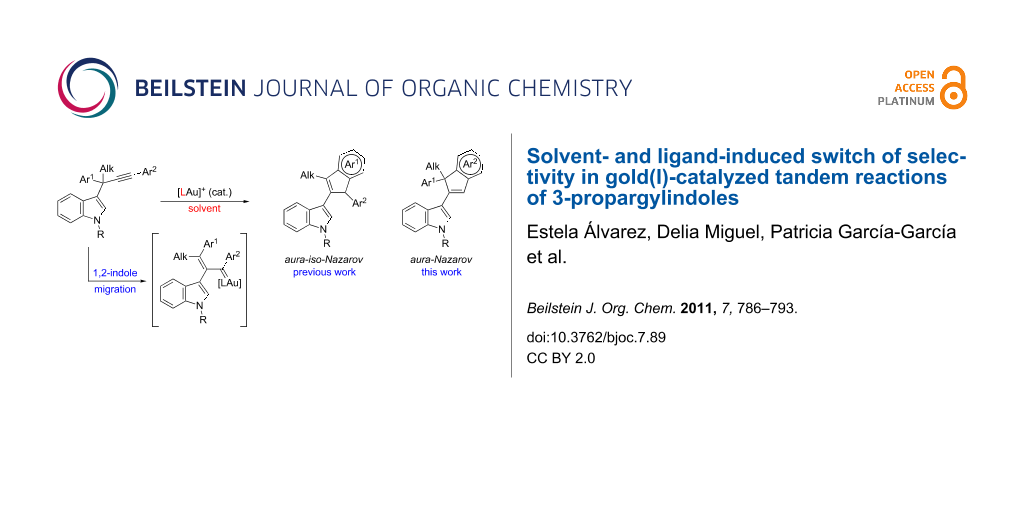
Abstract
The selectivity of our previously described gold-catalyzed tandem reaction, 1,2-indole migration followed by aura-iso-Nazarov cyclization, of 3-propargylindoles bearing (hetero)aromatic substituents at both the propargylic and terminal positions, was reversed by the proper choice of the catalyst and the reaction conditions. Thus, 3-(inden-2-yl)indoles, derived from an aura-Nazarov cyclization (instead of an aura-iso-Nazarov cyclization), were obtained in moderate to good yields from a variety of 3-propargylindoles.

Graphical Abstract
Introduction
Catalysis with gold complexes as carbophilic π-acids has become a highly developed area in the last decade [1-7]. In particular, 1,2-acyl migration reactions of propargylic esters have been extensively investigated. In these processes the gold-carbenoid species generated are able to undergo a wide variety of further transformations [8-11]. In addition, propargylic sulfides have also been reported as useful substrates for this type of process, participating in related 1,2-sulfur migrations [12]. Within this area we have reported the first examples of gold-catalyzed migration reactions in propargylic systems that involve a carbon-centered moiety, implying that carbon–carbon bonds are broken and formed instead of carbon–heteroatom bonds [13,14]. Based on the nucleophilic nature of indoles [15], which are known to react with gold-activated alkynes or allenes [16,17], and by taking advantage of our reported methodology for the synthesis of 3-propargylindoles [18,19], we have shown that the indole nucleus is able to participate in gold-catalyzed 1,2-migration reactions of propargylic systems. Thus, 3-propargylindoles 1 give rise to α,β-unsaturated gold-carbenoid intermediates 2 that evolve through different pathways depending on the substituents at the propargylic and terminal positions of the alkyne moiety (Scheme 1). If (hetero)aromatic substituents are present at either of these positions, they undergo further cyclizations to afford 3-(inden-2-yl)indoles 3 or 4 (Scheme 1). An analysis of the aromaticity of the transition state structures for these cyclizations by DFT calculations revealed that these electrocyclic ring closures could be considered as gold variants of the Nazarov (cyclization from 2 to 4) or iso-Nazarov reactions (cyclization from 2 to 3) [14]. These theoretical calculations also showed that in those cases where both cyclization pathways are possible (for example in 2a arising from 1a; Scheme 1), the calculated energy barriers for the two cyclization modes favored the iso-Nazarov-product 3a (9.42 kcal/mol vs 11.71 kcal/mol for the Nazarov cyclization assuming that the initial gold coordination to the alkyne is anti to the indole). This is in complete agreement with the experimental data, as we always observed the selective formation of cyclization products 3 in those cases where both 3 and 4 could be obtained. However, for the model compound 1a, similar energy profiles were obtained for the corresponding iso-Nazarov and Nazarov pathways (ΔE = 2.29 kcal/mol) [20]. Since there are several examples reported in the literature that show that the reactivity and selectivity of reactions catalyzed by gold complexes can be appropriately tuned [21-27], we thought that it should be possible to reverse the selectivity of our tandem reaction in favor of the iso-Nazarov pathway, to obtain compounds 4 by a proper setting of the reaction conditions (modulation of the electronic properties of the ligands, counter ion, solvent, substitution pattern of the substrates, etc.). Herein, we report our efforts to control the two competing pathways in the evolution of gold-carbenoid intermediates generated by an initial 1,2-indole migration in 3-propargylindoles.
