
Abstract
The impact of the level of complexity in self-sorting was elaborated through the fabrication of various scalene triangles. It turned out that the self-sorting system with a higher level of complexity was far superior to less complex sorting algorithms.
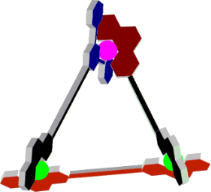
Graphical Abstract
Introduction
Self-assembly guided by self-sorting algorithms has received considerable attention over the past two decades as such protocols pave the way for intricate supramolecular assemblies [1-19]. However, despite its wide use, the definition of self-sorting remains vague with no precise guidelines provided in the literature. It has been widely reported that self-sorting operates when the numerical outcome of a chemical system is lower than the plausible number of potential aggregates (assemblies) estimated on the basis of statistical, chemical, and geometrical arguments [20,21]. This definition has drawbacks, and levelling effects [22] have been observed among various self-sorting systems. To numerically grasp the difference between different self-sorting processes, we defined the degree of self-sorting M as M = P/P0 with P representing the number of possibilities and P0 representing the number of experimentally observed aggregates in the mixture [8]. For example, the degree of the self-sorting process realised with ligands 1–4 in the presence of both Cu+ and Zn2+, as described in Scheme 1, is M = 10, as only two complexes formed out of twenty possible ones [8].
![[1860-5397-7-183-i1]](/bjoc/content/inline/1860-5397-7-183-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Self-sorting in a six-component library [8].
Scheme 1: Self-sorting in a six-component library [8].
In contrast, when ligands 1–4 were treated with either Cu+ or Zn2+, the observed experimental outcome was four complexes (Supporting Information File 1, Figures S1 and S2), which has to be evaluated in light of the ten possible products (Scheme 2). Thus, for this process M = 2.5. Is this difference in M of any relevance, for example in the fabrication of intricate entities, or not? Herein, we investigate the utility of both self-sorting algorithms, from Scheme 1 and Scheme 2, for the fabrication of supramolecular scalene triangles. Importantly, we are able to demonstrate that the clean formation of a scalene triangle is only possible with the algorithm exhibiting the higher degree of self-sorting.
![[1860-5397-7-183-i2]](/bjoc/content/inline/1860-5397-7-183-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Self-sorting in a five-component library. We used 2 in a slightly different form with Zn2+ (R = 4-C6H4I) and Cu+ (R = H).
Scheme 2: Self-sorting in a five-component library. We used 2 in a slightly different form with Zn2+ (R = 4-C6...
Among the triangular assemblies, the scalene triangle is found to be the most difficult to fabricate. Hence, it comes as no surprise that there is only one report on a scalene triangle so far [16]. To design a further scalene triangle we modified a design already probed in the preparation of a geometrically isosceles triangle [15], and we implemented the coordination motifs of 1–4 into the three different multitopic ligands 5–7 [15,23], integrating twice the [Cu(3)(4)]+ [24-26] and once the [Zn(1)(2)]2+ motif [8,16] (Figure 1). The coordination behaviour of molecular component 3 was integrated into 5, the latter being synthesised in a Sonogashira homocoupling reaction [27]. The information stored in 1 and 4 was instated in the unsymmetrical bisphenanthroline 6, readily accessible by stepwise Sonogashira cross-coupling reactions [15]. A known procedure was followed to prepare the terpyridine–phenanthroline hybrid 7 [8]. The lengths of the ligands were chosen in such a way that they provide the geometrically different sides of a scalene triangle.
![[1860-5397-7-183-1]](/bjoc/content/figures/1860-5397-7-183-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Ligands used in the present study.
Figure 1: Ligands used in the present study.
