Abstract
Neutral organic electron donors, featuring pyridinylidene–imidazolylidene, pyridinylidene–benzimidazolylidene and imidazolylidene–benzimidazolylidene linkages are reported. The pyridinylidene–benzimidazolylidene and imidazolylidene–benzimidazolylidene hybrid systems were designed to be the first super electron donors to convert iodoarenes to aryl radicals at room temperature, and indeed both show evidence for significant aryl radical formation at room temperature. The stronger pyridinylidene–imidazolylidene donor converts iodoarenes to aryl anions efficiently under appropriate conditions (3 equiv of donor). The presence of excess sodium hydride base has a very important and selective effect on some of these electron-transfer reactions, and a rationale for this is proposed.

Graphical Abstract
Introduction
Alkenes that are substituted by four heteroatoms are notable for their ease of oxidation. Whereas tetrathiafulvalenes and analogues [1-5] have principally found widespread applications in materials science, tetraazaalkenes and related compounds are much more reactive and are of potential or actual interest as reagents in synthesis [6-33]. Among the tetraazaalkenes, those that are converted to aromatic molecules upon oxidation, e.g., tetraazafulvalenes 1 and 2, are extremely reactive, and their electrochemical properties have been studied in some depth [13-16].
Neutral organic donors that can reduce aryl halides have been termed “super-electron-donors”. Our recent research has examined the remarkable chemical reactivity of such donors 1 and 2 as well as the related electron-donors 4 and 5 (Figure 1), with organic substrates [17-27]. Benzimidazole-derived donor 1 converted aryl iodides to aryl radicals by transfer of a single electron at 110 °C [17], and was the first neutral organic ground-state molecule to achieve this. Later, the more powerful reagents 2 [18], 4 [20] and 5 [21] and related compounds [25] afforded aryl anions from the same substrates by transfer of two electrons at room temperature, and also cleaved selected sulfonamides [19], bis-sulfones [19], Weinreb amides [22], acyloin derivatives [24], triflate esters and triflamides [26]. Most recently, we announced the synthesis of the unstable compound 3 [16,27].
![[1860-5397-8-112-1]](/bjoc/content/figures/1860-5397-8-112-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Super-electron-donors and related compounds.
Figure 1: Super-electron-donors and related compounds.
Cyclic voltammetry (CV) studies showed that the more powerful donors, e.g., 2 [15,16,18] and 4 [19], lose their second electrons at almost the same potential as their first electron (Figure 2 includes the CV of 4, showing a single two-electron redox wave, while Figure 4a includes that of 2). The differential strengths of the donors (2, 4 versus 1) correlated with the expected relative driving force resulting from aromatisation following the loss of electrons. In the case of the respective oxidised forms, i.e., the dications 6–8, the newly aromatic rings are represented in blue in Figure 1, while the pre-existing aromatic rings in 6 are represented in red. The driving force for oxidation arising through aromatisation is greater for the imidazole- and pyridine-derived motifs 7 and 8, which are associated with the strongest donors, than it is for the benzimidazole-derived motif, 6, which marks a weaker donor.
To extend the capabilities of such reagents, we now set out to design the first neutral, organic ground-state donor that could, at room temperature, reduce aryl iodides to aryl radicals (thereby acting as a single-electron donor) as opposed to aryl anions. Hybrid organic electron donors incorporating a "stronger" donor component and a "weaker" component, e.g., 9 or 10 would be prime candidates, as the driving force for the loss of their first electron should exceed that for the loss of their second. The electrochemical properties of some hybrid imidazolium–benzimidazolium-derived donors have been reported [15]. For comparison, donor 11 is also of interest, although it features two "stronger" donor components. As indicated below, our work has found remarkable effects of excess base in reactions of some hybrid donors.
Results and Discussion
Compounds 9–11 were adopted as targets for synthesis. Of these, 10 and 11 are imidazolylidenes derived from an imidazolium salt. Donors derived from imdazolium salts are highly reactive and unstable; CV studies in MeCN have shown [15] that two-electron reduction of 13, bearing a single trimethylene bridge, which was intended to afford 3, does not lead to a stable product; moreover, Taton and Chen [16] did not observe formation of 3 (by deprotonation of 14 in DMSO or by reduction of 13), reporting instead the formation of bis-carbene 15. In the recent synthesis of 3 [27], its decomposition was noted over a period of hours in ultradry conditions under argon. Within the series of imidazole-derived tetraazafulvalenes, Taton and Chen established [16] that the only member that remained stable on storage under inert conditions was the bis-trimethylene bridged donor 2, and its greater stability was attributed to the two trimethylene tethers. As compounds 10 and 11 are derived from imidazolium precursors, we were keen to explore their reactivity.
The redox properties of the donors were first measured by cyclic voltammetry. Either the electron donors or their oxidized salts could, in principle, be used as a starting point for the CV studies; however, the oxidised disalts were routinely chosen as they can be conveniently weighed out under air, while the donors are extremely air-sensitive. The oxidized salts, derived from the donors, were prepared as shown in Scheme 1. Reaction of 1-(3-bromopropyl)-4-dimethylaminopyridinium bromide (16) [34] with N-methylimidazole (17) afforded disalt 19. Deprotonation with NaH (15 equiv) in DMF then afforded the electron donor 11 in situ; this was reacted with iodine to afford the oxidised diiodide salt, and this was subjected to anion exchange to afford the bis(hexafluorophosphate) salt 21 for analysis. (Anion exchange to bis(hexafluorophosphate) salts was required since the iodide anions within diiodide salts would be electrochemically active in CV studies). To verify the intermediacy of 11, its formation from 19 was repeated in DMF-d7, and the 1H and 13C NMR spectra of 11 were determined. The 1H NMR spectrum showed the characteristic upfield shift of proton signals for nonaromatic electron-rich donors.