![[1860-5397-7-89-i1]](/bjoc/content/inline/1860-5397-7-89-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of 3-(inden-2-yl)indoles 3 and 4 from 3-propargylindoles. Energy barriers (kcal/mol) for the cyclization reactions of gold-carbenoid intermediate 2a.
Scheme 1: Formation of 3-(inden-2-yl)indoles 3 and 4 from 3-propargylindoles. Energy barriers (kcal/mol) for ...
Results and Discussion
It is an intriguing possibility that the aura-Nazarov reaction may also take place with substrates bearing aromatic substituents at both the propargylic and terminal positions, and thus allow access to new functionalized indole derivatives (Scheme 2). It should be remarked that, until now, only the aura-Nazarov cyclization to give products 4 from substrates 1 (without an aromatic substituent at the propargylic position, see R2, R3 in Scheme 1) has been observed.
![[1860-5397-7-89-i2]](/bjoc/content/inline/1860-5397-7-89-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Tandem 1,2-indole migration/aura-Nazarov cyclization from 3-propargylindoles bearing an aromatic substituent at the propargylic position.
Scheme 2: Tandem 1,2-indole migration/aura-Nazarov cyclization from 3-propargylindoles bearing an aromatic su...
For the initial selectivity control experiments, 1-methyl-3-(1-methyl-1,3-diphenylprop-2-ynyl)-1H-indole (1a) was selected as the model compound and was treated with several gold catalysts under different reaction conditions (Table 1). As expected, under our standard reported conditions ((Ph3P)AuCl/AgSbF6 in CH2Cl2 at room temperature), the 3-(inden-2-yl)indole 3a was obtained as the major product. However, a minor isomer 4a was also isolated along with 3a in a ca. 3.5/1 ratio (Table 1, entry 1). The structure of the minor compound 4a was established by X-ray diffraction (Figure 1), confirming that the gold-carbenoid intermediate 2a could also undergo the aura-Nazarov cyclization [28,29].