Results and Discussion
We tested both self-sorting algorithms as described earlier (Scheme 1 and Scheme 2). In a first round of experiments, we combined the ligands 5, 6, and 7 in equimolar ratio and made them react with one equivalent of Zn2+ and two equivalents of Cu+ in acetonitrile at 60 °C for 3 h. At the end, a clear red solution was furnished, which was characterised as received, by means of mass spectrometry, 1H NMR, diffusion-ordered spectroscopy (DOSY), differential pulse voltammetry (DPV) and elemental analysis. The electrospray ionisation mass spectrum (ESI-MS) of the reaction mixture suggests clean formation of the triangular species T = [Cu2Zn(5)(6)(7)](OTf)2(PF6)2 (Scheme 3). In the accessible spectral region of m/z = 150–2000 only three intense peaks were observed, all of them corresponding to triangle T (Figure 2). The most abundant peak at m/z = 666.8 can be assigned to [Cu2Zn(5)(6)(7)]4+, whereas the triply charged one at m/z = 938.5 is attributed to [Cu2Zn(5)(6)(7)](OTf)3+, and the doubly charged one at m/z = 1481.1 to [Cu2Zn(5)(6)(7)](OTf)22+. All peaks were isotopically resolved, showing full agreement with the theoretically expected isotopic distribution.
![[1860-5397-7-183-i3]](/bjoc/content/inline/1860-5397-7-183-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the triangular assembly T (only syn shown).
Scheme 3: Synthesis of the triangular assembly T (only syn shown).
![[1860-5397-7-183-2]](/bjoc/content/figures/1860-5397-7-183-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ESI-MS of triangle T in acetonitrile along with the isotopically resolved peak at 666.8 (black: Experimental; red: Calculated for [Cu2Zn(5)(6)(7)]4+). See also Figure S5 in Supporting Information File 1.
Figure 2: ESI-MS of triangle T in acetonitrile along with the isotopically resolved peak at 666.8 (black: Exp...
To corroborate the clean self-assembly process, we carefully examined the DOSY and 1H NMR of T. As in the ESI-MS, both sets of data unambiguously supported the presence of only one species, i.e., the DOSY spectrum showed only a single diffusion coefficient (Supporting Information File 1, Figure S4). Additional information was derived from the 1H NMR signals of the methoxy protons, as these appear in a diagnostic region. In T, up to eight singlets are expected for the four methoxy groups due to their constitutional differences and the occurrence of two diastereomers (syn and anti). Diastereomers form as a result of two stereogenic heteroleptic copper(I) complex motifs [Cu(3)(4)]+ in T [8,28,29]. The 1H NMR of the assembly indeed showed seven singlets (one peak is merged with the others) between 2.73–3.10 ppm (Figure 3c). From NMR integration the ratio of the diastereomers was found to be approximately 3:1. The finding of further characteristic 1H NMR shifts for protons of 6 (H–a and H–b) additionally supports the formation of T as a mixture of two diastereomers. Four triplets (two for each diastereomer) were observed for protons H–a between 3.53–3.97 ppm, and the same number of triplets was seen for H–b in the region of 0.57–1.00 ppm (Supporting Information File 1). Elemental analysis of the assembly also confirmed the exclusive formation of T.
![[1860-5397-7-183-3]](/bjoc/content/figures/1860-5397-7-183-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Partial 1H NMR (400 MHz, 298 K, CD3CN) spectra of an equimolar mixture of 5, 6, and 7 in the presence of (a) 3 equivalents of Cu+, (b) 3 equivalents of Zn2+ and (c) 3 equivalents of a metal-salt mixture (Cu+:Zn2+ = 2:1).
Figure 3: Partial 1H NMR (400 MHz, 298 K, CD3CN) spectra of an equimolar mixture of 5, 6, and 7 in the presen...
We further evaluated the structure using DPV, because this analytical method provides valuable information about redox active units (here Cu+). It is well known that copper(I) shows distinct oxidation potentials in different complex environments. Thus, the DPV measurements should allow us to analyse the number and ligand sphere of copper(I) centres in T. In the mononuclear complex [Cu(1)(4)](PF6) the copper(I) oxidation is observed at +0.29 VSCE, whereas for [Cu(3)(4)](PF6) and [Cu(1)(2)](PF6) the oxidation is placed at +0.44 VSCE and −0.21 VSCE, respectively [8]. In T, only one type of copper(I) complex is present. As two values were expected for the two diastereomers, the broad peak was deconvoluted for two copper(I) oxidation waves (Figure 4) resulting in two values at +0.59 and +0.64 VSCE. The values agree with those reported for a similar copper(I) complex (+0.61 and +0.67 VSCE) in a recently reported supramolecular trapezoid [8]. The population of the two diastereomers as determined from the deconvoluted DPV spectrum (Supporting Information File 1) was roughly 3:1, in full agreement with the ratio derived from 1H NMR results.
