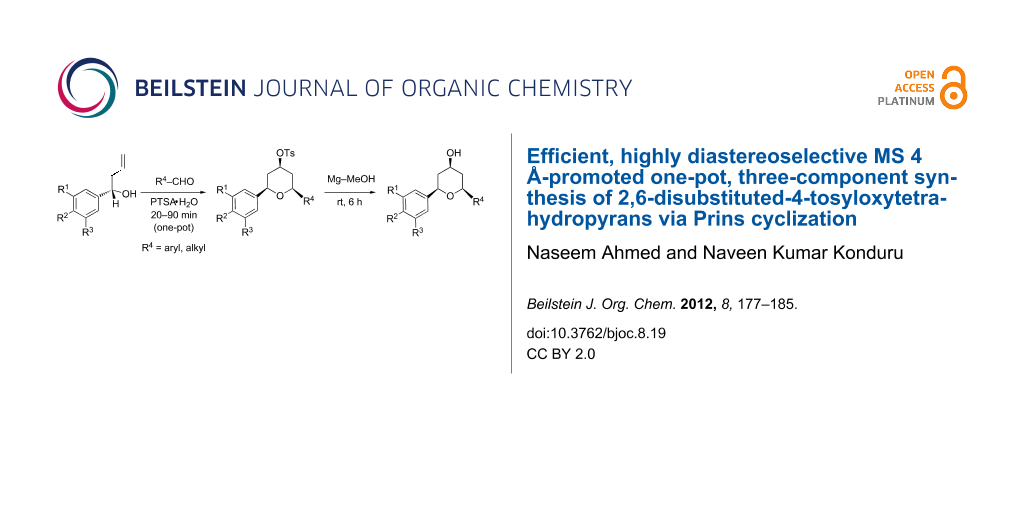
Abstract
A simple, efficient and highly diastereoselective one-pot three-component synthesis of functionalized 2,6-disubstituted-4-tosyloxytetrahydropyrans was performed. The synthesis features an optimized Prins cyclization in which an aromatic homoallylic alcohol, an aromatic/aliphatic aldehyde, and p-toluenesulfonic acid (catalyst and reagent) are reacted in the presence of molecular sieves (MS) 4 Å at reflux in dichloromethane to afford excellent yields (72–96%) within short reaction times (20–90 min). The MS 4 Å-promoted synthesis proved to be versatile enough to provide an array of symmetrical and unsymmetrical tetrahydropyran derivatives in economical manner. Furthermore, cleavage of the 4-tosyl group under mild conditions afforded 4-hydroxytetrahydropyran in excellent yields (95–96%).

Graphical Abstract
Introduction
Substituted tetrahydropyrans are common structural motifs in numerous biological molecules and natural products that include phorboxazoles (A and B) [1], (−)-centrolobine [2], GEX1A/herboxidiene [3], bryostatins [4], and pheromones [5] (Figure 1). Tetrahydropyran derivatives are also used as materials in photographic films [6] and host–guest chemistry [7]. In particular, 2,4,6-trisubstituted tetrahydropyrans have tremendous applications in pharmaceuticals and are widely present in biologically active core structures such as 4-oxygenated, 4-halogenated, 4-sulfonyl- and 4-azido/amidotetrahydropyrans [8-11]. Various applications of tetrahydropyran derivatives have inspired organic chemists to develop their efficient, economical and stereoselective synthesis.
![[1860-5397-8-19-1]](/bjoc/content/figures/1860-5397-8-19-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Tetrahydropyran ring containing natural products.
Figure 1: Tetrahydropyran ring containing natural products.
To introduce the desired substituents and stereochemistry at the 2, 4 and 6-positions of the tetrahydropyran ring, Prins cyclization has been considered to be the best approach. The reaction can be conducted either with Brønsted acid catalysts – namely TFA [12], AcOH, MeSO3H [13], Sc(OTf)3 [14], O3ReOSiPh3 [15], TMSBr [16] – or with Lewis acid catalysts such as AlCl3 [17], InCl3 [18,19], TiCl4, HBF4·OEt2 [20], InBr3 [21], TiBr4 [22], Fe(III) compounds [23], BF3·OEt2 [24], polymer supported [25], microwave accelerated with BiCl3 [26], and Pd0/SnII mediated by a three component cascade coupling (3-C3) [27]. However, these procedures suffer from drawbacks like moderate yields, high/low reaction temperatures, extended reaction times, and/or strong acid conditions [28,29]. Therefore, Wills et al. reported an efficient synthetic route for 2,4,6-trisubstituted tetrahydropyrans [30]; however, this method is restricted to electron withdrawing groups containing aromatic homoallylic alcohols. Similarly, She et al. recently have reported a new approach for the synthesis of 2,4,6-trisubstituted tetrahydropyrans via a Prins cyclization [31]. With this method, both symmetrical and unsymmetrical tetrahydropyrans can be synthesized.
p-Toluenesulfonic acid (PTSA) is reported as a versatile Brønsted acid catalyst in various organic transformations [32-34]. Previously, PTSA has been used as a catalyst in Prins cyclizations but the product yields were low even under extended reaction times [10].