Table 1: Effect of the catalyst and reaction conditions on the reactivity of 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-89-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | Solvent |
Ratiob
3a/4a |
|---|---|---|---|
| 1 | (Ph3P)AuCl/AgSbF6 | CH2Cl2 | 3.5/1 |
| 2 | (Ph3P)AuNTf2 | CH2Cl2 | 3.3/1 |
| 3 | SPhosAuNTf2c | CH2Cl2 | 2.5/1 |
| 4 | (Et3P)AuCl/AgSbF6 | CH2Cl2 | 3.3/1d |
| 5 | IMeAuCle/AgSbF6 | CH2Cl2 | 2.5/1 |
| 6 | IPrAuClf/AgSbF6 | CH2Cl2 | 3/1 |
| 7 | [(PhO)3P]AuCl/AgSbF6 | CH2Cl2 | 2.2/1 |
| 8 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgSbF6 | CH2Cl2 | 1.5/1 |
| 9 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgBF4 | CH2Cl2 | 1.5/1 |
| 10 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgNTf2 | CH2Cl2 | 2/1 |
| 11 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgOTs | CH2Cl2 | 1.5/1 |
| 12 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgOTf | CH2Cl2 | 1.4/1 |
| 13 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgSbF6 | DME | 1.4/1 |
| 14 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgSbF6 | THF | 1.4/1 |
| 15 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgSbF6 | toluene | 1/1.8 |
| 16 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgSbF6 | tolueneg | 1/2.3 |
| 17 | [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgSbF6 | tolueneh | 1/4i |
aReactions carried out until complete consumption of the starting material 1a, as judged by GC-MS and/or TLC analysis, unless otherwise stated. bDetermined by 1H NMR analysis of the crude reaction mixture. cSPhos = 2-dicyclohexylphosphino-2´,6´-dimethoxybiphenyl. d66% of conversion after 24 h. eIMe = 1,3,4,5-tetramethylimidazol-2-ylidene. fIPr = 1,3-bis(2,6-di-isopropylphenyl)imidazol-2-ylidene. gConducted at 0 °C. A similar result was obtained by using AgOTf as a silver salt. hCarried out at −20 °C. i50% conversion after 24 h.
![[1860-5397-7-89-1]](/bjoc/content/figures/1860-5397-7-89-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP diagram for 4a. Ellipsoids are shown at 30% level (hydrogen atoms are omitted for clarity).
Figure 1: ORTEP diagram for 4a. Ellipsoids are shown at 30% level (hydrogen atoms are omitted for clarity).
The use of cationic gold complexes bearing different types of phosphane ligands always provided the iso-Nazarov product 3a as the major isomer, with a small increase in the competing Nazarov product 4a on switching the ligand to SPhos (Table 1, entries 1–4). The use of complexes bearing N-heterocyclic carbene ligands [30] also produced 3a as the major compound of the corresponding mixtures (Table 1, entries 5 and 6). It was decided to increase the π-acceptor character of the ligand [31], and, in this case, the employment of a triphenylphosphite–gold(I) complex led to a slight increase in the ratio of 4a (Table 1, entry 7). Finally, the use of the bulky phosphite ligand tris(2,4-di-t-butylphenyl)phosphite, gave rise to a 1.5/1 ratio of 3a/4a (Table 1, entry 8) [32].
Once tris(2,4-di-t-butylphenyl)phosphite was selected as the best ligand to favor the desired tandem process, the influence of the metal counter ion was then studied. Thus, several silver salts were employed for the generation of the cationic catalytic active gold(I) complex, and it was concluded that the effect on the selectivity is almost negligible (Table 1, entries 9–12). Nevertheless, it should be noted that no reaction occurred when AgOBz was employed whilst the reactions with AgBF4, AgNTf2 and AgOTs were relatively slow. Therefore, AgOTf and AgSbF6 were selected as silver salts, due to their availability and higher reactivity, and subsequently the effect of the solvent was studied. Ethereal solvents, such as DME and THF, led to similar results as CH2Cl2 (Table 1, entries 13 and 14), whereas acetonitrile proved to be unsuitable for the reaction. Gratifyingly, it was found that the use of toluene as the solvent reverses the selectivity of the reaction, and, with this solvent, the Nazarov product 4a became the major isomer in the mixture (Table 1, entry 15). Finally, the effect of the temperature in toluene was investigated: It was found that carrying out the reaction at 0 °C afforded a 2.3/1 ratio of isomers in favor of 4a (Table 1, entry 16). If the temperature is lowered to −20 °C the ratio in favor of 4a was even higher, although only a 50% conversion was observed after 24 h (Table 1, entry 17). Under the optimized and synthetically useful reaction conditions, i.e., toluene at 0 °C, with [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgOTf as the catalytic system, 4a was obtained in 60% isolated yield.