![[1860-5397-7-183-4]](/bjoc/content/figures/1860-5397-7-183-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Differential pulse voltammogram of T in acetonitrile (0.1 M n-Bu4NPF6 as electrolyte, Ag wire as a quasi-reference electrode, 1,1'-dimethylferrocene as internal standard, scan rate = 20 mV s−1 and a pulse height of 2 mV).
Figure 4: Differential pulse voltammogram of T in acetonitrile (0.1 M n-Bu4NPF6 as electrolyte, Ag wire as a ...
We then focused on the self-sorting protocol mentioned in Scheme 2. We reacted three equivalents of copper(I) ions with an equimolar mixture of all three ligands 5–7. After 3 h, at similar conditions as for T, a dark red solution was afforded, which was characterised by 1H NMR without any further purification. The 1H NMR spectrum was found to be broad (Figure 3a). The broadening of the signals is partly due to the presence of a phenanthroline–Cu+–terpyridine complex. Due to the tetrahedral coordination behaviour of Cu+, one pyridine nitrogen atom of the terpyridine unit is left uncoordinated [30], and thus it undergoes rapid exchange leading to broad NMR signals. The experiment was also carried out in the presence of three equivalents of Zn2+. A clear yellow solution was produced after exposure to similar reaction conditions. Unlike the other experiment with Cu+, only sharp signals were observed in the 1H NMR (Figure 3b), but the many signals in the region 2.5–4.0 ppm suggest formation of several species. Thus, a comparison among the NMR spectra nicely demonstrated that the self-assembly process was only clean in the case of the mixed-metal scenario, whereas the situation turned out to be complicated in both homometallic cases.
The observations can be rationalised in the following way. In the homometallic cases selectivity was less, because the ligands may organise into ≥2 competing triangular arrays with different connectivities (constitutions). In the all-copper situation, the linkage between 5 and 6 is only possible by [Cu(35)(46)]+ coordination [31]. The triangle is completed through a bridging with 7. However, the connectivity of 7 is not defined. The ligand may arrange itself in either of the two possible ways, [Cu(35)(47)]+ and [Cu(16)(27)]+ or [Cu(27)(35)]+ and [Cu(16)(47)]+, as demonstrated in Scheme 2, resulting in the formation of two different triangular species. Hence, the number of constitutional isomers increases in the case of the all-copper triangle. A related explanation may be given for the all-zinc triangle. The hindered phenanthroline of 6, i.e., unit 1, has four methoxy groups available for coordination in addition to its two bisimine nitrogens. Thus, unit 1 may either act as a strong bidentate, tridentate or tetradentate binding site for zinc(II) ions, and there is no large thermodynamic difference between a [Zn(16)(27)]+-type connection and a [Zn(16)(47)]+-type link. The outcomes are similar to those observed with copper(I) ions. On the other hand, in the mixed-metal scenario the terpyridine prefers to connect with terminus 1 as embedded in 6, which is nicely illustrated from the self-sorting described in Scheme 1. Thus, the self-assembly process was constitutionally clean when self-sorting occurred along the algorithm with the higher level of complexity (Scheme 1). Due to the beauty of self-sorting, the five-component assembly (five different starting materials, mixed metal scenario) was flawless as compared to the four-component assembly (four different starting materials, homometallic cases).
As all attempts to obtain a crystal structure of T were unsuccessful, MM+ force-field computations and molecular dynamics on T (Hyperchem 7.52®, Hypercube, Inc.) provided some insight to their structure as scalene triangles. Taking the metal–metal distance as a measure, the three metal corners of T (syn) are separated by 1.27, 1.58 and 1.63 nm in the energy-minimised structure (Figure 5) and by 1.36, 1.58 and 1.63 nm in T (anti) (Supporting Information File 1), nicely illustrating the scalene triangle arrangement of T.
