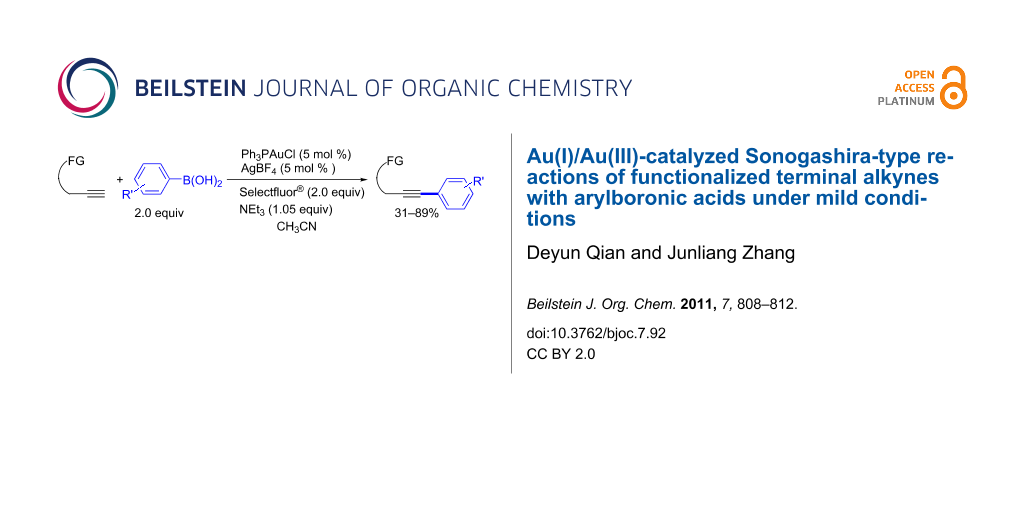
Abstract
A straightforward, efficient, and reliable redox catalyst system for the Au(I)/Au(III)-catalyzed Sonogashira cross-coupling reaction of functionalized terminal alkynes with arylboronic acids under mild conditions has been developed.

Graphical Abstract
Introduction
The Sonogashira reaction has become the most important and widely used method for the synthesis of arylalkynes and conjugated enynes, which are precursors for natural products, pharmaceuticals and other materials [1-3]. In the past decade, considerable efforts have been made to enhance the efficiency and generality of this reaction. All kinds of palladium catalyst systems [4-10] and other metal catalyst systems [3] have been developed for facilitating the Sonogashira cross-coupling, as well as expanding the substrate scope [11-14]. Various examples have recently been reported for the palladium-catalyzed Sonagashira-type cross-coupling of terminal alkynes with arylboronic acids [13], and the Pd(0)/Pd(II) catalytic cycles have been well studied. Nevertheless, this transformation catalyzed by gold, involving Au(I)/Au(III) catalytic cycles has, as yet, been less explored [15-22]. In the few examples already documented some conditions, such as rather high reaction temperatures (130 °C), high catalysis loading or special reagents were required [23]. Herein, we report a straightforward, efficient and robust catalyst system for the Sonogashira-type cross-coupling, in which Au(I)/Au(III) catalyzed Csp2–Csp bond formation of terminal alkynes from arylboronic acids under mild conditions.
By analogy with other d10 species, Au(I) has the same electronic structure as Pd(0) and can easily interact with the acetylenic group, and has the ability to undergo the Au(I)/Au(III) redox cycles [24,25]. In addition, with an increasing interest in the chemistry of gold(I) and gold(III) compounds, more and more studies have provided strong evidence for the existence of Au(I)/Au(III) catalytic cycles [26-32]. For instance, Zhang and co-workers have developed a gold-catalyzed oxidative cross-coupling reaction of arylboronic acids with propargyl esters [27], and Selectfluor® – a source of electrophilic fluorine – was used to oxidize the resulting Au(I) intermediate to Au(III) species. Recently, Toste reported the first experimental evidence for alkylgold(III) fluorides undergoing C–C bond forming reactions with arylboronic acids [32]. Inspired by these results, we envisioned that terminal alkynes would react with arylboronic acids in the presence of oxidant (Selectfluor®) and base, and undergo a Au(I)/Au(III)-catalyzed Sonogashira-type cross-coupling reaction (Scheme 1).