At this point it was unclear whether the observed change of selectivity in favor of the Nazarov product 4a was mainly a solvent effect, or if the nature of the ligand also exerted an influence on the selectivity. To clear up this point the initial catalyst [(Ph3P)AuCl/AgOTf] was revisited, and 1a was treated with this catalytic system in toluene. Since the reaction was very slow at 0 °C, the temperature was increased to rt. Under these conditions the observed 3a/4a ratio was 1/1.5. By comparing this result with that in entry 1 of Table 1 led to the conclusion that the change of solvent is the main factor responsible for the selectivity switch in favor of the Nazarov product. Nevertheless, the beneficial effect of the bulky phosphite ligand is also significant factor with regards to both reactivity and selectivity.
To examine further the scope of this switch of selectivity in favor of the Nazarov pathway in tandem gold-catalyzed reactions of 3-propargylindoles initiated by 1,2-indole migrations, a selection of substrates 1a–i, bearing a methyl group at one of the propargylic positions and different (hetero)aromatic groups at both the other propargylic and terminal positions, were reacted under the established conditions (Table 2). From the results obtained, the selectivity in favor of the Nazarov products 4 seems to be general for the selected indoles 1a–h (Table 2, entries 1–8). N-unsubstituted indole 1b also showed a preference for the corresponding Nazarov product 4b, although in this case the selectivity was slightly lower compared to 1a (Table 2, entry 2), and the reaction gave a poorer overall yield. When an electron-withdrawing substituent was present on the aryl group at the propargylic position, selectivity in favor of Nazarov products 4 appeared to be slightly increased (Table 2, entry 3) [33]. Substrate 1d, with a bulky electron-withdrawing substituent at one of the ortho positions of the aromatic propargylic group, afforded almost exclusively the Nazarov product 4d in high yield (Table 2, entry 4). Similarly, the presence of a π-electron rich heteroaromatic group or an electron-rich aromatic group at the terminal position of the triple bond also favors the Nazarov pathway (Table 2, entries 5–7). On the other hand, the use of 3-propargylindole 1h as starting material, bearing an electron-withdrawing substituent on the aromatic ring at the terminal position, led to a slight decrease in the selectivity (Table 2, entry 8). Moreover, the introduction of an electron-donating group on the aromatic ring at the propargylic position gave rise to the almost exclusive formation of the iso-Nazarov product 3i (Table 2, entry 9). A comparison of these selectivities with that obtained for the parent indole 1a, leads to the conclusion that the electronic nature of the aryl groups at both the propargylic and terminal positions also has a significant influence on the preferred cyclization pathway. The Nazarov products 4 seem to be more favored when electron-withdrawing groups are present at the propargylic position and electron-donating substituents are present at the terminal position. Under these optimized conditions, new and interesting 3-(inden-2-yl)indoles 4a–h were isolated in good yields.
Table 2: Synthesis of 3-(inden-2-yl)indoles 4 by gold-catalyzed tandem 1,2-indole migration/Nazarov-type cyclization of 3-propargylindoles 1.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-89-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate | Ratio Nazarov (4)/ iso-Nazarov (3)a | Product | Yield (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-89-i7.svg?max-width=637&scale=1.0)
|
2.3/1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-89-i8.svg?max-width=637&scale=1.0)
|
60 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-89-i9.svg?max-width=637&scale=1.0)
|
1.8/1 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-89-i10.svg?max-width=637&scale=1.0)
|
41 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-89-i11.svg?max-width=637&scale=1.0)
|
3/1 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-89-i12.svg?max-width=637&scale=1.0)
|
67 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-89-i13.svg?max-width=637&scale=1.0)
|
>10/1 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-89-i14.svg?max-width=637&scale=1.0)
|
86 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-89-i15.svg?max-width=637&scale=1.0)
|
3/1 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-89-i16.svg?max-width=637&scale=1.0)
|
62 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-89-i17.svg?max-width=637&scale=1.0)
|
4/1 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-89-i18.svg?max-width=637&scale=1.0)
|
71 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-89-i19.svg?max-width=637&scale=1.0)
|
3/1 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-89-i20.svg?max-width=637&scale=1.0)
|
60 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-89-i21.svg?max-width=637&scale=1.0)
|
1.2/1 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-89-i22.svg?max-width=637&scale=1.0)
|
47c |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-89-i23.svg?max-width=637&scale=1.0)
|
<1/10 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-89-i24.svg?max-width=637&scale=1.0)
|
88d |
aDetermined by 1H NMR analysis of the crude reaction mixture. bIsolated yield of compounds 4 after column chromatography, unless otherwise stated. cDetermined by NMR from the mixture of 3h and 4h.dCombined yield for 3i and 3´i, in which the double bond has isomerized. Both compounds have been isolated and characterized. See Supporting Information File 1 (Experimental and analytical data) and Supporting Information File 2 (NMR spectra).