![[1860-5397-7-183-5]](/bjoc/content/figures/1860-5397-7-183-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Two representations of the energy-minimised structure of the scalene triangle T (anti); copper(I) ions – green, zinc(II) ion – white.
Figure 5: Two representations of the energy-minimised structure of the scalene triangle T (anti); copper(I) i...
Conclusion
We have been able to establish that our strategy to fabricate an isosceles triangle [15] can equally be applied to the preparation of a supramolecular scalene triangle and thus is tolerant to changes at the angles (of the vertices) and to variations of the lengths of the sides. Moreover, we have demonstrated with a study on homo- versus heterometallic scalene triangles that the level of complexity of self-sorting is important for the fabrication of intricate supramolecular assemblies [32,33].
Experimental
General
All commercial reagents were used without further purification. The solvents were dried with appropriate desiccants and distilled prior to use. NMR measurements were carried out on a Bruker Avance 400 MHz spectrometer with the deuterated solvent as the lock and residual solvent as the internal reference. Electrospray ionisation mass spectra (ESI-MS) were recorded on a Thermo-Quest LCQ Deca. Differential pulse voltammetry (DPV) was measured on a Parstat 2273 in dry acetonitrile. The melting point was measured on a Büchi SMP-20 and is uncorrected. The infrared spectrum was recorded on a Varian 1000 FT-IR instrument and the elemental analysis measurement was performed with a EA 3000 CHNS. Compound 5 [27], 6 [15], and 7 [8] were synthesised according to known procedures.
Synthesis of scalene triangle T
6 (1.49 mg, 1.65 μmol), 5 (1.76 mg, 1.65 μmol), 7 (0.85 mg, 1.65 μmol), Zn(OTf)2 (0.60 mg, 1.66 μmol) and [Cu(MeCN)4]PF6 (1.23 mg, 3.31 μmol) were heated under reflux in a mixture of dichloromethane (10 mL) and acetonitrile (25 mL) for 2 h. The solvents were evaporated under reduced pressure and the solid was characterised as such. Yield quantitative; mp >260 °C; IR (KBr) ν: 3448, 3068, 2953, 2931, 2869, 2362, 2209, 1617, 1602, 1589, 1549, 1499, 1475, 1427, 1406, 1383, 1277, 1255, 1223, 1159, 1111, 1030, 1019, 912, 843, 791, 767, 725, 639; ESI-MS m/z (%): 666.8 (100) [M − 2PF6, 2OTf]4+, 938.5 (70) [M − 2PF6, OTf]3+, 1481.1 (20) [M − 2PF6]2+; Anal. calcd for C161H127Br2Cu2F18N13O12P2S2Zn·3CH2Cl2: C, 56.10; H, 3.82; N, 5.19; S, 1.83; found: C, 56.36; H, 3.27; N, 5.35; S, 1.91.