![[1860-5397-8-112-i1]](/bjoc/content/inline/1860-5397-8-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the oxidised disalts.
Scheme 1: Preparation of the oxidised disalts.
Figure 2 shows the cyclic voltammogram of 21 (blue trace) and a comparison with 8 (X = PF6) (red trace). As seen, 21 undergoes reversible redox chemistry [E1½ (DMF) = −1.75 V, E2½ (DMF) = −1.63 V versus Fc/Fc+; this corresponds to E1½ (DMF) = −1.30 V, E2½ (DMF) = −1.18 V versus SCE]. The cyclic voltammogram, together with the NMR determination above, shows that 11 is a stable imidazole-derived donor (i.e., it does not decompose under the conditions used for its formation) [15], and so its capability as an electron donor was tested.
![[1860-5397-8-112-2]](/bjoc/content/figures/1860-5397-8-112-2.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 2: Cyclic voltammograms in DMF of 8/4 (red) and 21/11 (blue). Current plotted vs V (relative to Fc/Fc+ as standard).
Figure 2: Cyclic voltammograms in DMF of 8/4 (red) and 21/11 (blue). Current plotted vs V (relative to Fc/Fc+...
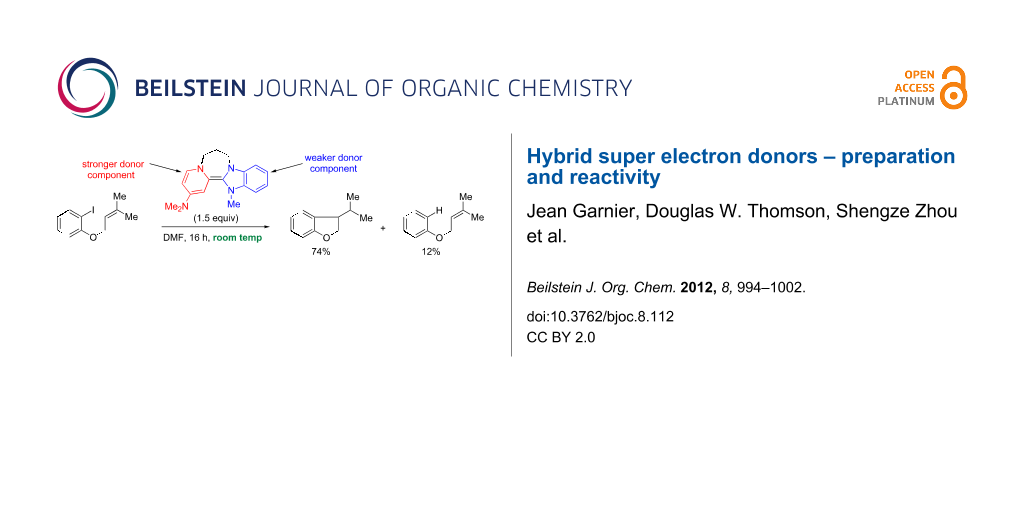
To test reactivity, donor 11 was prepared in situ and treated with the substrates 28 and 30 at room temperature (Scheme 2). Simple substrate 28 [35] was added to 11, prepared by adding disalt 19 (1.5 equiv) to excess sodium hydride (15 equiv). As expected, it behaved as a strong donor, affording 29 [20] in 74% yield. (A blank experiment, in which substrate 28 was treated at room temperature for 16 h with NaH in DMF, led to quantitative recovery of 28). Substrate 30 [35] was designed to test whether a single electron or two electrons are transferred to an iodoarene; single electron transfer would afford an aryl radical that would undergo cyclisation efficiently [17], while two-electron transfer to afford an aryl anion would afford an aryl anion that would not cyclise in DMF as solvent [18]. The reaction with 30 was conducted under slightly different conditions than with 28. Donor 11 was prepared by using disalt 19 (3 equiv) added to the excess sodium hydride (15 equiv), and the resulting donor solution was filtered to remove excess NaH before substrate 30 was added. (Previous experience had raised suspicion that the excess NaH could deprotonate the aliphatic side-chain of allyloxy substrates). The sole isolated product, i.e., the de-iodinated but uncyclised compound 31 (59%) [36], is consistent with 11 donating two electrons.
![[1860-5397-8-112-i2]](/bjoc/content/inline/1860-5397-8-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reductive reactions of donor 11 [17,18].
Scheme 2: Reductive reactions of donor 11 [17,18].
Donor 9 was prepared by a route analogous to that used for 11, and was then oxidised and converted to the bis(hexafluorophosphate) salt 22. Cyclic voltammetry, starting with its oxidised disalt 22 (Figure 3, blue trace) shows that its redox activity occurs as two separate steps at potentials intermediate between those for compounds 6 and 8. [E1½ (DMF) = −1.54 V, E2½ (DMF) = −1.42 V versus Fc/Fc+; this corresponds to E1½ (DMF) = −1.09 V, E2½ (DMF) = −0.97 V versus SCE]. The cyclic voltammetry studies on oxidised disalt 22 show two reversible one-electron transitions on its reduction to donor 9. The redox potential in the oxidation trace for the removal of the first electron from 9 shows that the molecule is not as strong a donor as 4, while the transfer of the second electron occurs at a more negative potential than for the first electron from 1.