By contrast, it was previously observed that substitution at C-2 of the starting 3-propargylindole led almost exclusively to the formation of iso-Nazarov products 3 [13,14]. For instance, indoles 1j and 1k, bearing a methyl and a phenyl group at C-2, respectively, provided the corresponding indole derivatives 3j and 3k with high selectivity when the reaction was conducted in CH2Cl2 with (Ph3P)AuCl/AgSbF6 as catalyst (Scheme 3). Interestingly, under the new conditions developed herein, i.e., treatment with a cationic phosphite–gold complex in toluene, the reaction of 1j afforded a ca. 3.5/1 mixture of 3j/4j, whereas 1k gave rise to a ca. 1.6/1 mixture of 3k/4k (Scheme 3). These results again show that the change of selectivity in the competitive iso-Nazarov/Nazarov pathways could be induced by a change of ligand and solvent, although complete reversal of selectivity was not achieved for these substrates.
![[1860-5397-7-89-i3]](/bjoc/content/inline/1860-5397-7-89-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Comparison of the reactivity of C-2 substituted indoles 1j and 1k. Conditions: a) (Ph3P)AuCl/AgSbF6 (5 mol %), CH2Cl2, rt; b) [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgOTf (5 mol %), toluene, 0 °C.
Scheme 3: Comparison of the reactivity of C-2 substituted indoles 1j and 1k. Conditions: a) (Ph3P)AuCl/AgSbF6...
It has also been observed that reactions of 3-propargylindoles bearing alkyl substituents bulkier than methyl at the propargylic position, such as 1l and 1m, almost exclusively produced the corresponding iso-Nazarov products 3l and 3m with (Ph3P)AuCl/AgSbF6 as catalyst in CH2Cl2 (Scheme 4) [13,14]. Again, the use of the phosphite–gold complex as catalyst and toluene as solvent slightly favored the Nazarov pathway: Approximately 3/1 ratios of the corresponding indole derivatives 3l, m/4l, m were obtained (Scheme 4) [32]. In addition, we were able to isolate the new Nazarov compounds 4l and 4m, albeit in low yields (Scheme 4). Finally, when the more sterically demanding isopropyl group was present at the propargylic position, the corresponding iso-Nazarov product was produced exclusively irrespective of the conditions employed. Although these results show that the change of the methyl group at the propargylic position of the starting indole 1 to a bulkier alkyl group strongly favors the iso-Nazarov pathway, they also show that our new reported conditions make the Nazarov pathway more accessible.
![[1860-5397-7-89-i4]](/bjoc/content/inline/1860-5397-7-89-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions of 3-propargylindoles 1l and 1m with bulky alkyl substituents at the propargylic position. Conditions: a) (Ph3P)AuCl/AgSbF6 (5 mol %), CH2Cl2, rt; b) [(2,4-(t-Bu)2C6H3O)3P]AuCl/AgOTf (5 mol %), toluene, 0 °C.
Scheme 4: Reactions of 3-propargylindoles 1l and 1m with bulky alkyl substituents at the propargylic position...