In continuation of our interest in acid catalysis [35-39], we report herein an efficient, economical and highly diastereoselective one-pot three-component synthesis for both symmetrical and unsymmetrical 2,4,6-trisubstituted tetrahydropyrans using aromatic homoallyl alcohols, aromatic/aliphatic aldehydes and p-toluenesulfonic acid (both as reagent and catalyst) in the presence of molecular sieves (MS 4 Å) in dichloromethane at reflux. The molecular sieves (MS 4 Å) had a significant effect on improving product yields (72–96%) under short reaction times (20–90 min). Further, cleavage of the 4-tosyl group under mild conditions afforded 4-hydroxytetrahydropyran in excellent yields (95–96%).
Results and Discussion
The starting materials, aromatic homoallylic alcohols, were readily prepared by treatment of aromatic aldehydes with allylic Grignard reagents under a nitrogen atmosphere at −78 °C for 2 h [40]. Their structures were assigned by 1H, 13C NMR, IR, and GC–MS data and compared with reported values in the literature.
Initially, we carried out the reaction with homoallylic alcohols, aromatic aldehydes and PTSA at room temperature to afford 2,4,6-trisubstituted tetrahydropyrans. After 22 h stirring more side-products than the desired product were observed. This might be due to the formation of an oxo-carbenium intermediate, which further reacted in a [3,3]sigmatropic rearrangement to give another oxo-carbenium ion (Scheme 1).
![[1860-5397-8-19-i1]](/bjoc/content/inline/1860-5397-8-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Plausible side products mechanism.
Scheme 1: Plausible side products mechanism.
To optimize the reaction yield, we varied the reaction conditions such as enhancing the catalytic loading (1.2 equiv to 1.4 equiv), varying solvents and temperature, rearranging the order of reagent addition and adding MS 4 Å as drying agent (Table 1).
Table 1: Optimization of reaction yields.
| Entrya | Brønsted acid | Solvent |
Temp
(°C) |
Time |
Yield
(%)b |
|---|---|---|---|---|---|
| 1 | PTSA (1.2 equiv) | Toluene | rt | 20 h | 25 |
| 2 | PTSA (1.2 equiv) | EtOAc | rt | 20 h | 22 |
| 3 | PTSA (1.2 equiv) | THF | rt | 28 h | 26 |
| 4 | PTSA (1.2 equiv) | DCE | rt | 18 h | 25 |
| 5 | PTSA (1.2 equiv) | DCM | rt | 22 h | 30 |
| 6 | PTSA (1.4 equiv) | DCM | rt | 20 h | 35 |
| 7 | PTSA (1.4 equiv) | THF | 66 | 22 h | 28 |
| 8 | PTSA (1.4 equiv) | DCM | rt | 40 min | 45 |
| 9 | PTSA (1.4 equiv) | DCM | 40 | 20 min | 75 |
| 10c | PTSA (1.4 equiv) | DCM | 40 | 20 min | 94 |
aAll reactions were carried out with homoallylic alcohol (1 equiv), aldehyde (1 equiv) and PTSA (1.2–1.4 equiv). bIsolated yield. cMS 4 Å (30 mg/equiv) was used.
Among different solvents (toluene, THF, DCM, DCE and diethyl ether) that were used, DCM was found as the solvent of choice at reflux temperature (Table 1, entries 5, 6, 8–10). Similarly, various acids like trifluoroacetic acid (TFA), benzoic acid, acetic acid, benzylphosphonic acid, and O,O’-diethyl dithiophosphate were tried as catalysts but all of them failed to give the desired product. We expected that the order of reagent addition would have significant influence on the yield. For example, the addition of PTSA (1.4 equiv) to a stirred solution of homoallylic alcohol and benzaldehyde in dichloromethane (DCM) at room temperature gave the product in 35% yield (Table 1, entry 6). Compared to that, the addition of homoallylic alcohol to a stirred mixture of PTSA (1.4 equiv) and aromatic aldehyde in DCM improved the yield (45%) at the same temperature (Table 1, entry 8). Following the later addition order at reflux (40 °C) in DCM (Table 1, entry 9), the yield was enhanced up to 75% within 20 min. Furthermore, the addition of molecular sieves (MS 4 Å) to the above mentioned reaction mixture within 20 min resulted in an essential improvement of the yield (94%, Table 1, entry 10). In the presence of MS 4 Å, the yield was unexpectedly enhanced from 75% to 94% under the same reaction conditions (Table 1, entry 9 vs entry 10). The significant improvements in product yields, reaction time and/or diastereoselectivity might be due to the prevention of the [3,3]sigmatropic rearrangement along with the dehydrating activity of MS 4 Å. In addition, we studied the stoichiometric ratio of MS 4 Å with respect to the substrate and found that 30 mg/mmol of MS 4 Å are necessary to receive optimal yields.
