Abstract
Triazole–Au (TA–Au) catalysts were employed in several transformations involving propargyl ester rearrangement. Good chemoselectivity was observed, which allowed the effective activation of the alkyne without affecting the reactivity of the allene ester intermediates. These results led to the investigation of the preparation of allene ester intermediates with TA–Au catalysts under anhydrous conditions. As expected, the desired 3,3-rearrangement products were obtained in excellent yields (generally >90% yields with 1% loading). Besides the typical ester migrating groups, carbonates and carbamates were also found to be suitable for this transformation, which provided a highly efficient, practical method for the preparation of substituted allenes.

Graphical Abstract
Introduction
The past decade has seen rapid growth in the use of homogeneous gold catalysis for conducting powerful organic transformations [1-9]. Like many other transition metal complexes, the reactivity of gold catalysts greatly depends on the nature of the ligands coordinating with the metal cations [10-15]. Of the two typical oxidation states, Au(I) and Au(III), more studies have been focused on the former cation due to the easier preparation of the catalyst and better pre-catalyst stability. It is currently accepted by the research community that Au(I) complexes adopt one of two coordination sites with 180° linear geometry (Scheme 1) (although some exceptions exist). The actual catalysts involved in alkyne and alkene activation are of the type [L–Au]+, with the open coordination site on the opposite side of the ligand (L) for substrate binding [5,6]. The recent success in obtaining the complexes of the alkyne-coordinated [L–Au]+, reported by Toste and coworkers, greatly supported this mechanistic model [16]. Generally, the PR3 compounds can be applied as the ligand in Au(I) catalysis. The recent development of N-heterocyclic carbene (NHC) derivatives has significantly expanded the choice of ligands by improving the catalyst stability through metal-ligand backbonding [17-20]. To access the active catalyst [L–Au]+, stable precursors L–Au–X or [L–Au–X]+·A− were typically used. While the ligands (L) are certainly considered critical in gold catalysis, more and more attention have been paid to the evaluation of whether the choice of counter ligand “X” can be used to adjust the overall catalyst reactivity.
![[1860-5397-7-115-i1]](/bjoc/content/inline/1860-5397-7-115-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The counter ligands, an important factor in Au(I) catalysis.
Scheme 1: The counter ligands, an important factor in Au(I) catalysis.
The propargyl ester rearrangement was considered as one of the most important reaction modes in the Au(I) promoted transformation [21-31]. Recent experimental and computational mechanistic studies revealed the 3,3-rearrangement to form the allene ester intermediate [32,33] as the key step in this transformation (Scheme 2a) [34]. Both experimental and theoretical investigations confirmed the reversibility between allene and propargyl ester due to effective activation of both functional groups by the Au(I) catalysts. As a result, it was extremely challenging to obtain the allene intermediates with good yields. Many strategies have been developed to make the Au(I)-activated allene esters react with other proper substrates, forming interesting new products in a cascade fashion. The indene synthesis (Scheme 2b), reported by Nolan and coworkers, is one good example highlighting the importance of the cascade process [35].
![[1860-5397-7-115-i2]](/bjoc/content/inline/1860-5397-7-115-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The challenge of the synthesis of allenes through gold activated alkynes.
Scheme 2: The challenge of the synthesis of allenes through gold activated alkynes.