Conclusion
We have studied the effect of the ligands and counter ion of the catalyst, as well as the electronic nature of the aryl substituents and the reaction conditions (solvent, temperature), in the gold(I)-catalyzed tandem reactions of 3-propargylindoles initiated by 1,2-indole migrations. We have been able to switch the preference of 3-propargylindoles, bearing (hetero)aromatic substituents at both propargylic and terminal positions of the alkyne moiety, from undergoing an aura-iso-Nazarov cyclization in favor of an aura-Nazarov cyclization. The two competitive pathways are influenced mainly by the electronic and steric properties of the aryl substituent at the propargylic position, as well as the ligand of the catalyst and the solvent used. In this way, new and interesting 3-(inden-2-yl)indoles were obtained in good yields.
Supporting Information
Experimental procedures and spectroscopic data for all new compounds. Copies of 1H NMR and 13C NMR spectra for new compounds.
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 171.5 KB | Download |
| Supporting Information File 2: NMR spectra. | ||
| Format: PDF | Size: 2.4 MB | Download |
Acknowledgements
We acknowledge MICINN (CTQ2010-15358 and CTQ2009-09949) and Junta de Castilla y León (BU021A09 and GR-172) for financial support. We are also grateful to MEC (FPU predoctoral fellowship to D.M., “Ramón y Cajal” contract to M. A. F. R. and “Juan de la Cierva” contract to P. G. G.). Many thanks are due to Prof. Dr. A. R. de Lera (Universidade de Vigo) for his helpful comments.
References
-
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] -
Michelet, V.; Toullec, P. Y.; Genét, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. doi:10.1002/anie.200701589
Return to citation in text: [1] -
Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j
Return to citation in text: [1] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369
Return to citation in text: [1] -
Wang, S.; Zhang, G.; Zhang, L. Synlett 2010, 692–706. doi:10.1055/s-0029-1219527
Return to citation in text: [1] -
Das, A.; Sohel, S. M. A.; Liu, R.-S. Org. Biomol. Chem. 2010, 8, 960–979. doi:10.1039/b923510h
Return to citation in text: [1] -
Bandini, M. Chem. Soc. Rev. 2011, 40, 1358–1367. doi:10.1039/c0cs00041h
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773
Return to citation in text: [1] -
Li, G.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2008, 130, 3740–3741. doi:10.1021/ja800001h
Return to citation in text: [1] -
Watson, I. D. G.; Ritter, S.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2056–2057. doi:10.1021/ja8085005
Return to citation in text: [1] -
Uemura, M.; Watson, I. D. G.; Katsukawa, M.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 3464–3465. doi:10.1021/ja900155x
Return to citation in text: [1] -
Peng, L.; Zhang, X.; Zhang, S.; Wang, J. J. Org. Chem. 2007, 72, 1192–1197. doi:10.1021/jo0618674
Return to citation in text: [1] -
Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660
Return to citation in text: [1] [2] [3] -
Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162
Return to citation in text: [1] [2] [3] [4] -
Lakhdar, S.; Westermaier, M.; Terrier, F.; Goumont, R.; Boubaker, T.; Ofial, A. R.; Mayr, H. J. Org. Chem. 2006, 71, 9088–9095. doi:10.1021/jo0614339
Return to citation in text: [1] -
Ferrer, C.; Amijs, C. H. M.; Echavarren, A. M. Chem.–Eur. J. 2007, 13, 1358–1373. doi:10.1002/chem.200601324
Return to citation in text: [1] -
Liu, C.; Widenhoefer, R. A. Org. Lett. 2007, 9, 1935–1938. doi:10.1021/ol070483c
Return to citation in text: [1] -
Sanz, R.; Miguel, D.; Álvarez-Gutiérrez, J. M.; Rodríguez, F. Synlett 2008, 975–978. doi:10.1055/s-2008-1072584
Return to citation in text: [1] -
Sanz, R.; Miguel, D.; Martínez, A.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; Álvarez, E.; Rodríguez, F. Eur. J. Org. Chem. 2010, 7027–7039. doi:10.1002/ejoc.201001055
Return to citation in text: [1] -
Considering an initial gold coordination to the alkyne syn to the indole heterocycle, the calculated energy barriers for the cyclizations favored the Nazarov cyclization (10.88 kcal/mol vs. 13.09 kcal/mol for the iso-Nazarov cyclization).