![[1860-5397-7-92-i1]](/bjoc/content/inline/1860-5397-7-92-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previous work and our projected gold-catalyzed Sonogashira-type cross-coupling.
Scheme 1: Previous work and our projected gold-catalyzed Sonogashira-type cross-coupling.
Results and Discussion
We embarked on developing a general protocol for Sonogashira-type cross-coupling by using propargyl tosylamide (1a) and phenylboronic acid as the model substrates because of their stability, availability, and broad spectrum of nucleophilicity (Table 1). With commercially available Ph3PAuCl as the catalyst, we treated 1a and phenylboronic acid in CH3CN at room temperature for 18 h (Table 1, entry 1). Disappointedly, only trace amounts of product was observed under the above conditions. Taking into account the counter ion effects in gold-catalyzed reactions [33-35], Ph3PAuOTf, produced from a combination of Ph3PAuCl and AgOTf in a 1:1 ratio, was investigated. To our delight, the expected product 2a could be isolated in 56% yield (Table 1, entry 2). Moreover, a control experiment was carried out to determine whether AgOTf by itself could catalyze this transformation. However, no 2a was produced in the absence of Ph3PAuCl (Table 1, entry 3). Without the phosphine ligand, both AuCl/AgOTf or AuCl3/AgOTf could catalyze this reaction, but the efficiency was quite low (Table 1, entries 4, 5). Without Selectfluor® as additive, the cross-coupling reaction did not occur (Table 1, entry 6), indicating that it was crucial for this transformation via oxidation of gold(I) to the gold(III) species [26-32]. Reducing the amount of phenylboronic acid resulted in a lower yield (Table 1, entry 7). The counter ion effect was then examined on cross-coupling reaction by variation of the silver salts. Alternative catalyst systems led to better yields (Table 1, entries 8, 9 and notably 11 vs entry 2). To optimize the reaction conditions, commonly employed well recognized and commercially available phosphine ligands [17,18], such as dppm, dppe, dppp, dppf, and XPhos, were screened to test the feasibility of this gold-catalyzed cross-coupling (Table 1, entries 10–12, see also Supporting Information File 1). In addition, a series of inorganic and organic bases was also investigated: K2CO3, K3PO4, K3PO4·3H2O, NaHCO3, NaOAc were substantially less effective, whilst organic bases such as iPr2NH, Et2NH, TMEDA, Bu3N, PhNMe2 were not effective bases in this catalytic system (see Supporting Information File 1). These results indicated that Et3N might also play an important role in this process. Furthermore, it was also found that neither slow addition nor a significant excess of alkyne was required to obtain selective and almost quantitative conversion (see Supporting Information File 1). When the reaction was carried out under an atmosphere of nitrogen (N2) there was a 10% increase in yield (Table 1, entry 8 vs 14). Although the conditions (Table 1, entry 11) seemed the best, this was not the general case for other substrates. Therefore, the optimum conditions were chosen as Ph3PAuCl/AgBF4 as the catalyst, Et3N as the base and under an atmosphere of nitrogen (Table 1, entry 14) .
