000.2067
Return to citation in text: [1] -
Jaroniec, C. P.; Filip, C.; Griffin, R. G. J. Am. Chem. Soc. 2002, 124, 10728–10742. doi:10.1021/ja026385y
Return to citation in text: [1] -
Jaroniec, C. P.; Tounge, B. A.; Herzfeld, J.; Griffin, R. G. J. Am. Chem. Soc. 2001, 123, 3507–3519. doi:10.1021/ja003266e
Return to citation in text: [1] -
Wi, S.; Sinha, N.; Hong, M. J. Am. Chem. Soc. 2004, 126, 12754–12755. doi:10.1021/ja0462732
Return to citation in text: [1] -
McDermott, A. E. Curr. Opin. Struct. Biol. 2004, 14, 554–561. doi:10.1016/j.sbi.2004.09.007
Return to citation in text: [1] -
Lopez, J. J.; Kaiser, C.; Shastri, S.; Glaubitz, C. J. Biomol. NMR 2008, 41, 97–104. doi:10.1007/s10858-008-9245-3
Return to citation in text: [1] -
Tanaka, F.; Jones, T.; Kubitz, D.; Lerner, R. A. Bioorg. Med. Chem. Lett. 2007, 17, 1943–1945. doi:10.1016/j.bmcl.2007.01.082
Return to citation in text: [1] -
Showell, H. J.; Freer, R. J.; Zigmond, S. H.; Schiffmann, E.; Aswanikumar, S.; Corcoran, B.; Becker, E. L. J. Exp. Med. 1976, 143, 1154–1169. doi:10.1084/jem.143.5.1154
Return to citation in text: [1] [2] [3] -
Sheehan, J. C.; Yang, D.-D. H. J. Am. Chem. Soc. 1958, 80, 1154–1158. doi:10.1021/ja01538a036
Return to citation in text: [1] -
Blankemeyer-Menge, B.; Nimtz, M.; Frank, R. Tetrahedron Lett. 1990, 31, 1701–1704. doi:10.1016/S0040-4039(00)88858-9
Return to citation in text: [1] -
Burkett, B. A.; Brown, R. C. D.; Meloni, M. M. Tetrahedron Lett. 2001, 42, 5773–5775. doi:10.1016/S0040-4039(01)01102-9
Return to citation in text: [1] [2] -
Chan, W. C.; White, P. D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: New York, 2004.
Return to citation in text: [1] -
Kaiser, E.; Colescott, R. L.; Bossinger, C. D.; Cook, P. I. Anal. Biochem. 1970, 34, 595–598. doi:10.1016/0003-2697(70)90146-6
Return to citation in text: [1] [2] -
Sarin, V. K.; Kent, S. B. H.; Tam, J. P.; Merrifield, R. B. Anal. Biochem. 1981, 117, 147–157. doi:10.1016/0003-2697(81)90704-1
Return to citation in text: [1] [2] -
Jacobsen, E. J.; Stelzer, L. S.; Belonga, K. L.; Carter, D. B.; Im, W. B.; Sethy, V. H.; Tang, A. H.; VonVoigtlander, P. F.; Petke, J. D. J. Med. Chem. 1996, 39, 3820–3836. doi:10.1021/jm960070+
Return to citation in text: [1] -
Billault, I.; Courant, F.; Pasquereau, L.; Derrien, S.; Robins, R. J.; Naulet, N. Anal. Chim. Acta 2007, 593, 20–29. doi:10.1016/j.aca.2007.04.039
Return to citation in text: [1] -
Jung, S. H.; Ahn, J. H.; Park, S. K.; Choi, J.-K. Bull. Korean Chem. Soc. 2002, 23, 149–150.
Return to citation in text: [1] -
Wang, S.-S. J. Am. Chem. Soc. 1973, 95, 1328–1333. doi:10.1021/ja00785a602
Return to citation in text: [1] -
Lu, G.-s.; Mojsov, S.; Tam, J. P.; Merrifield, R. B. J. Org. Chem. 1981, 46, 3433–3436. doi:10.1021/jo00330a009
Return to citation in text: [1] -
Krebs, A.; Ludwig, V.; Pfizer, J.; Dürner, G.; Göbel, M. W. Chem.–Eur. J. 2004, 10, 544–553. doi:10.1002/chem.200305421
Return to citation in text: [1] -
Suhartono, M.; Weidlich, M.; Stein, T.; Karas, M.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 1608–1614. doi:10.1002/ejoc.200701124
Return to citation in text: [1] -
Carpino, L. A.; Han, G. Y. J. Org. Chem. 1972, 37, 3404–3409. doi:10.1021/jo00795a005
Return to citation in text: [1]
| 1. | Rienstra, C. M.; Hohwy, M.; Hong, M.; Griffin, R. G. J. Am. Chem. Soc. 2000, 122, 10979–10990. doi:10.1021/ja001092v |
| 2. | Rienstra, C. M.; Hohwy, M.; Mueller, L. J.; Jaroniec, C. P.; Reif, B.; Griffin, R. G. J. Am. Chem. Soc. 2002, 124, 11908–11922. doi:10.1021/ja020802p |
| 3. | Rienstra, C. M.; Tucker-Kellogg, L.; Jaroniec, C. P.; Hohwy, M.; Reif, B.; McMahon, M. T.; Tidor, B.; Lozano-Pérez, T.; Griffin, R. G. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10260–10265. doi:10.1073/pnas.152346599 |