![[1860-5397-8-112-3]](/bjoc/content/figures/1860-5397-8-112-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: Cyclic voltammograms in DMF of 8/4 (red), 6/1 (green) and 22/9 (blue). Current plotted vs V relative to Fc/Fc+.
Figure 3: Cyclic voltammograms in DMF of 8/4 (red), 6/1 (green) and 22/9 (blue). Current plotted vs V relativ...
In situ generation of 9 from 20 (1.5 equiv, Scheme 3) and reaction with iodoarenes 28 and 30 was again carried out at room temperature. As for the reactions with donor 11, the excess NaH was filtered prior to the addition of substrate 30. Reaction of iodoarene 28 led to an inseparable mixture of 29 and 28 in a 2:1 ratio; based on the mass recovered, this corresponded to 29 (46%) and 28 (22%). By comparison, reaction with aryl iodide 30, again at room temperature, afforded a mixture of 32 [35], the product of aryl radical cyclization, (74%), together with recovered 30 (6%) and deiodinated but uncyclised product 31 (12%). This is the first observation of efficient aryl radical generation at room temperature from a super-electron-donor. For comparison, less than 1% yield of 32 was observed when repeating the reaction with donor 1, also generated in situ. Hence 9 reacts at room temperature with iodoarenes and functions as the strongest known neutral organic ground-state one-electron donor to iodoarenes.
![[1860-5397-8-112-i3]](/bjoc/content/inline/1860-5397-8-112-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reductive reactions of donor 9.
Scheme 3: Reductive reactions of donor 9.
Hybrid donor 10 was next prepared from the known chloropropylbenzimidazole 23 [37] (Scheme 1), then oxidised and converted to its bis(hexafluorophosphate) salt, 27, for analysis. Cyclic voltammetry on 27 is shown in Figure 4a (blue trace), in comparison with salts 7 and 6. Looking at the blue trace in Figure 4a, it is immediately clear that the oxidative sweep provides a very low current relative to the initial reductive sweep, suggesting decomposition of the reduced species on the timeframe of the CV studies. Repeating the experiment at different scan rates (Figure 4b) shows that at low scan speeds the effect is even more pronounced. Note that the CV traces reproduced by Ames et al. [15] demonstrate the same effect.
![[1860-5397-8-112-4]](/bjoc/content/figures/1860-5397-8-112-4.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 4: (a) c.v. in DMF of 7/2 (red), 6/1 (green) and 27/10 (blue); (b) c.v. in DMF of 27/10 at different scan rates. Current plotted vs V relative to Fc/Fc+.
Figure 4: (a) c.v. in DMF of 7/2 (red), 6/1 (green) and 27/10 (blue); (b) c.v. in DMF of 27/10 at different s...
This instability suggested that it should be difficult to obtain complete reaction when using 26 as a precursor of 10 in the preparative-scale reduction of aryl iodides. The standard two iodides 28 and 30 were tested under slightly different conditions, as mentioned above for donors 9 and 11. Here a surprising outcome was seen. Complete reduction was observed for iodide 28, affording a good isolated yield of 29 (70%) (Scheme 4). However, iodide 30 was reduced by 10 to give the products 32/31//30 in a 2:1.4:1 ratio (1H NMR) with a poor overall recovery of 54%.
![[1860-5397-8-112-i4]](/bjoc/content/inline/1860-5397-8-112-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Use of hybrid donor 10 in reduction of iodoarenes.
Scheme 4: Use of hybrid donor 10 in reduction of iodoarenes.
In both cases, the donor had been prepared in situ by reacting the precursor salt 26 (1.5 equiv) with excess sodium hydride (15 equiv). However, whereas 28 was simply added to the resulting mixture, which included residual excess base, the excess base had been removed by filtration prior to addition of 30. This led us to question whether excess base could be helpful in such reactions and, if so, could the reported instability of other imidazole-based electron donors [15] also be addressed in the presence of base?
To test this, mono-trimethylene precursor 14 [38], was prepared. This compound is the precursor of donor 3. However, earlier CV studies to prepare 3 by reduction of 13 showed that 3 was not a stable compound, as discussed above [16,27]. Treating 14 with excess NaH, and then adding 28 to this reaction mixture pleasingly provided 29 (74%) exclusively (Scheme 5). However, repeating the same reaction, but filtering the excess NaH prior to addition of substrate 28 gave only 11% of reduced product 29, together with starting substrate 28 (84%). The same outcome was seen with a second substrate, 33 [39]. In the presence of excess NaH, reduced product 34 was isolated in 86% yield, whereas when the substrate was added after removal of excess NaH, a lower yield of 34 (9%) was isolated, together with starting substrate 33 (85%).
![[1860-5397-8-112-i5]](/bjoc/content/inline/1860-5397-8-112-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reductive chemistry from disalt 15.
Scheme 5: Reductive chemistry from disalt 15.
How can the base be assisting these reactions? Scheme 6 takes disalt 14 as an example.