Table 1: Initial screenings of Au(I)/Au(III)-catalyzed Sonogashira couplinga.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-92-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | AuL (5 mol %) | AgX (mol %) | Time (h) | Yield (%)b |
|---|---|---|---|---|
| 1 | Ph3PAuCl | — | 18 | trace |
| 2 | Ph3PAuCl | AgOTf (5) | 22 | 56 |
| 3 | — | AgOTf (5) | 24 | 0 |
| 4 | AuCl | AgOTf (5) | 24 | 21 |
| 5 | AuCl3 | AgOTf (15) | 24 | 37c |
| 6d | Ph3PAuCl | AgOTf (5) | 22 | 0 |
| 7e | Ph3PAuCl | AgOTf (5) | 22 | 41 |
| 8 | Ph3PAuCl | AgBF4 (5) | 10 | 65 |
| 9 | Ph3PAuCl | AgSbF4(5) | 10 | 62 |
| 10 | (XPhos)AuCl | AgOTf (5) | 24 | 0 |
| 11f | dppm(AuCl)2 | AgOTf (5) | 16 | 83c |
| 12f | dppm(AuBr)2 | — | 16 | trace |
| 13f,g | Ph3PAuCl | AgOTf (5) | 12 | 72 (73c) |
| 14f | Ph3PAuCl | AgBF4 (5) | 12 | 75 (80c) |
aReaction conditions: The reaction was carried out by using 1a (0.4 mmol) and phenylboronic acid (0.8 mmol, 2.0 equiv), and 1.05 equiv of Et3N in 2 mL of CH3CN stirred at room temperature. bIsolated yields. cYield determined by 1H NMR with dibromomethane as an internal standard. dSelectfluor® (0 equiv). ePhB(OH)2 (1.5 equiv). fUnder an atmosphere of nitrogen (N2). gThe reaction temperature was 50 °C.
By using the above optimized conditions, the reaction scope was next studied by varying arylboronic acids. As shown in Scheme 2, functional groups including methyl, chloro, fluoro and ester in the para positions of the phenyl ring were tolerated. Reduced yields were observed with both electron-rich and electron-poor coupling partners (Scheme 2, 2a–f). Arylboronic acids with electron-withdrawing groups afforded 2c–2e in moderate to good yields. Notably, the slightly electron-deficient 4-chlorophenylboronic acid gave the best yield. On the other hand, the more electron-rich 4-methoxyphenylboronic acid produced the lowest yield, likely due to a competing oxidation of the boronic acid by Selectfluor® [27,30]. Gratifyingly, a number of potentially reactive functionalities, such as tertiary amine, 4-methylbenzenesulfonate, 1,6-enyne and 1,6-diyne, were compatible and remained unaffected, which illustrated the robustness of the catalyst system (Scheme 2, 3–8).
![[1860-5397-7-92-i2]](/bjoc/content/inline/1860-5397-7-92-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the Sonogashira-type cross-coupling reaction (isolated yield). aAgOTf in place of AgBF4. bYield determined by 1H NMR, the prouduct could not be separated from the unreacted starting material. cAgOTf in place of AgBF4, RT, 36 h.
Scheme 2: Scope of the Sonogashira-type cross-coupling reaction (isolated yield). aAgOTf in place of AgBF4. b...
On the basis of these results, a plausible mechanism is outlined in Scheme 3. Initially, unlike traditional C–X oxidative addition as shown by Pd in cross-coupling reactions, the active cationic gold(I) species A is oxidized by Selectfluor® to give a gold(III) species B [26-32,36]. With the aid of base, the reaction between B and alkyne affords intermediate C. The weak Au–F bond and the strong B–F bond drive the trans-metalation to produce intermediate D [29,36,37]. Finally, D undergoes reductive elimination to give the desired product and gold(I) species A. In addition, C also could experience a five-centered transition state D', which leads to the C–C bond-forming reaction through a bimolecular reductive elimination [30-32]. Notably, we believed that the key step of this mechanism is the generation of cationic gold species B by Selectfluor®. In the absence of Selectfluor®, no coupling is possible (Table 1, entry 6). In a current study, Xu and co-workers have provided strong evidence for the oxidation of Au(I) to Au(III) by Selectfluor® in their XPS measurements [36].