Under optimal reaction conditions we reacted a wide selection of aromatic homoallylic alcohols and aldehydes. The experimental results are summarized in Table 2. In all cases, the corresponding tetrahydropyrans were obtained in high diastereoselectivity and excellent yields without side products (Table 2). A high degree of diastereoselectivity was determined from the 1H NMR spectra without purification (crude product). We observed that substituents on the aromatic rings influenced the reaction rates and yields. For example, strong electron-donating groups such as methoxy or trimethoxy at homoallylic alcohols afforded the corresponding tetrahydropyrans in lower yields (72–75%) but in a faster rate (Table 2, entries 7 and 9). Similarly, the presence of electron-withdrawing substituents – such as chlorine or bromine atoms at homoallylic alcohols – gave the corresponding tetrahydropyrans in high yields (85–96%) but the reaction times were longer (Table 2, entries 2–4, 6, 8, 10). However, substituents on aldehydes have no significant effect on the reaction time or yields (Table 2, entries 2, 4, 6, 12–15). We further extended our method to aliphatic aldehydes (e.g., acetaldehyde) and hetero aromatic aldehydes (e.g., pyrrole aldehyde, furfural). Under optimal reaction conditions they reacted smoothly with homoallylic alcohols to afford the corresponding tetrahydropyran derivatives which show almost the same distereoselectivity and product yields (83–89%, Table 2, entries 16–18).
Table 2: Preparation of tetrahydropyrans from aromatic homoallylic alcohols, aldehydes and PTSA·H2O.
| Entry | Homoallylic alcohol | Aldehyde | Product | Reaction time (min) | Yield (%) |
|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-19-i4.svg?max-width=637&scale=1.0)
1a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-19-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-19-i6.svg?max-width=637&scale=1.0)
1b |
90 | 94 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-19-i7.svg?max-width=637&scale=1.0)
4a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-19-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-19-i9.svg?max-width=637&scale=1.0)
2b |
20 | 82 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-19-i10.svg?max-width=637&scale=1.0)
2a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-19-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-19-i12.svg?max-width=637&scale=1.0)
3b |
90 | 92 |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-19-i13.svg?max-width=637&scale=1.0)
4a |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-19-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-19-i15.svg?max-width=637&scale=1.0)
4b |
25 | 88 |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-19-i16.svg?max-width=637&scale=1.0)
1a |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-19-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-19-i18.svg?max-width=637&scale=1.0)
5b |
90 | 90 |
| 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-19-i19.svg?max-width=637&scale=1.0)
4a |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-19-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-19-i21.svg?max-width=637&scale=1.0)
6b |
35 | 85 |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-19-i22.svg?max-width=637&scale=1.0)
5a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-19-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-19-i24.svg?max-width=637&scale=1.0)
7b |
23 | 72 |
| 8 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-19-i25.svg?max-width=637&scale=1.0)
4a |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-19-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-19-i27.svg?max-width=637&scale=1.0)
8b |
90 | 95 |
| 9 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-19-i28.svg?max-width=637&scale=1.0)
3a |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-19-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-19-i30.svg?max-width=637&scale=1.0)
9b |
25 | 75 |
| 10 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-8-19-i31.svg?max-width=637&scale=1.0)
2a |
![[Graphic 29]](/bjoc/content/inline/1860-5397-8-19-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-8-19-i33.svg?max-width=637&scale=1.0)
10b |
90 | 95 |
| 11 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-8-19-i34.svg?max-width=637&scale=1.0)
2a |
![[Graphic 32]](/bjoc/content/inline/1860-5397-8-19-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-8-19-i36.svg?max-width=637&scale=1.0)