![[1860-5397-8-112-i6]](/bjoc/content/inline/1860-5397-8-112-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Rationalisation of effect of excess NaH base.
Scheme 6: Rationalisation of effect of excess NaH base.
Treatment of 14 with two equivalents of NaH would afford donor 3 as shown. This should then react with an iodoarene 37 to afford the dication salt 38 featuring an aryl anion and an iodide as counterions. In these circumstances, we suggest that the aryl anion can abstract a proton rapidly from the periphery of 38 to form reduced arene 39, consistent with our previous studies on deprotonation of pyridinium salts [21]. However, 38 is a dication, and, to attain neutrality, could lose two protons. Compound 3 could be a strong base (in support of this, we have witnessed complete conversion of the analogous donor 2 to form 40 by rapid exchange in CD3CN as solvent; see Supporting Information File 1). We also note that in the previously reported electrochemical studies, irreversible behaviour was always observed in acetonitrile, consistent with a role of this solvent as a proton donor in the decomposition, whereas it was much more rarely reported in the much less acidic solvent DMF [14,15], and if the experiment were conducted with no excess of NaH base, 3 could itself act as a base. Protonation of 3 would afford 36, capable of undergoing spontaneous fragmentation to 35 [40-43] thereby lowering the concentration of donor. However, excess sodium hydride can inhibit the protonation of 3 by competing for protons. (Notably, in earlier studies on the reversibility of formation of imidazoline-based donors, Liu and Lemal inhibited dissociation by adding KH as base [43]).
In the cyclic voltammetry case, 3 would be generated from disalt 38. As 3 starts to be generated, it can deprotonate 38, lowering the concentrations of 3 and therefore lowering the cathodic current in the CV, as observed for couple 27/10 in Figure 4.
It would then remain to explain why some imidazole-derived donors, e.g., 2 and 11, apparently are not affected, or are much less affected by this problem. Protonation of 2 leads to 41, and it is likely that the equilibrium fragmentation of this compound to 42 is less favourable than the fragmentation of 36 to 35 because of the restriction imposed by the second trimethylene bridge [16]. (Compound 41 has not previously been reported, but its existence is clear from its preparation here by deprotonation of 12 with one equivalent of NaH (see Supporting Information File 1). For protonated forms of other tetraaza donors, see [44,45]).
Compound 11 is likely to deprotonate dication 21 analogously to the previous examples. If 43 results from this protonation, then it should undergo easy fragmentation to 45, featuring a pyridinium salt and an imidazolylidene, and in these circumstances, it would be difficult to understand why this electron-donor system works well. However, if isomeric compound 44 is the product of protonation, then its fragmentation to 46, featuring an imidazolium salt and a pyridinylidene may well be relatively disfavoured. The pyridinylidene carbene in 46 should be less stabilised than the imidazolylidene carbene in 45, since in the former case, the carbene is stabilised by only one neighbouring N atom. Keeping the inter-ring C–C bond in 44 could make reversion to donor 11 much more straightforward (than for 45/43). This would then fit with our observations. Computational studies show indeed that 44 lies 87 kJ/mol below 43, and so the preferred protonated form is 44. Furthermore, fragmentation of 44 to 46 is indeed difficult, being uphill by 75 kJ/mol. This may explain why donor 11 is not significantly affected when excess base is absent.
Conclusion
Hybrid organic super-electron-donors have been prepared, and their reactivity with aryl iodides tested. The donors show evidence for transfer of one electron or two electrons, dependent on their structure. Excess sodium hydride has a very beneficial effect on yields of products in certain cases, and a rationale for this has been proposed.
Supporting Information
| Supporting Information File 1: Experimental and computational details. | ||
| Format: PDF | Size: 557.7 KB | Download |
References
-
Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4946. doi:10.1021/cr030666m
Return to citation in text: [1] -
Wudl, F.; Kaplan, M. L.; Engler, E. M.; Patel, V. V. 2,2′-Bi-1,3-Dithiolylidene (Tetrathiafulvalene, TTF) and its Radical Cation Salts. In Inorganic Syntheses; Shriver, D. F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 1979; Vol. 19, pp 27–34. doi:10.1002/9780470132500.ch7
Return to citation in text: [1] -
Frère, P.; Skabara, P. J. Chem. Soc. Rev. 2005, 34, 69–98. doi:10.1039/b316392j
Return to citation in text: [1] -
Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Chem. Commun. 1993, 295–297. doi:10.1039/c39930000295
See for uses of TTF in synthesis.
Return to citation in text: [1] -
Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995–1002. doi:10.1039/a900335e
See for uses of TTF in synthesis.