![[1860-5397-7-92-i3]](/bjoc/content/inline/1860-5397-7-92-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the Au(I)/Au(III)-catalyzed Sonogashira-type cross-coupling.
Scheme 3: Proposed mechanism for the Au(I)/Au(III)-catalyzed Sonogashira-type cross-coupling.
Conclusion
In conclusion, we have developed an unprecedented Au(I)/Au(III)-catalyzed Sonogashira-type cross-coupling reaction of terminal alkynes and arylboronic acids under mild conditions. Selectfluor® and counter ion effects play a significant role in the development of an exceptionally mild catalyst system. This chemistry strongly suggests the feasibility of Au(I) and Au(III) catalytic redox cycles, which would substantially broaden the field of gold catalysis and offer more functionalized products. Furthermore, the good tolerance toward many functional groups of substrates considerably extends the scope of a number of organic transformations and performs modular Csp2–Csp bond constructions at appropriate stages in the whole synthetic sequence.
Supporting Information
Supporting information features experimental procedure and spectroscopic data.
| Supporting Information File 1: Experimental details and spectra of new compounds. | ||
| Format: PDF | Size: 811.2 KB | Download |
References
-
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] -
Doucet, H.; Hierso, J.-C. Angew. Chem., Int. Ed. 2007, 46, 834–871. doi:10.1002/anie.200602761
Return to citation in text: [1] -
Plenio, H. Angew. Chem., Int. Ed. 2008, 47, 6954–6956. doi:10.1002/anie.200802270
Return to citation in text: [1] [2] -
Tougerti, A.; Negri, S.; Jutand, A. Chem.–Eur. J. 2007, 13, 666–676. doi:10.1002/chem.200600574
Return to citation in text: [1] -
Ahlquist, M.; Norrby, P.-O. Organometallics 2007, 26, 550–553. doi:10.1021/om0604932
Return to citation in text: [1] -
Liang, Y.; Xie, Y.; Li, J. J. Org. Chem. 2006, 71, 379–381. doi:10.1021/jo051882t
Return to citation in text: [1] -
Yi, C.; Hua, R. J. Org. Chem. 2006, 71, 2535–2537. doi:10.1021/jo0525175
Return to citation in text: [1] -
Liang, B.; Dai, M.; Chen, J.; Yang, Z. J. Org. Chem. 2005, 70, 391–393. doi:10.1021/jo048599z
Return to citation in text: [1] -
Urgaonkar, S.; Verkade, J. G. J. Org. Chem. 2004, 69, 5752–5755. doi:10.1021/jo049325e
Return to citation in text: [1] -
Finke, A. D.; Elleby, E. C.; Boyd, M. J.; Weissman, H.; Moore, J. S. J. Org. Chem. 2009, 74, 8897–8900. doi:10.1021/jo902015w
Return to citation in text: [1] -
R’kyek, O.; Halland, N.; Lindenschmidt, A.; Alonso, J.; Lindemann, P.; Urmann, M.; Nazaré, M. Chem.–Eur. J. 2010, 16, 9986–9989. doi:10.1002/chem.201001524
Return to citation in text: [1] -
Choy, P. Y.; Chow, W. K.; So, C. M.; Lau, C. P.; Kwong, F. K. Chem.–Eur. J. 2010, 16, 9982–9985. doi:10.1002/chem.201001269
Return to citation in text: [1] -
Zhou, M.-B.; Wei, W.-T.; Xie, Y.-X.; Lei, Y.; Li, J.-H. J. Org. Chem. 2010, 75, 5635–5642. doi:10.1021/jo101063p
Return to citation in text: [1] [2] -
Mitsudo, K.; Shiraga, T.; Mizukawa, J.; Suga, S.; Tanaka, H. Chem. Commun. 2010, 46, 9256–9258. doi:10.1039/C0CC02633F
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] [2] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] [2] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232–5241. doi:10.1002/anie.200907078
Return to citation in text: [1] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369
Return to citation in text: [1] -
Garcia, P.; Malacria, M.; Aubert, C.; Gandon, V.; Fensterbank, L. ChemCatChem 2010, 2, 493–497. doi:10.1002/cctc.200900319
Return to citation in text: [1] -
Li, P.; Wang, L.; Wang, M.; You, F. Eur. J. Org. Chem. 2008, 5946–5951. doi:10.1002/ejoc.200800765
Return to citation in text: [1] -
