
Abstract
Aurachins are myxobacterial 3-farnesyl-4(1H)-quinolone derived compounds initially described as respiratory chain inhibitors, more specifically as inhibitors of various cytochrome complexes. They are also known as potent antibiotic compounds. We describe herein the first synthesis of aurachin D through a key Conrad–Limpach reaction. The same strategy was used to reach some ring as opposed to chain analogues, allowing for the description of structure–activity relationships. Biological screening of the analogues showed antiparasitic, cytotoxic, antibacterial and antifungal activities, and depletion of the mitochondrial membrane potential. The strongest activity was found on Plasmodium falciparum with a selectivity index of 345, compared to Vero cells, for the natural product and its geranyl analogue. The loss of mitochondrial membrane potential induced by aurachins in human U-2 OS osteosarcoma cells was studied, showing the best activity for aurachin D and a naphthalene analogue, yet without totally explaining the observed cytotoxic activity of the compounds. Finally, a synthetic entry is given to the complete carboheterocyclic core of aurachin H through the N-oxidation/epoxidation of aurachin D and a shorter chain analogue, followed by subsequent biomimetic cyclization.
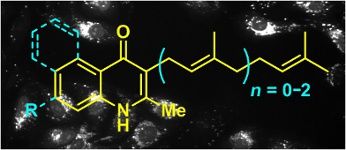
Graphical Abstract
Introduction
The four aurachins A–D (1–4) were first isolated from the myxobacterium Stigmatella aurantiaca strain Sg a15 in 1987 (Figure 1) [1]. This class of compounds is characterized by a quinolone or quinoline nucleus substituted in position 3 or 4 by a farnesyl chain. In addition, aurachins can further be functionalized by biosynthetic oxidative processes [2-5]. Since then, additional members of this natural product family were discovered, namely aurachins E–R, from the same strain [3,6], from Stigmatella erecta [7] or from Rhodococcus sp. Acta 2259 [8].
![[1860-5397-9-176-1]](/bjoc/content/figures/1860-5397-9-176-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The 2-methyl-4(1H)-quinolone compounds: aurachins and endochin.
Figure 1: The 2-methyl-4(1H)-quinolone compounds: aurachins and endochin.
Initially, aurachins were found to block NADH oxidation in mammalian submitochondrial particles (aurachins C and D being the most active) [1] by the inhibition of complexes I and III of the mitochondrial respiratory chain [9,10]. They are indeed analogues of the physiological ubiquinol and vitamins K. They also affect the photosystem II and the cytochrome bf complex of thylakoids in photosynthetic microorganisms [11]. Deeper studies led to the conclusion that these compounds are powerful inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd, with dissociation constants in the range of 10 nM. Aurachin D (4) was shown to selectively inhibit the cytochrome bd complex [12].
With such biological properties, aurachins and their analogues are also strong antimicrobial agents. Aurachins C and D (3 and 4) have been described as better inhibitors against Gram-positive bacteria compared to aurachins A and B (1 and 2) [1]. All were cytotoxic on L929 mouse fibroblasts with IC50 values in the range of 1–3 µg/mL and some of them strongly inhibited Plasmodium falciparum at an IC50 around 20 ng/mL (i.e., aurachins B, C and E, whereas aurachin D was 100- to 200-fold less active) [6]. This antimalarial activity was comparable to that of endochin (5) [13,14] and 2-methyl-4(1H)-quinolone analogues as determined in the course of important structure–activity optimization work [15]. In addition, many N-hydroxyquinolone derivatives with variable side chains displayed strong antiplasmodial activity at IC50 of ca. 1 nM [16].
In fact, owing to their occurrence in many natural products [17,18] and to the high frequency of their biological activities, compounds containing the 4(1H)-quinolone nucleus may be considered as important privileged structures [19-21] for medicinal purposes. This motivated the work therein, dealing with the first synthesis of aurachin D (4). On this occasion, the preliminary synthesis of ring versus chain analogues was undertaken, focusing on variations of the chain length or on the modulation of electron density of the aromatic ring including surface extension. Oxidative conditions were finally considered en route to N-oxide analogues, eventually leading to the heterocyclic core of aurachin H (6) (Figure 1).