| 4. | Reif, B.; Hohwy, M.; Jaroniec, C. P.; Rienstra, C. M.; Griffin, R. G. J. Magn. Reson. 2000, 145, 132–141. doi:10.1006/jmre.2000.2067 |
| 5. | Jaroniec, C. P.; Filip, C.; Griffin, R. G. J. Am. Chem. Soc. 2002, 124, 10728–10742. doi:10.1021/ja026385y |
| 6. | Jaroniec, C. P.; Tounge, B. A.; Herzfeld, J.; Griffin, R. G. J. Am. Chem. Soc. 2001, 123, 3507–3519. doi:10.1021/ja003266e |
| 7. | Wi, S.; Sinha, N.; Hong, M. J. Am. Chem. Soc. 2004, 126, 12754–12755. doi:10.1021/ja0462732 |
| 8. | McDermott, A. E. Curr. Opin. Struct. Biol. 2004, 14, 554–561. doi:10.1016/j.sbi.2004.09.007 |
| 9. | Lopez, J. J.; Kaiser, C.; Shastri, S.; Glaubitz, C. J. Biomol. NMR 2008, 41, 97–104. doi:10.1007/s10858-008-9245-3 |
| 13. | Blankemeyer-Menge, B.; Nimtz, M.; Frank, R. Tetrahedron Lett. 1990, 31, 1701–1704. doi:10.1016/S0040-4039(00)88858-9 |
| 14. | Burkett, B. A.; Brown, R. C. D.; Meloni, M. M. Tetrahedron Lett. 2001, 42, 5773–5775. doi:10.1016/S0040-4039(01)01102-9 |
| 11. | Showell, H. J.; Freer, R. J.; Zigmond, S. H.; Schiffmann, E.; Aswanikumar, S.; Corcoran, B.; Becker, E. L. J. Exp. Med. 1976, 143, 1154–1169. doi:10.1084/jem.143.5.1154 |
| 12. | Sheehan, J. C.; Yang, D.-D. H. J. Am. Chem. Soc. 1958, 80, 1154–1158. doi:10.1021/ja01538a036 |
| 16. | Kaiser, E.; Colescott, R. L.; Bossinger, C. D.; Cook, P. I. Anal. Biochem. 1970, 34, 595–598. doi:10.1016/0003-2697(70)90146-6 |
| 17. | Sarin, V. K.; Kent, S. B. H.; Tam, J. P.; Merrifield, R. B. Anal. Biochem. 1981, 117, 147–157. doi:10.1016/0003-2697(81)90704-1 |
| 11. | Showell, H. J.; Freer, R. J.; Zigmond, S. H.; Schiffmann, E.; Aswanikumar, S.; Corcoran, B.; Becker, E. L. J. Exp. Med. 1976, 143, 1154–1169. doi:10.1084/jem.143.5.1154 |
| 11. | Showell, H. J.; Freer, R. J.; Zigmond, S. H.; Schiffmann, E.; Aswanikumar, S.; Corcoran, B.; Becker, E. L. J. Exp. Med. 1976, 143, 1154–1169. doi:10.1084/jem.143.5.1154 |
| 10. | Tanaka, F.; Jones, T.; Kubitz, D.; Lerner, R. A. Bioorg. Med. Chem. Lett. 2007, 17, 1943–1945. doi:10.1016/j.bmcl.2007.01.082 |
| 25. | Carpino, L. A.; Han, G. Y. J. Org. Chem. 1972, 37, 3404–3409. doi:10.1021/jo00795a005 |
| 18. | Jacobsen, E. J.; Stelzer, L. S.; Belonga, K. L.; Carter, D. B.; Im, W. B.; Sethy, V. H.; Tang, A. H.; VonVoigtlander, P. F.; Petke, J. D. J. Med. Chem. 1996, 39, 3820–3836. doi:10.1021/jm960070+ |
| 21. | Wang, S.-S. J. Am. Chem. Soc. 1973, 95, 1328–1333. doi:10.1021/ja00785a602 |
| 22. | Lu, G.-s.; Mojsov, S.; Tam, J. P.; Merrifield, R. B. J. Org. Chem. 1981, 46, 3433–3436. doi:10.1021/jo00330a009 |
| 16. | Kaiser, E.; Colescott, R. L.; Bossinger, C. D.; Cook, P. I. Anal. Biochem. 1970, 34, 595–598. doi:10.1016/0003-2697(70)90146-6 |
| 17. | Sarin, V. K.; Kent, S. B. H.; Tam, J. P.; Merrifield, R. B. Anal. Biochem. 1981, 117, 147–157. doi:10.1016/0003-2697(81)90704-1 |
| 23. | Krebs, A.; Ludwig, V.; Pfizer, J.; Dürner, G.; Göbel, M. W. Chem.–Eur. J. 2004, 10, 544–553. doi:10.1002/chem.200305421 |
| 24. | Suhartono, M.; Weidlich, M.; Stein, T.; Karas, M.; Dürner, G.; Göbel, M. W. Eur. J. Org. Chem. 2008, 1608–1614. doi:10.1002/ejoc.200701124 |
| 15. | Chan, W. C.; White, P. D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: New York, 2004. |
| 14. | Burkett, B. A.; Brown, R. C. D.; Meloni, M. M. Tetrahedron Lett. 2001, 42, 5773–5775. doi:10.1016/S0040-4039(01)01102-9 |
| 19. | Billault, I.; Courant, F.; Pasquereau, L.; Derrien, S.; Robins, R. J.; Naulet, N. Anal. Chim. Acta 2007, 593, 20–29. doi:10.1016/j.aca.2007.04.039 |
| 20. | Jung, S. H.; Ahn, J. H.; Park, S. K.; Choi, J.-K. Bull. Korean Chem. Soc. 2002, 23, 149–150. |
© 2008 Breitung et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)