Carrettin, S.; Guzman, J.; Corma, A. Angew. Chem., Int. Ed. 2005, 44, 2242–2245. doi:10.1002/anie.200462560
Return to citation in text: [1] -
González-Arellano, C.; Abad, A.; Corma, A.; García, H.; Iglesias, M.; Sánchez, F. Angew. Chem., Int. Ed. 2007, 46, 1536–1538. doi:10.1002/anie.200604746
Return to citation in text: [1] -
Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. J. Organomet. Chem. 2009, 694, 592–597. doi:10.1016/j.jorganchem.2008.11.054
Return to citation in text: [1] [2] [3] -
Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585
Return to citation in text: [1] [2] [3] [4] [5] -
Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w
Return to citation in text: [1] [2] [3] -
Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d
Return to citation in text: [1] [2] [3] [4] -
Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739
Return to citation in text: [1] [2] [3] [4] [5] -
Melhado, A. D.; Brenzovich, W. E.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123
Return to citation in text: [1] [2] [3] [4] -
Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n
Return to citation in text: [1] [2] [3] [4] [5] -
LaLonde, R. L.; Sherry, B. D.; Kang, E. J.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 2452–2453. doi:10.1021/ja068819l
Return to citation in text: [1] -
Hamilton, G. L.; Kang, E. J.; Mba, M.; Toste, F. D. Science 2007, 317, 496–499. doi:10.1126/science.1145229
Return to citation in text: [1] -
LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 598–601. doi:10.1002/anie.200905000
Return to citation in text: [1] -
Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593
Return to citation in text: [1] [2] [3] -
Okabayashi, T.; Nakaoka, Y.; Yamazaki, E.; Tanimoto, M. Chem. Phys. Lett. 2002, 366, 406–411. doi:10.1016/S0009-2614(02)01586-5
Return to citation in text: [1]
| 36. | Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593 |
| 29. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 36. | Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593 |
| 37. | Okabayashi, T.; Nakaoka, Y.; Yamazaki, E.; Tanimoto, M. Chem. Phys. Lett. 2002, 366, 406–411. doi:10.1016/S0009-2614(02)01586-5 |
| 30. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 31. | Melhado, A. D.; Brenzovich, W. E.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 32. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 1. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 2. | Doucet, H.; Hierso, J.-C. Angew. Chem., Int. Ed. 2007, 46, 834–871. doi:10.1002/anie.200602761 |
| 3. | Plenio, H. Angew. Chem., Int. Ed. 2008, 47, 6954–6956. doi:10.1002/anie.200802270 |
| 13. | Zhou, M.-B.; Wei, W.-T.; Xie, Y.-X.; Lei, Y.; Li, J.-H. J. Org. Chem. 2010, 75, 5635–5642. doi:10.1021/jo101063p |
| 27. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
| 30. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 11. | R’kyek, O.; Halland, N.; Lindenschmidt, A.; Alonso, J.; Lindemann, P.; Urmann, M.; Nazaré, M. Chem.–Eur. J. 2010, 16, 9986–9989. doi:10.1002/chem.201001524 |
| 12. | Choy, P. Y.; Chow, W. K.; So, C. M.; Lau, C. P.; Kwong, F. K. Chem.–Eur. J. 2010, 16, 9982–9985. doi:10.1002/chem.201001269 |
| 13. | Zhou, M.-B.; Wei, W.-T.; Xie, Y.-X.; Lei, Y.; Li, J.-H. J. Org. Chem. 2010, 75, 5635–5642. doi:10.1021/jo101063p |
| 14. | Mitsudo, K.; Shiraga, T.; Mizukawa, J.; Suga, S.; Tanaka, H. Chem. Commun. 2010, 46, 9256–9258. doi:10.1039/C0CC02633F |
| 26. | Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. J. Organomet. Chem. 2009, 694, 592–597. doi:10.1016/j.jorganchem.2008.11.054 |
| 27. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
| 28. | Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w |