Results and Discussion
Synthesis of aurachin D (4) and analogues with chain-length variations
Aurachin D (4) was synthesized in a three-step sequence from ethyl acetoacetate (7) (Scheme 1). Alkylation of the sodium salt of 7 in the presence of farnesyl bromide furnished the ethyl 2-farnesyl(acetoacetate) 8a in 82% yield. Upon treatment with aniline, a Conrad–Limpach process [22-25] gave aurachin D (4) in 72% yield. This transformation was performed in toluene under reflux over 3 Å molecular sieves by formation of an imine intermediate, which was cyclized at 250 °C by Claisen condensation. Precipitation of the product by addition of pentane and subsequent filtration gave the pure synthetic aurachin D (4) which shared the same spectral properties as the natural product [3]. Analogous compounds bearing a shorter side chain (geranyl: 9 [26], prenyl: 10, or methyl: 11) were obtained by the same reaction sequence through the respective 2-alkyl(acetoacetate) 8b–8d, yet with unexpectedly lower yields for the shorter-chain analogues 10 and 11.
![[1860-5397-9-176-i1]](/bjoc/content/inline/1860-5397-9-176-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of aurachin D (4) and geranyl (9), prenyl (10) and methyl (11) analogues.
Scheme 1: Synthesis of aurachin D (4) and geranyl (9), prenyl (10) and methyl (11) analogues.
This method allowed for the rapid gram-scale synthesis of aurachin D (max. 1.1 g made in this work) with good yields and in a minimum of steps, which has to be compared to the extraction yield of the natural product. Indeed from a culture batch of 60 L, the production of compound 4 by Stigmatella aurantiaca Sg a15 under basal conditions was described as less than 1 mg/L after about 6 days [1]. This production was improved by the addition of anthranilic acid (i.e., the aurachin biosynthetic precursor) to the culture medium, furnishing 40 mg of 4 from 120 L [3].
Aurachin D analogues with variations of the aromatic cycles
The same methodology was tentatively applied to other anilines such as 2-naphthylamine (12), p-anisidine (13), 3,4-(methylenedioxy)aniline (14), 3,4-difluoroaniline (15) and methyl 3-aminobenzoate (16), using the 2-farnesyl(acetoacetate) 8a. Not surprisingly, only the electron-rich anilines 12–14 were reactive enough to furnish the expected 3-farnesyl-4(1H)-quinolones 17–19 in moderate to good yields (Table 1). The electron-poor anilines 15 and 16 were unreactive under these experimental conditions, although the intermediate imine was supposedly formed during the reaction but not isolated.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-176-i3.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-176-i4.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-176-i5.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-176-i6.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-176-i7.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-176-i8.svg?max-width=637&scale=1.0)
The carbocyclic core of aurachin H
Aurachin H (6) was isolated from Stigmatella aurantiaca at the same time as the aurachin series A–L [3]. Biosynthetically, it may arise from the double oxidation of the quinolone nitrogen and the olefin epoxidation (2',3'-position) of the farnesyl chain, with the epoxide being ring-opened with 5-exo selectivity by the 4-hydroxy group of the resulting 4-hydroxyquinoline N-oxide. Taking the geranyl analogue 9 as a model compound (Scheme 2), after benzylic protection of the 4-hydroxyquinoline form into 20, oxidative conditions were applied to perform both N-oxidation and olefin epoxidations, in the presence of three equivalents of m-chloroperbenzoic acid (mCPBA). The use of smaller quantities of the oxidant only led to complex mixtures of nonselectively oxidized products. The crude product 21a was then submitted to benzyl group hydrogenolysis. As expected upon completion of the reaction, the 4,5-dihydrofuro[3,2-c]quinoline N-oxide ring system 22 was obtained as a racemic pair of diastereoisomers in a 1:1 ratio, supposedly formed through the spontaneous cyclization of intermediate 21b, which was not isolated. We also performed the same reaction sequence from aurachin D (4), giving the aurachin-H diepoxide with comparable yields [27]. After optimizing the selectivity of the epoxidation, this reaction may furnish a rapid strategy toward aurachin H (6) and related compounds in a bio-inspired manner.
![[1860-5397-9-176-i2]](/bjoc/content/inline/1860-5397-9-176-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Strategy toward the heterocyclic core of aurachin H.
Scheme 2: Strategy toward the heterocyclic core of aurachin H.