Return to citation in text: [1] -
Médebielle, M.; Dolbier, W. R., Jr. J. Fluorine Chem. 2008, 129, 930–942. doi:10.1016/j.jfluchem.2008.06.029
Return to citation in text: [1] -
Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 1998, 63, 5385–5394. doi:10.1021/jo980201+
Return to citation in text: [1] -
Takechi, N.; Ait-Mohand, S.; Médebielle, M.; Dolbier, W. R., Jr. Tetrahedron Lett. 2002, 43, 4317–4319. doi:10.1016/S0040-4039(02)00800-6
Return to citation in text: [1] -
Since, M.; Terme, T.; Vanelle, P. Tetrahedron 2009, 65, 6128–6134. doi:10.1016/j.tet.2009.05.036
Return to citation in text: [1] -
Juspin, J.; Giuglio-Tonolo, G.; Terme, T.; Vanelle, P. Synthesis 2010, 844–848. doi:10.1055/s-0029-1218590
Return to citation in text: [1] -
Wang, H.-J.; Shi, J.; Fang, M.; Li, Z.; Guo, Q.-X. J. Phys. Org. Chem. 2010, 23, 75–83. doi:10.1002/poc.1590
Return to citation in text: [1] -
Mahesh, M.; Murphy, J. A.; LeStrat, F.; Wessel, H. P. Beilstein J. Org. Chem. 2009, 5, No. 1. doi:10.3762/bjoc.5.1
Return to citation in text: [1] -
Hünig, S.; Scheutzow, D.; Schlaf, H. Justus Liebigs Ann. Chem. 1973, 765, 126–132. doi:10.1002/jlac.19727650113
Return to citation in text: [1] [2] -
Shi, Z.; Thummel, R. P. J. Org. Chem. 1995, 60, 5935–5945. doi:10.1021/jo00123a034
Return to citation in text: [1] [2] [3] -
Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Murphy, J. A.; Khan, T. A.; Zhou, S.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038
Return to citation in text: [1] [2] [3] [4] [5] -
Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Schoenebeck, F.; Murphy, J. A.; Zhou, S.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368–13369. doi:10.1021/ja074417h
Return to citation in text: [1] [2] [3] [4] [5] -
Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g
Return to citation in text: [1] [2] [3] [4] -
Garnier, J.; Murphy, J. A.; Zhou, S.-Z.; Turner, A. T. Synlett 2008, 2127–2131. doi:10.1055/s-2008-1078242
Return to citation in text: [1] [2] [3] [4] -
Cutulic, S. P. Y.; Murphy, J. A.; Farwaha, H.; Zhou, S.-Z.; Chrystal, E. Synlett 2008, 2132–2136. doi:10.1055/s-2008-1078240
Return to citation in text: [1] [2] [3] -
Murphy, J. A.; Schoenebeck, F.; Findlay, N. J.; Thomson, D. W.; Zhou, S.; Garnier, J. J. Am. Chem. Soc. 2009, 131, 6475–6479. doi:10.1021/ja8092746
Return to citation in text: [1] [2] -
Cutulic, S. P. Y.; Findlay, N. J.; Zhou, S.; Chrystal, E. J. T.; Murphy, J. A. J. Org. Chem. 2009, 74, 8713–8718. doi:10.1021/jo901815t
Return to citation in text: [1] [2] [3] -
Garnier, J.; Kennedy, A. R.; Berlouis, L. E. A.; Turner, A. T.; Murphy, J. A. Beilstein J. Org. Chem. 2010, 6, No. 73. doi:10.3762/bjoc.6.73
Return to citation in text: [1] [2] [3] -
Jolly, P. I.; Fleary-Roberts, N.; O'Sullivan, S.; Doni, E.; Zhou, S.; Murphy, J. A. Org. Biomol. Chem. 2012, 10, 100000–111111. doi:10.1039/c2ob25116g
Return to citation in text: [1] [2] [3] -
Jolly, P. I.; Zhou, S.; Thomson, D. W.; Garnier, J.; Parkinson, J. A.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2012, 3, 1675–1679. doi:10.1039/C2SC20054F
Return to citation in text: [1] [2] [3] [4] [5] -
Porter, W. W., III; Vaid, T. P.; Rheingold, A. L. J. Am. Chem. Soc. 2005, 127, 16559–16566. doi:10.1021/ja053084q
Return to citation in text: [1] -
Porter, W. W., III; Vaid, T. P. J. Org. Chem. 2005, 70, 5028–5035. doi:10.1021/jo050328g
Return to citation in text: [1] -
Vaid, T. P.; Lytton-Jean, A. K.; Barnes, B. C. Chem. Mater. 2003, 15, 4292–4299. doi:10.1021/cm034646c
Return to citation in text: [1] -
Peters, A.; Kaifer, E.; Himmel, H.-J. Eur. J. Org. Chem. 2008, 5907–5914. doi:10.1002/ejoc.200800900
Return to citation in text: [1] -
Peters, A.; Trumm, C.; Reinmuth, M.; Emeljanenko, D.; Kaifer, E.; Himmel, H.-J. Eur. J. Inorg. Chem. 2009, 3791–3800. doi:10.1002/ejic.200900399
Return to citation in text: [1] -
Vitske, V.; König, C.; Hübner, O.; Kaifer, E.; Himmel, H.-J. Eur. J. Inorg. Chem. 2010, 115–126. doi:10.1002/ejic.200900724
Return to citation in text: [1] -
Cid, M. H. B.; Holzgrabe, U.; Kostenis, E.; Mohr, K.; Traenkle, C. J. Med. Chem. 1994, 37, 1439–1445. doi:10.1021/jm00036a008
Return to citation in text: [1] -