11b |
90 | 96 |
| 12 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-8-19-i37.svg?max-width=637&scale=1.0)
1a |
![[Graphic 35]](/bjoc/content/inline/1860-5397-8-19-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 36]](/bjoc/content/inline/1860-5397-8-19-i39.svg?max-width=637&scale=1.0)
12b |
20 | 83 |
| 13 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-8-19-i40.svg?max-width=637&scale=1.0)
2a |
![[Graphic 38]](/bjoc/content/inline/1860-5397-8-19-i41.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-8-19-i42.svg?max-width=637&scale=1.0)
13b |
30 | 88 |
| 14 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-8-19-i43.svg?max-width=637&scale=1.0)
1a |
![[Graphic 41]](/bjoc/content/inline/1860-5397-8-19-i44.svg?max-width=637&scale=1.0)
|
![[Graphic 42]](/bjoc/content/inline/1860-5397-8-19-i45.svg?max-width=637&scale=1.0)
14b |
90 | 95 |
| 15 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-8-19-i46.svg?max-width=637&scale=1.0)
2a |
![[Graphic 44]](/bjoc/content/inline/1860-5397-8-19-i47.svg?max-width=637&scale=1.0)
|
![[Graphic 45]](/bjoc/content/inline/1860-5397-8-19-i48.svg?max-width=637&scale=1.0)
15b |
60 | 93 |
| 16 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-8-19-i49.svg?max-width=637&scale=1.0)
1a |
![[Graphic 47]](/bjoc/content/inline/1860-5397-8-19-i50.svg?max-width=637&scale=1.0)
|
![[Graphic 48]](/bjoc/content/inline/1860-5397-8-19-i51.svg?max-width=637&scale=1.0)
16b |
50 | 83 |
| 17 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-8-19-i52.svg?max-width=637&scale=1.0)
1a |
![[Graphic 50]](/bjoc/content/inline/1860-5397-8-19-i53.svg?max-width=637&scale=1.0)
|
![[Graphic 51]](/bjoc/content/inline/1860-5397-8-19-i54.svg?max-width=637&scale=1.0)
17b |
50 | 86 |
| 18 |
![[Graphic 52]](/bjoc/content/inline/1860-5397-8-19-i55.svg?max-width=637&scale=1.0)
1a |
![[Graphic 53]](/bjoc/content/inline/1860-5397-8-19-i56.svg?max-width=637&scale=1.0)
|
![[Graphic 54]](/bjoc/content/inline/1860-5397-8-19-i57.svg?max-width=637&scale=1.0)
18b |
75 | 89 |
From a mechanistical point of view, these reactions are similar to the Prins cyclization [41]. First, the aldehyde got activated by PTSA protonation followed by a nucleophilic attack of the homoallylic alcohol and proton transfer to the hydroxy group. Then, a nucleophilic attack of PTSA resulted in α-tosyloxyether formation after losing a water molecule. In the α-tosyloxyether, the delocalization of lone-pair electrons on the oxygen atom led to the removal of the tosylate group and oxo-carbenium ion intermediate formation. Then, an intramolecular nucleophilic attack of the double bond in the oxo-carbenium ion led to cyclization and charge transfer complex formation with the tosylate group to afford the 2,6-disubstituted-4-tosyloxytetrahydropyran (Scheme 2).
![[1860-5397-8-19-i2]](/bjoc/content/inline/1860-5397-8-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible reaction mechanism via Prins cyclization.
Scheme 2: Plausible reaction mechanism via Prins cyclization.
All structures of the 2,6-disubstituted-4-tosyloxytetrahydropyrans were established by 1H, 13C NMR, IR, and GC–MS spectral data and elemental analysis. NOE studies of compound 3b (Table 2, entry 3) confirmed that all three substituents occupy equatorial positions on the tetrahydropyran ring. The NOE cross peaks H1/H3, H5/H3 reveal that H1, H5 and H3 are on the same side of the ring and take a diaxial orientation (Figure 2). 1H NMR experiments were performed at 500 MHz in CDCl3. The proposed structure of 3b (Table 1, entry 3) is further supported by the coupling constants JH1/H2b = 1.5 Hz, JH1/H2a = 11.5 Hz, JH2b/H3 = 4.5 Hz and JH2a/H3 = 11.5 Hz. Thus, H2a and H4a are antiperiplanar to H1, H3 and H5. All these findings confirm that the pyran ring takes a chair conformation, where the substituents at C1, C3 and C5 are present in equatorial position.
![[1860-5397-8-19-2]](/bjoc/content/figures/1860-5397-8-19-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Schematic NOE diagram of compound 3b.
Figure 2: Schematic NOE diagram of compound 3b.
The tosyl group at C4 got easily deprotected at room temperature with Mg–MeOH (Scheme 3) [42] to afford 2,6-disubstited-4-hydroxytetrahydropyrans with retention of the stereochemistry in quantitative yield (Table 3). The structures of 2,6-disubstituted-4-hydroxytetrahydropyrans were established by 1H, 13C NMR, IR, and GC–MS and elemental analysis and were compared with reported data [43].