Biological activities
The cytotoxic activity of aurachin D (4) and analogues 9–11 and 17–19 was evaluated in growth inhibition experiments on mammalian cell lines (Table 2). After 5 days treatment, half inhibitory concentrations (IC50) were determined for human HCT-116 colon carcinoma and human K562 myelogenous leukemia cells. The synthesized natural product 4 and its geranyl and methoxy analogues (9 and 18) were the most active amongst the tested derivatives with IC50 values in the low µg/mL range, whereas analogues 10, 11, 17, and 19 displayed no significant toxicity up to concentrations of 10 µg/mL.
Table 2: Cytotoxicity tests on HCT-116 human colon carcinoma and K562 myelogenous leukemia cells (showing IC50: half inhibitory concentrations, µg/mL)a,b.
| Compounds: | 4 | 9 | 10 | 11 | 17 | 18 | 19 |
|---|---|---|---|---|---|---|---|
| HCT-116 | 2.23 ± 0.76 | 2.52 ± 0.48 | >10 | >10 | >10 | 1.64 ± 0.47 | >10 |
| K562 | 0.65 ± 0.16 | 1.50 ± 0.31 | ca. 10 | >10 | ca. 10 | 1.15 ± 0.22 | >10 |
aActivities on Vero cells (nontumor cells) were all at IC50 >10 µg/mL, with aurachin D (4) being cytotoxic at 13.8 µg/mL, except compound 19 at 4.4 µg/mL (the aurachin H analogue 22 was also not active at any concentration up to 50 µg/mL); bAll tests performed in triplicate; values were calculated by sigmoidal curve fitting and are displayed as mean ±SD.
Since aurachins are described as electron-transport inhibitors of the mitochondrial respiratory chain (complex I and III) [28], we analyzed the synthetic analogues in high-content screening experiments with respect to a decrease of the mitochondrial membrane potential (MMP). Human U-2 OS osteosarcoma cells were treated for 16 hours with aurachins 4, 9–11, and 17–19 at concentrations between 1 and 10 µg/mL, and mitochondria were labeled with tetramethylrhodamine methyl ester (TMRM) in order to assess changes in MMP (Figure 2). Most prominent concentration-dependent effects were determined for aurachin D (4) and one of its analogues (17). At concentrations as low as 5 µg/mL, 4 and 17 significantly reduced the MMP, which was further diminished at higher concentrations (Figure 2A). Other analogues with aromatic variations (18 and 19) were essentially inactive in the tested concentration range, although aurachin analogue 18 was determined to have a cytotoxic activity (IC50) at ca. 1 µg/mL. This might be due to differential effects on different cell lines, a different time course of electron-transport inhibition, or a different mode of inducing apoptosis (e.g., via enhanced ROS formation) [29]. However, at the highest assay concentration also shorter chain analogues 9–11 reduced the MMP to approximately 60–80% of the control value as exemplarily shown for geranyl analogue 9 (Figure 2B).
![[1860-5397-9-176-2]](/bjoc/content/figures/1860-5397-9-176-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (A) Loss of mitochondrial membrane potential in human U-2 OS osteosarcoma cells that were treated with aurachin D (4) and analogue 17. Images were acquired on a BD Pathway855 automated microscope and subsequently processed in AttoVision v1.6.2. The relative MMP was calculated based on TMRM fluorescence intensity in the cytoplasmic segments. The value of the negative control (untreated cells) was set to 100% and the value of FCCP-treated cells (positive control) was set to 0%. Bars represent the mean ±SEM of all cellular segments within a well. (B) Representative HCS images of U-2 OS cells that were treated with aurachin D (4) or the geranyl analogue 9. Cells were labeled with TMRM and imaged in the rhodamine channel at 200× magnification.
Figure 2: (A) Loss of mitochondrial membrane potential in human U-2 OS osteosarcoma cells that were treated w...