Curran, D. P.; Totleben, M. J. J. Am. Chem. Soc. 1992, 114, 6050–6058. doi:10.1021/ja00041a024
Return to citation in text: [1] [2] [3] -
Vece, V.; Ricci, J.; Poulain-Martini, S.; Nava, P.; Carissan, Y.; Humbel, S.; Duñach, E. Eur. J. Org. Chem. 2010, 6239–6248. doi:10.1002/ejoc.201000738
Return to citation in text: [1] -
Aldabbagh, F.; Bowman, W. R. Tetrahedron 1999, 55, 4109–4122. doi:10.1016/S0040-4020(99)00104-0
Return to citation in text: [1] -
Khan, S. S.; Liebscher, J. Synthesis 2010, 2609–2615. doi:10.1055/s-0029-1218837
Return to citation in text: [1] -
Oldfield, M. F.; Chen, L.; Botting, N. P. Tetrahedron 2004, 60, 1887–1893. doi:10.1016/j.tet.2003.12.033
Return to citation in text: [1] -
Alder, R. W.; Blake, M. E.; Chaker, L.; Harvey, J. N.; Paolini, F.; Schütz, J. Angew. Chem., Int. Ed. 2004, 43, 5896–5911. doi:10.1002/anie.200400654
Return to citation in text: [1] -
Liu, Y.; Lindner, P. E.; Lemal, D. M. J. Am. Chem. Soc. 1999, 121, 10626–10627. doi:10.1021/ja9922678
Return to citation in text: [1] -
Hahn, F. E.; Wittenbecher, L.; Le Van, D.; Fröhlich, R. Angew. Chem., Int. Ed. 2000, 39, 541–544. doi:10.1002/(SICI)1521-3773(20000204)39:3<541::AID-ANIE541>3.0.CO;2-B
Return to citation in text: [1] -
Liu, Y.; Lemal, D. M. Tetrahedron Lett. 2000, 41, 599–602. doi:10.1016/S0040-4039(99)02161-9
Return to citation in text: [1] [2] -
Chen, Y. T.; Jordan, F. J. Org. Chem. 1991, 56, 5029–5038. doi:10.1021/jo00017a010
Return to citation in text: [1] -
Alder, R. W.; Chaker, L.; Paolini, F. P. V. Chem. Commun. 2004, 2172–2173. doi:10.1039/B409112D
Return to citation in text: [1]
| 17. | Murphy, J. A.; Khan, T. A.; Zhou, S.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038 |
| 18. | Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 36. | Vece, V.; Ricci, J.; Poulain-Martini, S.; Nava, P.; Carissan, Y.; Humbel, S.; Duñach, E. Eur. J. Org. Chem. 2010, 6239–6248. doi:10.1002/ejoc.201000738 |
| 1. | Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4946. doi:10.1021/cr030666m |
| 2. | Wudl, F.; Kaplan, M. L.; Engler, E. M.; Patel, V. V. 2,2′-Bi-1,3-Dithiolylidene (Tetrathiafulvalene, TTF) and its Radical Cation Salts. In Inorganic Syntheses; Shriver, D. F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 1979; Vol. 19, pp 27–34. doi:10.1002/9780470132500.ch7 |
| 3. | Frère, P.; Skabara, P. J. Chem. Soc. Rev. 2005, 34, 69–98. doi:10.1039/b316392j |
| 4. |
Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Chem. Commun. 1993, 295–297. doi:10.1039/c39930000295
See for uses of TTF in synthesis. |
| 5. |
Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995–1002. doi:10.1039/a900335e
See for uses of TTF in synthesis. |
| 17. | Murphy, J. A.; Khan, T. A.; Zhou, S.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038 |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 27. | Jolly, P. I.; Zhou, S.; Thomson, D. W.; Garnier, J.; Parkinson, J. A.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2012, 3, 1675–1679. doi:10.1039/C2SC20054F |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 27. | Jolly, P. I.; Zhou, S.; Thomson, D. W.; Garnier, J.; Parkinson, J. A.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2012, 3, 1675–1679. doi:10.1039/C2SC20054F |
| 17. | Murphy, J. A.; Khan, T. A.; Zhou, S.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038 |
| 18. | Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 19. | Schoenebeck, F.; Murphy, J. A.; Zhou, S.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368–13369. doi:10.1021/ja074417h |
| 20. | Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g |
| 21. | Garnier, J.; Murphy, J. A.; Zhou, S.-Z.; Turner, A. T. Synlett 2008, 2127–2131. doi:10.1055/s-2008-1078242 |
| 22. | Cutulic, S. P. Y.; Murphy, J. A.; Farwaha, H.; Zhou, S.-Z.; Chrystal, E. Synlett 2008, 2132–2136. doi:10.1055/s-2008-1078240 |
| 23. | Murphy, J. A.; Schoenebeck, F.; Findlay, N. J.; Thomson, D. W.; Zhou, S.; Garnier, J. J. Am. Chem. Soc. 2009, 131, 6475–6479. doi:10.1021/ja8092746 |
| 24. | Cutulic, S. P. Y.; Findlay, N. J.; Zhou, S.; Chrystal, E. J. T.; Murphy, J. A. J. Org. Chem. 2009, 74, 8713–8718. doi:10.1021/jo901815t |