![[1860-5397-8-19-i3]](/bjoc/content/inline/1860-5397-8-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deprotection of the hydroxy group.
Scheme 3: Deprotection of the hydroxy group.
Conclusion
In conclusion, we have reported a simple and efficient one-pot three-component synthesis of highly diastereoselective and functionalized 2,6-disubstituted-4-tosyloxytetrahydropyrans via Prins cyclization. An aromatic homoallylic alcohol, an aromatic/aliphatic aldehyde, and p-toluenesulfonic acid (catalyst and reagent) are reacted in the presence of MS 4 Å in dichloromethane at reflux to afford 2,6-disubstituted-4-tosyloxytetrahydropyrans in excellent yields (72–96%) within short reaction times (20–90 min). The MS 4 Å promoted synthesis proved to be versatile enough to provide an array of symmetrical and unsymmetrical tetrahydropyran derivatives in an economical manner. Moreover, it was observed that MS 4 Å might have a vital part in controlling the reversibility of the [3,3]sigmatropic rearrangement. Furthermore, cleavage of the 4-tosyl group under mild conditions afforded 4-hydroxytetrahydropyrans in high diastereoselectivity and excellent yields (95–96%).
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of synthesized compounds, 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Searle, P. A.; Molinski, T. F.; Brzezinski, L. J.; Leahy, J. W. J. Am. Chem. Soc. 1996, 118, 9422–9423. doi:10.1021/ja962092r
Return to citation in text: [1] -
Prasad, K. R.; Anbarasan, P. Tetrahedron 2007, 63, 1089–1092. doi:10.1016/j.tet.2006.11.062
Return to citation in text: [1] -
Hasegawa, M.; Miura, T.; Kuzuya, K.; Inoue, A.; Ki, S. W.; Horinouchi, S.; Yoshida, T.; Kunoh, T.; Koseki, K.; Mino, K.; Sasaki, R.; Yoshida, M.; Mizukami, T. ACS Chem. Biol. 2011, 6, 229–233. doi:10.1021/cb100248e
Return to citation in text: [1] -
Szallasi, Z.; Du, L.; Levine, R.; Lewin, N. E.; Nguyen, P. N.; Williams, M. D.; Pettit, G. R.; Blumberg, P. M. Cancer Res. 1996, 56, 2105–2111.
Return to citation in text: [1] -
Schwarz, M.; Graminski, G. F.; Waters, R. M. J. Org. Chem. 1986, 51, 260–263. doi:10.1021/jo00352a031
Return to citation in text: [1] -
Sakata, H.; Yasukawa, H. Heat-developable dry silver photographic films forming high-contrast images and development thereon. Jpn. Kokai Tokkyo Koho JP 2003121964 A, 2003; pp 42 ff.
Return to citation in text: [1] -
McGarvey, G. J.; Stepanian, M. W.; Bressette, A. R.; Sabat, M. Org. Lett. 2000, 2, 3453–3456. doi:10.1021/ol006468o
Return to citation in text: [1] -
Zhang, W.-C.; Li, C.-J. Tetrahedron 2000, 56, 2403–2411. doi:10.1016/S0040-4020(00)00152-6
Return to citation in text: [1] -
Hu, Y.; Skalitzky, D. J.; Rychnovsky, S. D. Tetrahedron Lett. 1996, 37, 8679–8682. doi:10.1016/S0040-4039(96)02008-4
Return to citation in text: [1] -
Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054
Return to citation in text: [1] [2] -
Sabitha, G.; Bhikshapathi, M.; Nayak, S.; Yadav, J. S. Synth. Commun. 2010, 41, 8–15. doi:10.1080/00397910903422534
Return to citation in text: [1] -
Hart, D. J.; Bennett, C. E. Org. Lett. 2003, 5, 1499–1502. doi:10.1021/ol0342756
Return to citation in text: [1] -
Jervis, P. J.; Kariuki, B. M.; Cox, L. R. Org. Lett. 2006, 8, 4649–4652. doi:10.1021/ol061957v
Return to citation in text: [1] -