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] -
Baskar, B.; Bae, H. J.; An, S. E.; Cheong, J. Y.; Rhee, Y. H.; Duschek, A.; Kirsch, S. F. Org. Lett. 2008, 10, 2605–2607. doi:10.1021/ol8008733
Return to citation in text: [1] -
Davies, P. W.; Martin, N. Org. Lett. 2009, 11, 2293–2296. doi:10.1021/ol900609f
Return to citation in text: [1] -
Dudnik, A. S.; Xia, Y.; Li, Y.; Gevorgyan, V. J. Am. Chem. Soc. 2010, 132, 7645–7655. doi:10.1021/ja910290c
Return to citation in text: [1] -
Seo, H.; Roberts, B. P.; Abboud, K. A.; Merz, K. M., Jr.; Hong, S. Org. Lett. 2010, 12, 4860–4863. doi:10.1021/ol102018z
Return to citation in text: [1] -
Klahn, P.; Kirsch, S. F. ChemCatChem 2011, 3, 649–652. doi:10.1002/cctc.201000366
Return to citation in text: [1] -
Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194
Return to citation in text: [1] -
CCDC-818023 contains the supplementary crystallographic data for compound 4a. These data can be obtained free of charge from The Cambridge Cyrstallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
In our first communication [13] the structure of the minor isomer obtained from an analogous 3-propargylindole bearing an ethyl group at the propargylic position was erroneously assigned to a 3-(inden-1-yl)indole derivative arising from a competitive hydroarylation of the triple bond by the propargylic phenyl group.
Return to citation in text: [1] -
Nolan, S. P. Acc. Chem. Res. 2011, 44, 91–100. doi:10.1021/ar1000764
Return to citation in text: [1] -
Mauleón, P.; Zeldin, R. M.; González, A. Z.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 6348–6349. doi:10.1021/ja901649s
Return to citation in text: [1] -
When [(2,4-(t-Bu)2C6H3O)3P]AuCl was used as catalyst the iso-Nazarov products were obtained as a mixture of 3 and 3´, in which the double bond is isomerized. So, the ratio of isomers 3/4 reflects the ratio of both the iso-Nazarov compounds 3 and 3´ against the Nazarov product 4.
Return to citation in text: [1] [2] -
We have also checked that 1b affords a ca. 2.2/1 mixture of 3b/4b when (Ph3P)AuCl/AgSbF6 was used as catalytic system in CH2Cl2, also proving the effect of the electron-withdrawing substituent on the phenyl group at the propargylic position.
Return to citation in text: [1]
| 13. | Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660 |
| 13. | Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660 |
| 14. | Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162 |
| 32. | When [(2,4-(t-Bu)2C6H3O)3P]AuCl was used as catalyst the iso-Nazarov products were obtained as a mixture of 3 and 3´, in which the double bond is isomerized. So, the ratio of isomers 3/4 reflects the ratio of both the iso-Nazarov compounds 3 and 3´ against the Nazarov product 4. |
| 1. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 2. | Michelet, V.; Toullec, P. Y.; Genét, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. doi:10.1002/anie.200701589 |
| 3. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j |
| 4. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 5. | Wang, S.; Zhang, G.; Zhang, L. Synlett 2010, 692–706. doi:10.1055/s-0029-1219527 |
| 6. | Das, A.; Sohel, S. M. A.; Liu, R.-S. Org. Biomol. Chem. 2010, 8, 960–979. doi:10.1039/b923510h |
| 7. | Bandini, M. Chem. Soc. Rev. 2011, 40, 1358–1367. doi:10.1039/c0cs00041h |
| 15. | Lakhdar, S.; Westermaier, M.; Terrier, F.; Goumont, R.; Boubaker, T.; Ofial, A. R.; Mayr, H. J. Org. Chem. 2006, 71, 9088–9095. doi:10.1021/jo0614339 |