Antiparasitic activities were evaluated on the chloroquine-resistant Plasmodium falciparum FcB1 and on Trypanosoma brucei gambiense. Aurachin D (4) and its geranyl analogue 9 showed a strong antiplasmodial activity at 0.04 and 0.07 µg/mL, respectively, while analogues 10, 11 and 17–19 were 10- to 100-fold less active (Table 3). Based on the simplest but less active 3-methyl analogue 11, we compared the activity with the commercial 2-methyl-4(1H)-quinolone (23), the 3-bromo-2-methyl-4(1H)-quinolone (24) and N-hydroxy-2,3-dimethyl-4(1H)-quinolone (25), showing the importance of a substituent in position 3. The N-hydroxy substitution of 25 had no effect on the activity of the 2,3-dimethylquinolone core (compared to 11). Due to the lack of the farnesyl chain, this comparison of the quinolone substitution effect yet diverges from previous observations showing an approximately 200-fold higher activity of aurachin C (3) compared to aurachin D (4) (which however displayed strong activity on our FcB1 strain) [6]. Comparatively, it is known from the literature that farnesol alone is poorly antiplasmodial with an IC50 of 64 µM [30], showing the crucial importance of the quinolone core for this strong activity.
Table 3: Antiparasitic activities on Plasmodium falciparum FcB1 and Trypanosoma brucei gambiense (IC50, µg/mL).a
| Compounds: | 4 | 9 | 10 | 11 | 17 | 18 | 19 | 23 | 24 | 25 |
|---|---|---|---|---|---|---|---|---|---|---|
| P. falciparum FcB1 | 0.04 ± 0.02 | 0.07 ± 0.02 | 0.55 ± 0.16 | 2.30 ± 1.40 | 0.51 ± 0.18 | 0.20 ± 0.04 | 0.42 ± 0.09 | 7.70 ± 2.50 | 1.10 | 2.70 ± 1.30 |
| T. brucei gambiense | 0.4 | 1.5 | 5.8 | >10 | 0.8 | 1.6 | >10 | >10 | 1.7 | >10 |
aFor P. falciparum FcB1, all tests performed in triplicate (except compound 24); values were calculated by sigmoidal curve fitting and are displayed as mean ±SD.
The antitrypanosomal activity was found much lower than the antiplasmodial activity, with the most active compounds being the natural product 4 and the aromatic analogue 17 at IC50 = 0.4 and 0.8 µg/mL, respectively (Table 3). The geranyl (9) and prenyl (10) analogues still retained some activity at 1.5 and 5.8 µg/mL, respectively. Overall, we found interesting antiparasitic activities for these compounds relative to the cytotoxicity on human Vero cells, with antiplasmodial and antitrypanosomal selectivity indexes of 345 and 35, respectively, for aurachin D (4), which still make it medicinally relevant for antiparasitic purposes [31].
In accordance with previous reports [1], antibacterial activities (Table 4) were mainly observed on Gram-positive bacteria while Gram-negative bacteria were not affected, except Escherichia coli TolC which is deficient in the corresponding multidrug efflux transporter. The tested compounds were neither active on yeast (Candida albicans) nor on molds (Mucor hiemalis, Fusarium oxysporum, Alternaria alternata). The natural compound 4 was one of the most active compounds, but also shorter chain analogues 9 and 10 inhibited, for instance, the growth of Staphylococcus aureus at concentrations in the low µg/mL range, whereas analogue 9 was approximately twice as active as 10. As the 3-methyl analogue 11, aurachin D analogues with variations of the aromatic cycle (17–19) were essentially inactive. Solely analogues 18 and 19 displayed a significant growth inhibitory effect on Bacillus subtilis.
Table 4: Antibacterial activities of 4(1H)-quinolone derivatives (MIC50, µg/mL).a
| Compounds: | 4 | 9 | 10 | 11 | 17 | 18 | 19 |
|---|---|---|---|---|---|---|---|
| Gram-positive | |||||||
| Bacillus subtilis DSM10 | <0.1 | <0.1 | 0.22 ± 0.06 | 49.04 ± 6.26 | >64 | 6.75 ± 0.60 | 4.66 ± 0.50 |
| Micrococcus luteus | 1.47 ± 0.12 | 2.59 ± 0.34 | 14.31 ± 1.29 | >64 | >64 | 49.17 ± 3.94 | >64 |
| Staphylococcus aureus Newman (MSSA) | 5.46 ± 1.05 | 3.96 ± 0.49 | 9.66 ± 2.39 | >64 | >64 | >64 | 34.56 ± 5.13 |
| Staphylococcus aureus DSM11822 (MRSA) | 2.10 ± 0.19 | 4.50 ± 0.74 | 11.3 ± 1.44 | >64 | >64 | >64 | >64 |
| Gram-negative | |||||||
| Escherichia coli DH5α (wt) | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| Escherichia coli (TolC) | 8.36 ± 1.00 | 1.41 ± 0.45 | 6.55 ± 1.36 | >64 | >64 | >64 | >64 |
| Klebsiella pneumoniae DSM30104 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
aAll tests performed in duplicate; values were calculated by sigmoidal curve fitting and are displayed as mean ±SD.