| 25. | Garnier, J.; Kennedy, A. R.; Berlouis, L. E. A.; Turner, A. T.; Murphy, J. A. Beilstein J. Org. Chem. 2010, 6, No. 73. doi:10.3762/bjoc.6.73 |
| 26. | Jolly, P. I.; Fleary-Roberts, N.; O'Sullivan, S.; Doni, E.; Zhou, S.; Murphy, J. A. Org. Biomol. Chem. 2012, 10, 100000–111111. doi:10.1039/c2ob25116g |
| 27. | Jolly, P. I.; Zhou, S.; Thomson, D. W.; Garnier, J.; Parkinson, J. A.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2012, 3, 1675–1679. doi:10.1039/C2SC20054F |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 18. | Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 39. | Oldfield, M. F.; Chen, L.; Botting, N. P. Tetrahedron 2004, 60, 1887–1893. doi:10.1016/j.tet.2003.12.033 |
| 13. | Hünig, S.; Scheutzow, D.; Schlaf, H. Justus Liebigs Ann. Chem. 1973, 765, 126–132. doi:10.1002/jlac.19727650113 |
| 14. | Shi, Z.; Thummel, R. P. J. Org. Chem. 1995, 60, 5935–5945. doi:10.1021/jo00123a034 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 24. | Cutulic, S. P. Y.; Findlay, N. J.; Zhou, S.; Chrystal, E. J. T.; Murphy, J. A. J. Org. Chem. 2009, 74, 8713–8718. doi:10.1021/jo901815t |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 6. | Médebielle, M.; Dolbier, W. R., Jr. J. Fluorine Chem. 2008, 129, 930–942. doi:10.1016/j.jfluchem.2008.06.029 |
| 7. | Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 1998, 63, 5385–5394. doi:10.1021/jo980201+ |
| 8. | Takechi, N.; Ait-Mohand, S.; Médebielle, M.; Dolbier, W. R., Jr. Tetrahedron Lett. 2002, 43, 4317–4319. doi:10.1016/S0040-4039(02)00800-6 |
| 9. | Since, M.; Terme, T.; Vanelle, P. Tetrahedron 2009, 65, 6128–6134. doi:10.1016/j.tet.2009.05.036 |
| 10. | Juspin, J.; Giuglio-Tonolo, G.; Terme, T.; Vanelle, P. Synthesis 2010, 844–848. doi:10.1055/s-0029-1218590 |
| 11. | Wang, H.-J.; Shi, J.; Fang, M.; Li, Z.; Guo, Q.-X. J. Phys. Org. Chem. 2010, 23, 75–83. doi:10.1002/poc.1590 |
| 12. | Mahesh, M.; Murphy, J. A.; LeStrat, F.; Wessel, H. P. Beilstein J. Org. Chem. 2009, 5, No. 1. doi:10.3762/bjoc.5.1 |
| 13. | Hünig, S.; Scheutzow, D.; Schlaf, H. Justus Liebigs Ann. Chem. 1973, 765, 126–132. doi:10.1002/jlac.19727650113 |
| 14. | Shi, Z.; Thummel, R. P. J. Org. Chem. 1995, 60, 5935–5945. doi:10.1021/jo00123a034 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 17. | Murphy, J. A.; Khan, T. A.; Zhou, S.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038 |
| 18. | Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 19. | Schoenebeck, F.; Murphy, J. A.; Zhou, S.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368–13369. doi:10.1021/ja074417h |
| 20. | Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g |
| 21. | Garnier, J.; Murphy, J. A.; Zhou, S.-Z.; Turner, A. T. Synlett 2008, 2127–2131. doi:10.1055/s-2008-1078242 |
| 22. | Cutulic, S. P. Y.; Murphy, J. A.; Farwaha, H.; Zhou, S.-Z.; Chrystal, E. Synlett 2008, 2132–2136. doi:10.1055/s-2008-1078240 |
| 23. | Murphy, J. A.; Schoenebeck, F.; Findlay, N. J.; Thomson, D. W.; Zhou, S.; Garnier, J. J. Am. Chem. Soc. 2009, 131, 6475–6479. doi:10.1021/ja8092746 |
| 24. | Cutulic, S. P. Y.; Findlay, N. J.; Zhou, S.; Chrystal, E. J. T.; Murphy, J. A. J. Org. Chem. 2009, 74, 8713–8718. doi:10.1021/jo901815t |
| 25. | Garnier, J.; Kennedy, A. R.; Berlouis, L. E. A.; Turner, A. T.; Murphy, J. A. Beilstein J. Org. Chem. 2010, 6, No. 73. doi:10.3762/bjoc.6.73 |
| 26. | Jolly, P. I.; Fleary-Roberts, N.; O'Sullivan, S.; Doni, E.; Zhou, S.; Murphy, J. A. Org. Biomol. Chem. 2012, 10, 100000–111111. doi:10.1039/c2ob25116g |
| 27. | Jolly, P. I.; Zhou, S.; Thomson, D. W.; Garnier, J.; Parkinson, J. A.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2012, 3, 1675–1679. doi:10.1039/C2SC20054F |
| 28. | Porter, W. W., III; Vaid, T. P.; Rheingold, A. L. J. Am. Chem. Soc. 2005, 127, 16559–16566. doi:10.1021/ja053084q |
| 29. | Porter, W. W., III; Vaid, T. P. J. Org. Chem. 2005, 70, 5028–5035. doi:10.1021/jo050328g |
| 30. | Vaid, T. P.; Lytton-Jean, A. K.; Barnes, B. C. Chem. Mater. 2003, 15, 4292–4299. doi:10.1021/cm034646c |
| 31. | Peters, A.; Kaifer, E.; Himmel, H.-J. Eur. J. Org. Chem. 2008, 5907–5914. doi:10.1002/ejoc.200800900 |