Supporting Information
| Supporting Information File 1: 1H NMR spectra of self-sorting mixtures and of T. DOSY, ESI and DPV spectra of T. | ||
| Format: PDF | Size: 372.3 KB | Download |
References
-
Krämer, R.; Lehn, J.-M.; Marquis-Rigault, A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394–5398. doi:10.1073/pnas.90.12.5394
Return to citation in text: [1] -
Caulder, D. L.; Raymond, K. N. Angew. Chem., Int. Ed. Engl. 1997, 36, 1440–1442. doi:10.1002/anie.199714401
Return to citation in text: [1] -
Lehn, J.-M. Science 2002, 295, 2400–2403. doi:10.1126/science.1071063
Return to citation in text: [1] -
Liu, S.; Ruspic, C.; Mukhopadhyay, P.; Chakrabarti, S.; Zavalij, P. Y.; Isaacs, L. J. Am. Chem. Soc. 2005, 127, 15959–15967. doi:10.1021/ja055013x
Return to citation in text: [1] -
Nitschke, J. R. Acc. Chem. Res. 2007, 40, 103–112. doi:10.1021/ar068185n
Return to citation in text: [1] -
Xu, S.; Giuseppone, N. J. Am. Chem. Soc. 2008, 130, 1826–1827. doi:10.1021/ja710248q
Return to citation in text: [1] -
Wang, F.; Han, C.; He, C.; Zhou, Q.; Zhang, J.; Wang, C.; Li, N.; Huang, F. J. Am. Chem. Soc. 2008, 130, 11254–11255. doi:10.1021/ja8035465
Return to citation in text: [1] -
Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Barboiu, M.; Dumitru, F.; Legrand, Y.-M.; Petit, E.; van der Lee, A. Chem. Commun. 2009, 2192–2194. doi:10.1039/b900155g
Return to citation in text: [1] -
Ulrich, S.; Lehn, J.-M. J. Am. Chem. Soc. 2009, 131, 5546–5559. doi:10.1021/ja809828g
Return to citation in text: [1] -
Zheng, Y.-R.; Yang, H.-B.; Ghosh, K.; Zhao, L.; Stang, P. J. Chem.–Eur. J. 2009, 15, 7203–7214. doi:10.1002/chem.200900230
Return to citation in text: [1] -
Ajami, D.; Hou, J.-L.; Dale, T. J.; Barrett, E.; Rebek, J., Jr. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10430–10434. doi:10.1073/pnas.0809903106
Return to citation in text: [1] -
Jiang, W.; Schalley, C. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10425–10429. doi:10.1073/pnas.0809512106
Return to citation in text: [1] -
Northrop, B. H.; Zheng, Y.-R.; Chi, K.-W.; Stang, P. J. Acc. Chem. Res. 2009, 42, 1554–1563. doi:10.1021/ar900077c
Return to citation in text: [1] -
Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Mahata, K.; Saha, M. L.; Schmittel, M. J. Am. Chem. Soc. 2010, 132, 15933–15935. doi:10.1021/ja108419k
Return to citation in text: [1] [2] [3] -
Johnson, A. M.; Hooley, R. J. Inorg. Chem. 2011, 50, 4671–4673. doi:10.1021/ic2001688
Return to citation in text: [1] -
Jiang, W.; Wang, Q.; Linder, I.; Klautzsch, F.; Schalley, C. A. Chem.–Eur. J. 2011, 17, 2344–2348. doi:10.1002/chem.201003194
Return to citation in text: [1] -
Jiang, W.; Sattler, D.; Rissanen, K.; Schalley, C. A. Org. Lett. 2011, 13, 4502–4505. doi:10.1021/ol201618f
Return to citation in text: [1] -
Chas, M.; Gil-Ramírez, G.; Ballester, P. Org. Lett. 2011, 13, 3402–3405. doi:10.1021/ol201146r
Return to citation in text: [1] -
Dumitru, F.; Petit, E.; van der Lee, A.; Barboiu, M. Eur. J. Inorg. Chem. 2005, 4255–4262. doi:10.1002/ejic.200500463
Return to citation in text: [1] -
No differences among various self-sorting processes.
Return to citation in text: [1] -
We use here a simplification to denote the ligand setting. While ligand 3 is not identical with the 2-bromoduryl-9-mesitylphenanthroline ligand site in 5, our investigations over 10 years have revealed an analogous coordination behaviour of both ligands.