| 33. | We have also checked that 1b affords a ca. 2.2/1 mixture of 3b/4b when (Ph3P)AuCl/AgSbF6 was used as catalytic system in CH2Cl2, also proving the effect of the electron-withdrawing substituent on the phenyl group at the propargylic position. |
| 13. | Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660 |
| 14. | Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162 |
| 13. | Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660 |
| 14. | Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162 |
| 12. | Peng, L.; Zhang, X.; Zhang, S.; Wang, J. J. Org. Chem. 2007, 72, 1192–1197. doi:10.1021/jo0618674 |
| 31. | Mauleón, P.; Zeldin, R. M.; González, A. Z.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 6348–6349. doi:10.1021/ja901649s |
| 8. | Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773 |
| 9. | Li, G.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2008, 130, 3740–3741. doi:10.1021/ja800001h |
| 10. | Watson, I. D. G.; Ritter, S.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2056–2057. doi:10.1021/ja8085005 |
| 11. | Uemura, M.; Watson, I. D. G.; Katsukawa, M.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 3464–3465. doi:10.1021/ja900155x |
| 32. | When [(2,4-(t-Bu)2C6H3O)3P]AuCl was used as catalyst the iso-Nazarov products were obtained as a mixture of 3 and 3´, in which the double bond is isomerized. So, the ratio of isomers 3/4 reflects the ratio of both the iso-Nazarov compounds 3 and 3´ against the Nazarov product 4. |
| 20. | Considering an initial gold coordination to the alkyne syn to the indole heterocycle, the calculated energy barriers for the cyclizations favored the Nazarov cyclization (10.88 kcal/mol vs. 13.09 kcal/mol for the iso-Nazarov cyclization). |
| 28. | CCDC-818023 contains the supplementary crystallographic data for compound 4a. These data can be obtained free of charge from The Cambridge Cyrstallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 29. | In our first communication [13] the structure of the minor isomer obtained from an analogous 3-propargylindole bearing an ethyl group at the propargylic position was erroneously assigned to a 3-(inden-1-yl)indole derivative arising from a competitive hydroarylation of the triple bond by the propargylic phenyl group. |
| 14. | Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162 |
| 18. | Sanz, R.; Miguel, D.; Álvarez-Gutiérrez, J. M.; Rodríguez, F. Synlett 2008, 975–978. doi:10.1055/s-2008-1072584 |
| 19. | Sanz, R.; Miguel, D.; Martínez, A.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; Álvarez, E.; Rodríguez, F. Eur. J. Org. Chem. 2010, 7027–7039. doi:10.1002/ejoc.201001055 |
| 16. | Ferrer, C.; Amijs, C. H. M.; Echavarren, A. M. Chem.–Eur. J. 2007, 13, 1358–1373. doi:10.1002/chem.200601324 |
| 17. | Liu, C.; Widenhoefer, R. A. Org. Lett. 2007, 9, 1935–1938. doi:10.1021/ol070483c |
| 21. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 22. | Baskar, B.; Bae, H. J.; An, S. E.; Cheong, J. Y.; Rhee, Y. H.; Duschek, A.; Kirsch, S. F. Org. Lett. 2008, 10, 2605–2607. doi:10.1021/ol8008733 |
| 23. | Davies, P. W.; Martin, N. Org. Lett. 2009, 11, 2293–2296. doi:10.1021/ol900609f |
| 24. | Dudnik, A. S.; Xia, Y.; Li, Y.; Gevorgyan, V. J. Am. Chem. Soc. 2010, 132, 7645–7655. doi:10.1021/ja910290c |
| 25. | Seo, H.; Roberts, B. P.; Abboud, K. A.; Merz, K. M., Jr.; Hong, S. Org. Lett. 2010, 12, 4860–4863. doi:10.1021/ol102018z |
| 26. | Klahn, P.; Kirsch, S. F. ChemCatChem 2011, 3, 649–652. doi:10.1002/cctc.201000366 |
| 27. | Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194 |
© 2011 Álvarez et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)