| 29. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 30. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 31. | Melhado, A. D.; Brenzovich, W. E.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 32. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 36. | Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593 |
| 3. | Plenio, H. Angew. Chem., Int. Ed. 2008, 47, 6954–6956. doi:10.1002/anie.200802270 |
| 26. | Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. J. Organomet. Chem. 2009, 694, 592–597. doi:10.1016/j.jorganchem.2008.11.054 |
| 27. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
| 28. | Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w |
| 29. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 30. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 31. | Melhado, A. D.; Brenzovich, W. E.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 32. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 4. | Tougerti, A.; Negri, S.; Jutand, A. Chem.–Eur. J. 2007, 13, 666–676. doi:10.1002/chem.200600574 |
| 5. | Ahlquist, M.; Norrby, P.-O. Organometallics 2007, 26, 550–553. doi:10.1021/om0604932 |
| 6. | Liang, Y.; Xie, Y.; Li, J. J. Org. Chem. 2006, 71, 379–381. doi:10.1021/jo051882t |
| 7. | Yi, C.; Hua, R. J. Org. Chem. 2006, 71, 2535–2537. doi:10.1021/jo0525175 |
| 8. | Liang, B.; Dai, M.; Chen, J.; Yang, Z. J. Org. Chem. 2005, 70, 391–393. doi:10.1021/jo048599z |
| 9. | Urgaonkar, S.; Verkade, J. G. J. Org. Chem. 2004, 69, 5752–5755. doi:10.1021/jo049325e |
| 10. | Finke, A. D.; Elleby, E. C.; Boyd, M. J.; Weissman, H.; Moore, J. S. J. Org. Chem. 2009, 74, 8897–8900. doi:10.1021/jo902015w |
| 17. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 18. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 26. | Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. J. Organomet. Chem. 2009, 694, 592–597. doi:10.1016/j.jorganchem.2008.11.054 |
| 27. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
| 28. | Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w |
| 29. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 30. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 31. | Melhado, A. D.; Brenzovich, W. E.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 32. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 32. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 24. | Carrettin, S.; Guzman, J.; Corma, A. Angew. Chem., Int. Ed. 2005, 44, 2242–2245. doi:10.1002/anie.200462560 |
| 25. | González-Arellano, C.; Abad, A.; Corma, A.; García, H.; Iglesias, M.; Sánchez, F. Angew. Chem., Int. Ed. 2007, 46, 1536–1538. doi:10.1002/anie.200604746 |
| 33. | LaLonde, R. L.; Sherry, B. D.; Kang, E. J.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 2452–2453. doi:10.1021/ja068819l |
| 34. | Hamilton, G. L.; Kang, E. J.; Mba, M.; Toste, F. D. Science 2007, 317, 496–499. doi:10.1126/science.1145229 |
| 35. | LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 598–601. doi:10.1002/anie.200905000 |
| 23. | Li, P.; Wang, L.; Wang, M.; You, F. Eur. J. Org. Chem. 2008, 5946–5951. doi:10.1002/ejoc.200800765 |
| 15. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 16. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 17. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 18. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 19. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 20. | Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232–5241. doi:10.1002/anie.200907078 |
| 21. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 22. | Garcia, P.; Malacria, M.; Aubert, C.; Gandon, V.; Fensterbank, L. ChemCatChem 2010, 2, 493–497. doi:10.1002/cctc.200900319 |
| 27. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
© 2011 Qian and Zhang; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)