Zhang, W.-C.; Viswanathan, G. S.; Li, C.-J. Chem. Commun. 1999, 291–292. doi:10.1039/a808960d
Return to citation in text: [1] -
Tadpetch, K.; Rychnovsky, S. D. Org. Lett. 2008, 10, 4839–4842. doi:10.1021/ol8019204
Return to citation in text: [1] -
Liu, F.; Loh, T.-P. Org. Lett. 2007, 9, 2063–2066. doi:10.1021/ol070506n
Return to citation in text: [1] -
Coppi, L.; Ricci, A.; Taddei, M. J. Org. Chem. 1988, 53, 911–913. doi:10.1021/jo00239a053
Return to citation in text: [1] -
Dobbs, A. P.; Guesne, S. J. J.; Martinovic, S.; Coles, S. J.; Hursthouse, M. B. ChemInform 2004, 35. doi:10.1002/chin.200405043
Return to citation in text: [1] -
Viswanathan, G. S.; Yang, J.; Li, C.-J. Org. Lett. 1999, 1, 993–995. doi:10.1021/ol990789m
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Anusha, B.; Reddy, U. V. S.; Reddy, V. V. B. Tetrahedron Lett. 2010, 51, 2872–2874. doi:10.1016/j.tetlet.2010.03.082
Return to citation in text: [1] -
Yang, J.; Viswanathan, G. S.; Li, C.-J. Tetrahedron Lett. 1999, 40, 1627–1630. doi:10.1016/S0040-4039(99)00027-1
Return to citation in text: [1] -
Patterson, B.; Marumoto, S.; Rychnovsky, S. D. Org. Lett. 2003, 5, 3163–3166. doi:10.1021/ol035303n
Return to citation in text: [1] -
Miranda, P. O.; Díaz, D. D.; Padrón, J. I.; Bermejo, J.; Martín, V. S. Org. Lett. 2003, 5, 1979–1982. doi:10.1021/ol034568z
Return to citation in text: [1] -
Launay, G. G.; Slawin, A. M. Z.; O'Hagan, D. Beilstein J. Org. Chem. 2010, 6, No. 41. doi:10.3762/bjoc.6.41
Return to citation in text: [1] -
Kumar, H. M. S.; Qazi, N. A.; Shafi, S.; Kumar, V. N.; Krishna, A. D.; Yadav, J. S. Tetrahedron Lett. 2005, 46, 7205–7207. doi:10.1016/j.tetlet.2005.08.070
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Venugopal, C.; Srinivas, R.; Ramalingam, T. Synth. Commun. 2002, 32, 1803–1808. doi:10.1081/SCC-120004059
Return to citation in text: [1] -
Roy, U. K.; Jana, P. K.; Roy, S. Tetrahedron Lett. 2007, 48, 1183–1186. doi:10.1016/j.tetlet.2006.12.080
Return to citation in text: [1] -
Chavre, S. N.; Choo, H.; Lee, J. K.; Pae, A. N.; Kim, Y.; Cho, Y. S. J. Org. Chem. 2008, 73, 7467–7471. doi:10.1021/jo800967p
Return to citation in text: [1] -
Jasti, R.; Rychnovsky, S. D. Org. Lett. 2006, 8, 2175–2178. doi:10.1021/ol0606738
Return to citation in text: [1] -
Crosby, S. R.; Harding, J. R.; King, C. D.; Parker, G. D.; Willis, C. L. Org. Lett. 2002, 4, 577–580. doi:10.1021/ol0102850
Return to citation in text: [1] -
Yu, B.; Jiang, T.; Li, J.; Su, Y.; Pan, X.; She, X. Org. Lett. 2009, 11, 3442–3445. doi:10.1021/ol901291w
Return to citation in text: [1] -
Filippi, J.-J.; Duñach, E.; Fernandez, X.; Meierhenrich, U. J. Tetrahedron 2008, 64, 9999–10003. doi:10.1016/j.tet.2008.07.088
Return to citation in text: [1] -
Karodia, N.; Liu, X.; Ludley, P.; Pletsas, D.; Stevenson, G. Tetrahedron 2006, 62, 11039–11043. doi:10.1016/j.tet.2006.09.052
Return to citation in text: [1] -
Yadav, J. S.; Chakravarthy, P. P.; Borkar, P.; Reddy, B. V. S.; Sarma, A. V. S. Tetrahedron Lett. 2009, 50, 5998–6000. doi:10.1016/j.tetlet.2009.08.023
Return to citation in text: [1] -
Ahmed, N.; Babu, B. V.; Kumar, H. Synthesis 2011, 2471–2477. doi:10.1055/s-0030-1260091
Return to citation in text: [1] -
Ahmed, N.; Babu, B. V.; Kumar, H. Synth. Commun. 2011, in press.