Return to citation in text: [1] -
Schmittel, M.; Ganz, A. Chem. Commun. 1997, 999–1000. doi:10.1039/a701509g
Return to citation in text: [1] -
Schmittel, M.; Lüning, U.; Meder, M.; Ganz, A.; Michel, C.; Herderich, M. Heterocycl. Commun. 1997, 3, 493–498. doi:10.1515/HC.1997.3.6.493
Return to citation in text: [1] -
Schmittel, M.; Ammon, H.; Kalsani, V.; Wiegrefe, A.; Michel, C. Chem. Commun. 2002, 2566–2567. doi:10.1039/b207801e
Return to citation in text: [1] -
Schmittel, M.; Michel, C.; Wiegrefe, A.; Kalsani, V. Synthesis 2001, 1561–1567. doi:10.1055/s-2001-16093
Return to citation in text: [1] [2] -
Schmittel, M.; Kalsani, V.; Fenske, D.; Wiegrefe, A. Chem. Commun. 2004, 490–491. doi:10.1039/b312807e
Return to citation in text: [1] -
Schmittel, M.; Mahata, K. Chem. Commun. 2008, 2550–2552. doi:10.1039/b801462k
Return to citation in text: [1] -
Medlycott, E. A.; Hanan, G. S. Chem. Commun. 2007, 4884–4886. doi:10.1039/b711765e
Return to citation in text: [1] -
We use here a short notation: [Cu(35)(46)]+-type linkage: The copper complex is made from the phenanthroline terminus 3 embedded in 5 and the phenanthroline terminus 4 embedded in 6.
Return to citation in text: [1] -
Funeriu, D. P.; Rissanen, K.; Lehn, J.-M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10546–10551. doi:10.1073/pnas.191362998
Return to citation in text: [1] -
Funeriu, D. P.; Lehn, J.-M.; Fromm, K. M.; Fenske, D. Chem.–Eur. J. 2000, 6, 2103–2111. doi:10.1002/1521-3765(20000616)6:12<2103::AID-CHEM2103>3.0.CO;2-S
Return to citation in text: [1]
| 31. | We use here a short notation: [Cu(35)(46)]+-type linkage: The copper complex is made from the phenanthroline terminus 3 embedded in 5 and the phenanthroline terminus 4 embedded in 6. |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 30. | Medlycott, E. A.; Hanan, G. S. Chem. Commun. 2007, 4884–4886. doi:10.1039/b711765e |
| 1. | Krämer, R.; Lehn, J.-M.; Marquis-Rigault, A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394–5398. doi:10.1073/pnas.90.12.5394 |
| 2. | Caulder, D. L.; Raymond, K. N. Angew. Chem., Int. Ed. Engl. 1997, 36, 1440–1442. doi:10.1002/anie.199714401 |
| 3. | Lehn, J.-M. Science 2002, 295, 2400–2403. doi:10.1126/science.1071063 |
| 4. | Liu, S.; Ruspic, C.; Mukhopadhyay, P.; Chakrabarti, S.; Zavalij, P. Y.; Isaacs, L. J. Am. Chem. Soc. 2005, 127, 15959–15967. doi:10.1021/ja055013x |
| 5. | Nitschke, J. R. Acc. Chem. Res. 2007, 40, 103–112. doi:10.1021/ar068185n |
| 6. | Xu, S.; Giuseppone, N. J. Am. Chem. Soc. 2008, 130, 1826–1827. doi:10.1021/ja710248q |
| 7. | Wang, F.; Han, C.; He, C.; Zhou, Q.; Zhang, J.; Wang, C.; Li, N.; Huang, F. J. Am. Chem. Soc. 2008, 130, 11254–11255. doi:10.1021/ja8035465 |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 9. | Barboiu, M.; Dumitru, F.; Legrand, Y.-M.; Petit, E.; van der Lee, A. Chem. Commun. 2009, 2192–2194. doi:10.1039/b900155g |
| 10. | Ulrich, S.; Lehn, J.-M. J. Am. Chem. Soc. 2009, 131, 5546–5559. doi:10.1021/ja809828g |
| 11. | Zheng, Y.-R.; Yang, H.-B.; Ghosh, K.; Zhao, L.; Stang, P. J. Chem.–Eur. J. 2009, 15, 7203–7214. doi:10.1002/chem.200900230 |
| 12. | Ajami, D.; Hou, J.-L.; Dale, T. J.; Barrett, E.; Rebek, J., Jr. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10430–10434. doi:10.1073/pnas.0809903106 |