| 32. | Peters, A.; Trumm, C.; Reinmuth, M.; Emeljanenko, D.; Kaifer, E.; Himmel, H.-J. Eur. J. Inorg. Chem. 2009, 3791–3800. doi:10.1002/ejic.200900399 |
| 33. | Vitske, V.; König, C.; Hübner, O.; Kaifer, E.; Himmel, H.-J. Eur. J. Inorg. Chem. 2010, 115–126. doi:10.1002/ejic.200900724 |
| 26. | Jolly, P. I.; Fleary-Roberts, N.; O'Sullivan, S.; Doni, E.; Zhou, S.; Murphy, J. A. Org. Biomol. Chem. 2012, 10, 100000–111111. doi:10.1039/c2ob25116g |
| 38. | Khan, S. S.; Liebscher, J. Synthesis 2010, 2609–2615. doi:10.1055/s-0029-1218837 |
| 25. | Garnier, J.; Kennedy, A. R.; Berlouis, L. E. A.; Turner, A. T.; Murphy, J. A. Beilstein J. Org. Chem. 2010, 6, No. 73. doi:10.3762/bjoc.6.73 |
| 19. | Schoenebeck, F.; Murphy, J. A.; Zhou, S.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368–13369. doi:10.1021/ja074417h |
| 37. | Aldabbagh, F.; Bowman, W. R. Tetrahedron 1999, 55, 4109–4122. doi:10.1016/S0040-4020(99)00104-0 |
| 21. | Garnier, J.; Murphy, J. A.; Zhou, S.-Z.; Turner, A. T. Synlett 2008, 2127–2131. doi:10.1055/s-2008-1078242 |
| 22. | Cutulic, S. P. Y.; Murphy, J. A.; Farwaha, H.; Zhou, S.-Z.; Chrystal, E. Synlett 2008, 2132–2136. doi:10.1055/s-2008-1078240 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 20. | Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g |
| 17. | Murphy, J. A.; Khan, T. A.; Zhou, S.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038 |
| 18. | Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 18. | Murphy, J. A.; Zhou, S.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 19. | Schoenebeck, F.; Murphy, J. A.; Zhou, S.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368–13369. doi:10.1021/ja074417h |
| 35. | Curran, D. P.; Totleben, M. J. J. Am. Chem. Soc. 1992, 114, 6050–6058. doi:10.1021/ja00041a024 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 19. | Schoenebeck, F.; Murphy, J. A.; Zhou, S.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368–13369. doi:10.1021/ja074417h |
| 21. | Garnier, J.; Murphy, J. A.; Zhou, S.-Z.; Turner, A. T. Synlett 2008, 2127–2131. doi:10.1055/s-2008-1078242 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 14. | Shi, Z.; Thummel, R. P. J. Org. Chem. 1995, 60, 5935–5945. doi:10.1021/jo00123a034 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 40. | Alder, R. W.; Blake, M. E.; Chaker, L.; Harvey, J. N.; Paolini, F.; Schütz, J. Angew. Chem., Int. Ed. 2004, 43, 5896–5911. doi:10.1002/anie.200400654 |
| 41. | Liu, Y.; Lindner, P. E.; Lemal, D. M. J. Am. Chem. Soc. 1999, 121, 10626–10627. doi:10.1021/ja9922678 |
| 42. | Hahn, F. E.; Wittenbecher, L.; Le Van, D.; Fröhlich, R. Angew. Chem., Int. Ed. 2000, 39, 541–544. doi:10.1002/(SICI)1521-3773(20000204)39:3<541::AID-ANIE541>3.0.CO;2-B |
| 43. | Liu, Y.; Lemal, D. M. Tetrahedron Lett. 2000, 41, 599–602. doi:10.1016/S0040-4039(99)02161-9 |
| 20. | Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g |
| 35. | Curran, D. P.; Totleben, M. J. J. Am. Chem. Soc. 1992, 114, 6050–6058. doi:10.1021/ja00041a024 |
| 15. | Ames, J. R.; Houghtaling, M. A.; Terrian, D. L.; Mitchell, T. P. Can. J. Chem. 1997, 75, 28–36. doi:10.1139/v97-004 |
| 35. | Curran, D. P.; Totleben, M. J. J. Am. Chem. Soc. 1992, 114, 6050–6058. doi:10.1021/ja00041a024 |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 44. | Chen, Y. T.; Jordan, F. J. Org. Chem. 1991, 56, 5029–5038. doi:10.1021/jo00017a010 |
| 45. | Alder, R. W.; Chaker, L.; Paolini, F. P. V. Chem. Commun. 2004, 2172–2173. doi:10.1039/B409112D |
| 34. | Cid, M. H. B.; Holzgrabe, U.; Kostenis, E.; Mohr, K.; Traenkle, C. J. Med. Chem. 1994, 37, 1439–1445. doi:10.1021/jm00036a008 |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
| 43. | Liu, Y.; Lemal, D. M. Tetrahedron Lett. 2000, 41, 599–602. doi:10.1016/S0040-4039(99)02161-9 |
| 27. | Jolly, P. I.; Zhou, S.; Thomson, D. W.; Garnier, J.; Parkinson, J. A.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2012, 3, 1675–1679. doi:10.1039/C2SC20054F |
| 16. | Taton, T. A.; Chen, P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1011–1013. doi:10.1002/anie.199610111 |
© 2012 Garnier et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)