Conclusion
During this work, not only the total synthesis of aurachin D (4) was accomplished, but also that of analogues with variations in chain length or aromatic cycle. The general strategy involved a key Conrad–Limpach synthesis of quinolones. Additionally, we were able to build the carboheterocyclic core of aurachin H (6) under epoxidation conditions, i.e., the 4,5-dihydrofuro[2,3-c]quinoline N-oxide, and thus to synthesize chain analogues of 6. Biological investigations demonstrated the superiority of the natural product 4 regarding its inhibitory effect on Gram-positive bacteria, human cancer cell lines, and above all on the parasites Plasmodium falciparum FcB1 (with an excellent selectivity index of 345) and Trypanosoma brucei gambiense. We found that the biological activities were mostly conserved with the geranyl analogue 9. Also other shorter chain analogues (10 and 11) retained some activities. Inhibitory concentrations were, however, by one or two orders of magnitude higher than those determined for aurachin D (4) and its geranyl analogue 9. Variants with modifications of the aromatic cycle (17, 18) were generally less potent although it was found that the methoxy variant 18 is cytotoxic on human cancer cell lines. Indeed, this cytotoxic effect might be rather unspecific or only in part linked to inhibition of the electron transport chain. Finally, the biological activity of the compounds was assessed in a high-content screen on mitochondrial dysfunction. For aurachin D (4) and its shorter-chain analogues (9–11) we found that the mitochondrial membrane potential was reduced to approximately 50% and 60–80% of the control, respectively. Analogues 18 and 19 with variations of the aromatic cycle were inactive up to concentrations of 10 µg/mL. However, analogue 17 strongly diminished the MMP, whereas it remains to be elucidated whether this effect is induced by selectively blocking the electron transport chain or by other apoptotic processes, which, in turn, induce a loss of MMP.
Supporting Information
Supporting information is available, featuring the synthetic procedures and spectral data for compounds 9, 10, 17–22.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 153.3 KB | Download |
References
-
Kunze, B.; Höfle, G.; Reichenbach, H. J. Antibiot. 1987, 40, 258–265. doi:10.7164/antibiotics.40.258
Return to citation in text: [1] [2] [3] [4] [5] -
Sandmann, A.; Dickschat, J.; Jenke-Kodama, H.; Kunze, B.; Dittmann, E.; Müller, R. Angew. Chem., Int. Ed. 2007, 46, 2712–2716. doi:10.1002/anie.200603513
Return to citation in text: [1] -
Höfle, G.; Kunze, B. J. Nat. Prod. 2008, 71, 1843–1849. doi:10.1021/np8003084
Return to citation in text: [1] [2] [3] [4] [5] -
Pistorius, D.; Li, Y.; Sandmann, A.; Müller, R. Mol. BioSyst. 2011, 7, 3308–3315. doi:10.1039/c1mb05328k
Return to citation in text: [1] -
Katsuyama, Y.; Harmrolfs, K.; Pistorius, D.; Li, Y.; Müller, R. Angew. Chem., Int. Ed. 2012, 51, 9437–9440. doi:10.1002/anie.201204138
Return to citation in text: [1] -
Höfle, G.; Böhlendorf, B.; Fecker, T.; Sasse, F.; Kunze, B. J. Nat. Prod. 2008, 71, 1967–1969. doi:10.1021/np8004612
Return to citation in text: [1] [2] [3] -
Höfle, G.; Irschik, H. J. Nat. Prod. 2008, 71, 1946–1948. doi:10.1021/np800325z
Return to citation in text: [1] -
Nachtigall, J.; Schneider, K.; Nicholson, G.; Goodfellow, M.; Zinecker, H.; Imhoff, J. F.; Süssmuth, R. D.; Fiedler, H.-P. J. Antibiot. 2010, 63, 567–569. doi:10.1038/ja.2010.79
Return to citation in text: [1] -
Oettmeier, W.; Masson, K.; Soll, M.; Reil, E. Biochem. Soc. Trans. 1994, 22, 213–216.