Return to citation in text: [1] -
Ahmed, N.; van Lier, J. E. Tetrahedron Lett. 2007, 48, 13–15. doi:10.1016/j.tetlet.2006.11.020
Return to citation in text: [1] -
Ahmed, N.; van Lier, J. E. Tetrahedron Lett. 2006, 47, 2725–2729. doi:10.1016/j.tetlet.2006.02.086
Return to citation in text: [1] -
Ahmed, N.; Ali, H.; van Lier, J. E. J. Porphyrins Phthalocyanines 2006, 10, 1172–1178. doi:10.1142/S1088424606000533
Return to citation in text: [1] -
Lee, K.; Kim, H.; Hong, J. Org. Lett. 2009, 11, 5202–5205. doi:10.1021/ol902125d
Return to citation in text: [1] -
Marumoto, S.; Jaber, J. J.; Vitale, J. P.; Rychnovsky, S. D. Org. Lett. 2002, 4, 3919–3922. doi:10.1021/ol026751i
Return to citation in text: [1] -
Sridhar, M.; Kumar, B. A.; Narender, G. Tetrahedron Lett. 1998, 39, 2847–2850. doi:10.1016/S0040-4039(98)00314-1
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Reddy, M. S.; Niranjan, N. J. Mol. Catal. A: Chem. 2004, 210, 99–103. doi:10.1016/j.molcata.2003.08.024
Return to citation in text: [1]
| 10. | Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054 |
| 35. | Ahmed, N.; Babu, B. V.; Kumar, H. Synthesis 2011, 2471–2477. doi:10.1055/s-0030-1260091 |
| 36. | Ahmed, N.; Babu, B. V.; Kumar, H. Synth. Commun. 2011, in press. |
| 37. | Ahmed, N.; van Lier, J. E. Tetrahedron Lett. 2007, 48, 13–15. doi:10.1016/j.tetlet.2006.11.020 |
| 38. | Ahmed, N.; van Lier, J. E. Tetrahedron Lett. 2006, 47, 2725–2729. doi:10.1016/j.tetlet.2006.02.086 |
| 39. | Ahmed, N.; Ali, H.; van Lier, J. E. J. Porphyrins Phthalocyanines 2006, 10, 1172–1178. doi:10.1142/S1088424606000533 |
| 40. | Lee, K.; Kim, H.; Hong, J. Org. Lett. 2009, 11, 5202–5205. doi:10.1021/ol902125d |
| 1. | Searle, P. A.; Molinski, T. F.; Brzezinski, L. J.; Leahy, J. W. J. Am. Chem. Soc. 1996, 118, 9422–9423. doi:10.1021/ja962092r |
| 5. | Schwarz, M.; Graminski, G. F.; Waters, R. M. J. Org. Chem. 1986, 51, 260–263. doi:10.1021/jo00352a031 |
| 18. | Dobbs, A. P.; Guesne, S. J. J.; Martinovic, S.; Coles, S. J.; Hursthouse, M. B. ChemInform 2004, 35. doi:10.1002/chin.200405043 |
| 19. | Viswanathan, G. S.; Yang, J.; Li, C.-J. Org. Lett. 1999, 1, 993–995. doi:10.1021/ol990789m |
| 4. | Szallasi, Z.; Du, L.; Levine, R.; Lewin, N. E.; Nguyen, P. N.; Williams, M. D.; Pettit, G. R.; Blumberg, P. M. Cancer Res. 1996, 56, 2105–2111. |
| 20. | Yadav, J. S.; Reddy, B. V. S.; Anusha, B.; Reddy, U. V. S.; Reddy, V. V. B. Tetrahedron Lett. 2010, 51, 2872–2874. doi:10.1016/j.tetlet.2010.03.082 |
| 3. | Hasegawa, M.; Miura, T.; Kuzuya, K.; Inoue, A.; Ki, S. W.; Horinouchi, S.; Yoshida, T.; Kunoh, T.; Koseki, K.; Mino, K.; Sasaki, R.; Yoshida, M.; Mizukami, T. ACS Chem. Biol. 2011, 6, 229–233. doi:10.1021/cb100248e |
| 2. | Prasad, K. R.; Anbarasan, P. Tetrahedron 2007, 63, 1089–1092. doi:10.1016/j.tet.2006.11.062 |
| 17. | Coppi, L.; Ricci, A.; Taddei, M. J. Org. Chem. 1988, 53, 911–913. doi:10.1021/jo00239a053 |
| 12. | Hart, D. J.; Bennett, C. E. Org. Lett. 2003, 5, 1499–1502. doi:10.1021/ol0342756 |
| 14. | Zhang, W.-C.; Viswanathan, G. S.; Li, C.-J. Chem. Commun. 1999, 291–292. doi:10.1039/a808960d |