| 13. | Jiang, W.; Schalley, C. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10425–10429. doi:10.1073/pnas.0809512106 |
| 14. | Northrop, B. H.; Zheng, Y.-R.; Chi, K.-W.; Stang, P. J. Acc. Chem. Res. 2009, 42, 1554–1563. doi:10.1021/ar900077c |
| 15. | Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k |
| 16. | Mahata, K.; Saha, M. L.; Schmittel, M. J. Am. Chem. Soc. 2010, 132, 15933–15935. doi:10.1021/ja108419k |
| 17. | Johnson, A. M.; Hooley, R. J. Inorg. Chem. 2011, 50, 4671–4673. doi:10.1021/ic2001688 |
| 18. | Jiang, W.; Wang, Q.; Linder, I.; Klautzsch, F.; Schalley, C. A. Chem.–Eur. J. 2011, 17, 2344–2348. doi:10.1002/chem.201003194 |
| 19. | Jiang, W.; Sattler, D.; Rissanen, K.; Schalley, C. A. Org. Lett. 2011, 13, 4502–4505. doi:10.1021/ol201618f |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 28. | Schmittel, M.; Kalsani, V.; Fenske, D.; Wiegrefe, A. Chem. Commun. 2004, 490–491. doi:10.1039/b312807e |
| 29. | Schmittel, M.; Mahata, K. Chem. Commun. 2008, 2550–2552. doi:10.1039/b801462k |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 15. | Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 20. | Chas, M.; Gil-Ramírez, G.; Ballester, P. Org. Lett. 2011, 13, 3402–3405. doi:10.1021/ol201146r |
| 21. | Dumitru, F.; Petit, E.; van der Lee, A.; Barboiu, M. Eur. J. Inorg. Chem. 2005, 4255–4262. doi:10.1002/ejic.200500463 |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 15. | Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k |
| 23. | We use here a simplification to denote the ligand setting. While ligand 3 is not identical with the 2-bromoduryl-9-mesitylphenanthroline ligand site in 5, our investigations over 10 years have revealed an analogous coordination behaviour of both ligands. |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 16. | Mahata, K.; Saha, M. L.; Schmittel, M. J. Am. Chem. Soc. 2010, 132, 15933–15935. doi:10.1021/ja108419k |
| 27. | Schmittel, M.; Michel, C.; Wiegrefe, A.; Kalsani, V. Synthesis 2001, 1561–1567. doi:10.1055/s-2001-16093 |
| 15. | Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k |
| 27. | Schmittel, M.; Michel, C.; Wiegrefe, A.; Kalsani, V. Synthesis 2001, 1561–1567. doi:10.1055/s-2001-16093 |
| 15. | Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k |
| 16. | Mahata, K.; Saha, M. L.; Schmittel, M. J. Am. Chem. Soc. 2010, 132, 15933–15935. doi:10.1021/ja108419k |
| 15. | Schmittel, M.; Mahata, K. Chem. Commun. 2010, 46, 4163–4165. doi:10.1039/c0cc00191k |
| 8. | Mahata, K.; Schmittel, M. J. Am. Chem. Soc. 2009, 131, 16544–16554. doi:10.1021/ja907185k |
| 24. | Schmittel, M.; Ganz, A. Chem. Commun. 1997, 999–1000. doi:10.1039/a701509g |
| 25. | Schmittel, M.; Lüning, U.; Meder, M.; Ganz, A.; Michel, C.; Herderich, M. Heterocycl. Commun. 1997, 3, 493–498. doi:10.1515/HC.1997.3.6.493 |
| 26. | Schmittel, M.; Ammon, H.; Kalsani, V.; Wiegrefe, A.; Michel, C. Chem. Commun. 2002, 2566–2567. doi:10.1039/b207801e |
| 32. | Funeriu, D. P.; Rissanen, K.; Lehn, J.-M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10546–10551. doi:10.1073/pnas.191362998 |
| 33. | Funeriu, D. P.; Lehn, J.-M.; Fromm, K. M.; Fenske, D. Chem.–Eur. J. 2000, 6, 2103–2111. doi:10.1002/1521-3765(20000616)6:12<2103::AID-CHEM2103>3.0.CO;2-S |
© 2011 Mahata and Schmittel; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