Return to citation in text: [1] -
Friedrich, T.; Ohnishi, T.; Forche, E.; Kunze, B.; Jansen, R.; Trowitzsch, W.; Höfle, G.; Reichenbach, H.; Weiss, H. Biochem. Soc. Trans. 1994, 22, 226–230.
Return to citation in text: [1] -
Oettmeier, W.; Dostatni, R.; Majewski, C.; Höfle, G.; Fecker, T.; Kunze, B.; Reichenbach, H. Z. Naturforsch. 1990, 45, 322–328.
Return to citation in text: [1] -
Meunier, B.; Madgwick, S. A.; Reil, E.; Oettmeier, W.; Rich, P. R. Biochemistry 1995, 34, 1076–1083. doi:10.1021/bi00003a044
Return to citation in text: [1] -
Salzer, W.; Timmler, H.; Andersag, H. Chem. Ber. 1948, 81, 12–19. doi:10.1002/cber.19480810103
Return to citation in text: [1] -
Cross, R. M.; Manetsch, R. J. Org. Chem. 2010, 75, 8654–8657. doi:10.1021/jo1014504
Return to citation in text: [1] -
Cross, R. M.; Monastyrskyi, A.; Mutka, T. S.; Burrows, J. N.; Kyle, D. E.; Manetsch, R. J. Med. Chem. 2010, 53, 7076–7094. doi:10.1021/jm1007903
Return to citation in text: [1] -
Tietze, L. F.; Ma, L. Heterocycles 2010, 82, 377–396. doi:10.3987/COM-10-S(E)9
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 1997, 14, 605–618. doi:10.1039/np9971400605
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2008, 25, 166–187. doi:10.1039/b612168n
Return to citation in text: [1] -
DeSimone, R. W.; Currie, K. S.; Mitchell, S. A.; Darrow, J. W.; Pippin, D. A. Comb. Chem. High Throughput Screening 2004, 7, 473–494. doi:10.2174/1386207043328544
Return to citation in text: [1] -
Duarte, C. D.; Barreiro, E. J.; Fraga, C. A. M. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. doi:10.2174/138955707782331722
Return to citation in text: [1] -
Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347–361. doi:10.1016/j.cbpa.2010.02.018
Return to citation in text: [1] -
Conrad, M.; Limpach, L. Ber. Dtsch. Chem. Ges. 1887, 20, 944–948. doi:10.1002/cber.188702001215
Return to citation in text: [1] -
Conrad, M.; Limpach, L. Ber. Dtsch. Chem. Ges. 1887, 20, 948–959. doi:10.1002/cber.188702001216
Return to citation in text: [1] -
Manske, R. H. Chem. Rev. 1942, 30, 113–144. doi:10.1021/cr60095a006
Return to citation in text: [1] -
For more recent examples involving the Conrad–Limpach reaction, the reader can refer to references [14-16].
Return to citation in text: [1] -
Abe, H.; Kawada, M.; Inoue, H.; Ohba, S.-i.; Nomoto, A.; Watanabe, T.; Shibasaki, M. Org. Lett. 2013, 15, 2124–2127. doi:10.1021/ol400587a
Intervenolin, a 2-geranyl analogous compound with antitumor activity, was very recently synthesized through a key Suzuki–Miyaura coupling.
Return to citation in text: [1] -
However, since the epoxidation was not stereoselective, a complex mixture of four racemic stereoisomers was obtained from aurachin D (4).
Return to citation in text: [1] -
Friedrich, T.; van Heek, P.; Leif, H.; Ohnishi, T.; Forche, E.; Kunze, B.; Jansen, R.; Trowitzsch-Kienast, W.; Höfle, G.; Reichenbach, H.; Weiss, H. Eur. J. Biochem. 1994, 219, 691–698. doi:10.1111/j.1432-1033.1994.tb19985.x
Return to citation in text: [1] -
Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J. A.; Robinson, J. P. J. Biol. Chem. 2003, 278, 8516–8525. doi:10.1074/jbc.M210432200
Return to citation in text: [1] -
Rodrigues Goulart, H. R.; Kimura, E. A.; Peres, V. J.; Couto, A. S.; Aquino Duarte, F. A.; Katzin, A. M. Antimicrob. Agents Chemother. 2004, 48, 2502–2509. doi:10.1128/AAC.48.7.2502-2509.2004
Return to citation in text: [1] -
The selectivity index (SI) is defined as the ratio between the cytotoxic concentration (Vero cells in this work) and the concentration active on the target. Compounds are medicinally relevant when the SI is high.