| 43. | Yadav, J. S.; Reddy, B. V. S.; Reddy, M. S.; Niranjan, N. J. Mol. Catal. A: Chem. 2004, 210, 99–103. doi:10.1016/j.molcata.2003.08.024 |
| 8. | Zhang, W.-C.; Li, C.-J. Tetrahedron 2000, 56, 2403–2411. doi:10.1016/S0040-4020(00)00152-6 |
| 9. | Hu, Y.; Skalitzky, D. J.; Rychnovsky, S. D. Tetrahedron Lett. 1996, 37, 8679–8682. doi:10.1016/S0040-4039(96)02008-4 |
| 10. | Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054 |
| 11. | Sabitha, G.; Bhikshapathi, M.; Nayak, S.; Yadav, J. S. Synth. Commun. 2010, 41, 8–15. doi:10.1080/00397910903422534 |
| 15. | Tadpetch, K.; Rychnovsky, S. D. Org. Lett. 2008, 10, 4839–4842. doi:10.1021/ol8019204 |
| 7. | McGarvey, G. J.; Stepanian, M. W.; Bressette, A. R.; Sabat, M. Org. Lett. 2000, 2, 3453–3456. doi:10.1021/ol006468o |
| 41. | Marumoto, S.; Jaber, J. J.; Vitale, J. P.; Rychnovsky, S. D. Org. Lett. 2002, 4, 3919–3922. doi:10.1021/ol026751i |
| 6. | Sakata, H.; Yasukawa, H. Heat-developable dry silver photographic films forming high-contrast images and development thereon. Jpn. Kokai Tokkyo Koho JP 2003121964 A, 2003; pp 42 ff. |
| 13. | Jervis, P. J.; Kariuki, B. M.; Cox, L. R. Org. Lett. 2006, 8, 4649–4652. doi:10.1021/ol061957v |
| 42. | Sridhar, M.; Kumar, B. A.; Narender, G. Tetrahedron Lett. 1998, 39, 2847–2850. doi:10.1016/S0040-4039(98)00314-1 |
| 23. | Miranda, P. O.; Díaz, D. D.; Padrón, J. I.; Bermejo, J.; Martín, V. S. Org. Lett. 2003, 5, 1979–1982. doi:10.1021/ol034568z |
| 21. | Yang, J.; Viswanathan, G. S.; Li, C.-J. Tetrahedron Lett. 1999, 40, 1627–1630. doi:10.1016/S0040-4039(99)00027-1 |
| 22. | Patterson, B.; Marumoto, S.; Rychnovsky, S. D. Org. Lett. 2003, 5, 3163–3166. doi:10.1021/ol035303n |
| 31. | Yu, B.; Jiang, T.; Li, J.; Su, Y.; Pan, X.; She, X. Org. Lett. 2009, 11, 3442–3445. doi:10.1021/ol901291w |
| 32. | Filippi, J.-J.; Duñach, E.; Fernandez, X.; Meierhenrich, U. J. Tetrahedron 2008, 64, 9999–10003. doi:10.1016/j.tet.2008.07.088 |
| 33. | Karodia, N.; Liu, X.; Ludley, P.; Pletsas, D.; Stevenson, G. Tetrahedron 2006, 62, 11039–11043. doi:10.1016/j.tet.2006.09.052 |
| 34. | Yadav, J. S.; Chakravarthy, P. P.; Borkar, P.; Reddy, B. V. S.; Sarma, A. V. S. Tetrahedron Lett. 2009, 50, 5998–6000. doi:10.1016/j.tetlet.2009.08.023 |
| 28. | Chavre, S. N.; Choo, H.; Lee, J. K.; Pae, A. N.; Kim, Y.; Cho, Y. S. J. Org. Chem. 2008, 73, 7467–7471. doi:10.1021/jo800967p |
| 29. | Jasti, R.; Rychnovsky, S. D. Org. Lett. 2006, 8, 2175–2178. doi:10.1021/ol0606738 |
| 30. | Crosby, S. R.; Harding, J. R.; King, C. D.; Parker, G. D.; Willis, C. L. Org. Lett. 2002, 4, 577–580. doi:10.1021/ol0102850 |
| 26. | Yadav, J. S.; Reddy, B. V. S.; Venugopal, C.; Srinivas, R.; Ramalingam, T. Synth. Commun. 2002, 32, 1803–1808. doi:10.1081/SCC-120004059 |
| 27. | Roy, U. K.; Jana, P. K.; Roy, S. Tetrahedron Lett. 2007, 48, 1183–1186. doi:10.1016/j.tetlet.2006.12.080 |
| 24. | Launay, G. G.; Slawin, A. M. Z.; O'Hagan, D. Beilstein J. Org. Chem. 2010, 6, No. 41. doi:10.3762/bjoc.6.41 |
| 25. | Kumar, H. M. S.; Qazi, N. A.; Shafi, S.; Kumar, V. N.; Krishna, A. D.; Yadav, J. S. Tetrahedron Lett. 2005, 46, 7205–7207. doi:10.1016/j.tetlet.2005.08.070 |
© 2012 Ahmed and Konduru; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)