Return to citation in text: [1]
| 31. | The selectivity index (SI) is defined as the ratio between the cytotoxic concentration (Vero cells in this work) and the concentration active on the target. Compounds are medicinally relevant when the SI is high. |
| 1. | Kunze, B.; Höfle, G.; Reichenbach, H. J. Antibiot. 1987, 40, 258–265. doi:10.7164/antibiotics.40.258 |
| 14. | Cross, R. M.; Manetsch, R. J. Org. Chem. 2010, 75, 8654–8657. doi:10.1021/jo1014504 |
| 15. | Cross, R. M.; Monastyrskyi, A.; Mutka, T. S.; Burrows, J. N.; Kyle, D. E.; Manetsch, R. J. Med. Chem. 2010, 53, 7076–7094. doi:10.1021/jm1007903 |
| 16. | Tietze, L. F.; Ma, L. Heterocycles 2010, 82, 377–396. doi:10.3987/COM-10-S(E)9 |
| 1. | Kunze, B.; Höfle, G.; Reichenbach, H. J. Antibiot. 1987, 40, 258–265. doi:10.7164/antibiotics.40.258 |
| 8. | Nachtigall, J.; Schneider, K.; Nicholson, G.; Goodfellow, M.; Zinecker, H.; Imhoff, J. F.; Süssmuth, R. D.; Fiedler, H.-P. J. Antibiot. 2010, 63, 567–569. doi:10.1038/ja.2010.79 |
| 17. | Michael, J. P. Nat. Prod. Rep. 1997, 14, 605–618. doi:10.1039/np9971400605 |
| 18. | Michael, J. P. Nat. Prod. Rep. 2008, 25, 166–187. doi:10.1039/b612168n |
| 7. | Höfle, G.; Irschik, H. J. Nat. Prod. 2008, 71, 1946–1948. doi:10.1021/np800325z |
| 19. | DeSimone, R. W.; Currie, K. S.; Mitchell, S. A.; Darrow, J. W.; Pippin, D. A. Comb. Chem. High Throughput Screening 2004, 7, 473–494. doi:10.2174/1386207043328544 |
| 20. | Duarte, C. D.; Barreiro, E. J.; Fraga, C. A. M. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. doi:10.2174/138955707782331722 |
| 21. | Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347–361. doi:10.1016/j.cbpa.2010.02.018 |
| 3. | Höfle, G.; Kunze, B. J. Nat. Prod. 2008, 71, 1843–1849. doi:10.1021/np8003084 |
| 6. | Höfle, G.; Böhlendorf, B.; Fecker, T.; Sasse, F.; Kunze, B. J. Nat. Prod. 2008, 71, 1967–1969. doi:10.1021/np8004612 |
| 15. | Cross, R. M.; Monastyrskyi, A.; Mutka, T. S.; Burrows, J. N.; Kyle, D. E.; Manetsch, R. J. Med. Chem. 2010, 53, 7076–7094. doi:10.1021/jm1007903 |
| 2. | Sandmann, A.; Dickschat, J.; Jenke-Kodama, H.; Kunze, B.; Dittmann, E.; Müller, R. Angew. Chem., Int. Ed. 2007, 46, 2712–2716. doi:10.1002/anie.200603513 |
| 3. | Höfle, G.; Kunze, B. J. Nat. Prod. 2008, 71, 1843–1849. doi:10.1021/np8003084 |
| 4. | Pistorius, D.; Li, Y.; Sandmann, A.; Müller, R. Mol. BioSyst. 2011, 7, 3308–3315. doi:10.1039/c1mb05328k |
| 5. | Katsuyama, Y.; Harmrolfs, K.; Pistorius, D.; Li, Y.; Müller, R. Angew. Chem., Int. Ed. 2012, 51, 9437–9440. doi:10.1002/anie.201204138 |
| 16. | Tietze, L. F.; Ma, L. Heterocycles 2010, 82, 377–396. doi:10.3987/COM-10-S(E)9 |
| 12. | Meunier, B.; Madgwick, S. A.; Reil, E.; Oettmeier, W.; Rich, P. R. Biochemistry 1995, 34, 1076–1083. doi:10.1021/bi00003a044 |
| 6. | Höfle, G.; Böhlendorf, B.; Fecker, T.; Sasse, F.; Kunze, B. J. Nat. Prod. 2008, 71, 1967–1969. doi:10.1021/np8004612 |
| 11. | Oettmeier, W.; Dostatni, R.; Majewski, C.; Höfle, G.; Fecker, T.; Kunze, B.; Reichenbach, H. Z. Naturforsch. 1990, 45, 322–328. |
| 13. | Salzer, W.; Timmler, H.; Andersag, H. Chem. Ber. 1948, 81, 12–19. doi:10.1002/cber.19480810103 |
| 14. | Cross, R. M.; Manetsch, R. J. Org. Chem. 2010, 75, 8654–8657. doi:10.1021/jo1014504 |
| 9. | Oettmeier, W.; Masson, K.; Soll, M.; Reil, E. Biochem. Soc. Trans. 1994, 22, 213–216. |
| 10. | Friedrich, T.; Ohnishi, T.; Forche, E.; Kunze, B.; Jansen, R.; Trowitzsch, W.; Höfle, G.; Reichenbach, H.; Weiss, H. Biochem. Soc. Trans. 1994, 22, 226–230. |
| 1. | Kunze, B.; Höfle, G.; Reichenbach, H. J. Antibiot. 1987, 40, 258–265. doi:10.7164/antibiotics.40.258 |
| 1. | Kunze, B.; Höfle, G.; Reichenbach, H. J. Antibiot. 1987, 40, 258–265. doi:10.7164/antibiotics.40.258 |
| 26. |
Abe, H.; Kawada, M.; Inoue, H.; Ohba, S.-i.; Nomoto, A.; Watanabe, T.; Shibasaki, M. Org. Lett. 2013, 15, 2124–2127. doi:10.1021/ol400587a
Intervenolin, a 2-geranyl analogous compound with antitumor activity, was very recently synthesized through a key Suzuki–Miyaura coupling. |
| 22. | Conrad, M.; Limpach, L. Ber. Dtsch. Chem. Ges. 1887, 20, 944–948. doi:10.1002/cber.188702001215 |
| 23. | Conrad, M.; Limpach, L. Ber. Dtsch. Chem. Ges. 1887, 20, 948–959. doi:10.1002/cber.188702001216 |
| 24. | Manske, R. H. Chem. Rev. 1942, 30, 113–144. doi:10.1021/cr60095a006 |
| 25. | For more recent examples involving the Conrad–Limpach reaction, the reader can refer to references [14-16]. |
| 6. | Höfle, G.; Böhlendorf, B.; Fecker, T.; Sasse, F.; Kunze, B. J. Nat. Prod. 2008, 71, 1967–1969. doi:10.1021/np8004612 |
| 30. | Rodrigues Goulart, H. R.; Kimura, E. A.; Peres, V. J.; Couto, A. S.; Aquino Duarte, F. A.; Katzin, A. M. Antimicrob. Agents Chemother. 2004, 48, 2502–2509. doi:10.1128/AAC.48.7.2502-2509.2004 |
| 28. | Friedrich, T.; van Heek, P.; Leif, H.; Ohnishi, T.; Forche, E.; Kunze, B.; Jansen, R.; Trowitzsch-Kienast, W.; Höfle, G.; Reichenbach, H.; Weiss, H. Eur. J. Biochem. 1994, 219, 691–698. doi:10.1111/j.1432-1033.1994.tb19985.x |
| 29. | Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J. A.; Robinson, J. P. J. Biol. Chem. 2003, 278, 8516–8525. doi:10.1074/jbc.M210432200 |
| 27. | However, since the epoxidation was not stereoselective, a complex mixture of four racemic stereoisomers was obtained from aurachin D (4). |
| 1. | Kunze, B.; Höfle, G.; Reichenbach, H. J. Antibiot. 1987, 40, 258–265. doi:10.7164/antibiotics.40.258 |
© 2013 Li et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