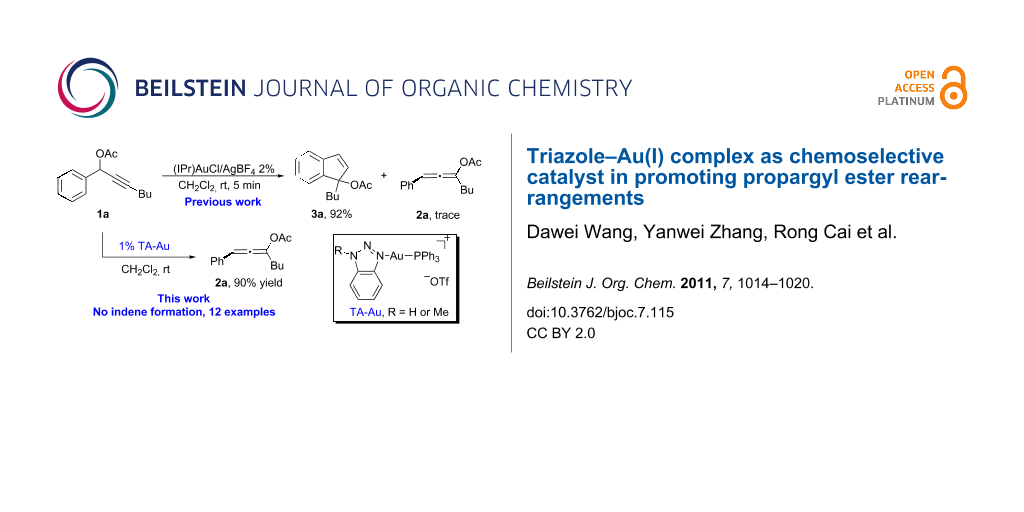
As shown in Scheme 2b, with the [IPr–Au]+ catalyst, only trace amount of the allene 2a was obtained. The major product derived from the Friedel–Crafts addition of the aromatic ring to the gold activated allene. Therefore, selective activation of the alkyne over the allene was considered as a significant challenge in gold catalysis.
Results and Discussions
Recently, our group reported the synthesis and characterization of the 1,2,3-triazole [36-40] coordinated gold(I) complexes. As revealed by the X-ray crystal structures (Scheme 3), both neutral and anionic triazoles can coordinate with Au(I) cation, forming stable TA–Au complexes [41].
![[1860-5397-7-115-i3]](/bjoc/content/inline/1860-5397-7-115-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: X-ray crystal structures of the two different types of 1,2,3-triazole–Au complexes.
Scheme 3: X-ray crystal structures of the two different types of 1,2,3-triazole–Au complexes.
The preparation of these complexes was very straightforward. Simply treating the NH-triazoles with PPh3AuCl in methanol under basic conditions (K2CO3, 1 equiv) at room temperature gave the neutral TA–Au-1 in >90% yield. The “cationic” complex TA–Au-2 was prepared either from the addition of HOTf to TA–Au-1 or by the reaction between PPh3Au–OTf (prepared from PPh3–Au–Cl and AgOTf) and benzotriazole. Both complexes were stable and could be further purified by recrystallization to ensure no extra Ag+ or acid in the catalysts. The crystal structures revealed nearly identical Au–P bond length for both the anionic and neutral triazole coordinated Au(I) complexes. The longer Au–N bond in TA–Au-2 implies that the neutral triazole dissociates more easily to release the coordination site for substrate activation. This new class of compounds offers improved thermal stability and substrate stability in the gold(I) promoted hydroamination and Hashmi phenol synthesis [42], which makes them interesting novel catalysts in the field of gold catalysis. One particular new development of the TA–Au catalysis that attracted our attention was the synthesis of α-iodoenone from propargyl esters (Scheme 4a) [43].
![[1860-5397-7-115-i4]](/bjoc/content/inline/1860-5397-7-115-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of α-iodoenone compounds from propargyl esters.
Scheme 4: Synthesis of α-iodoenone compounds from propargyl esters.
As indicated in Scheme 4a, the typical [L–Au]+ catalyst promoted the sequential rearrangement and iodination, giving the thermally, dynamically stable (Z)-isomer [44-46]. The cationic TA–Au catalyst, on the other hand, produced the kinetically favored (E)-isomer. Notably, treating the allene ester 2a with NIS gave the (E)-isomer as the dominant product. These results imply that the allene iodination should favor the formation of the (E)-isomer (Scheme 4b). The typical [L–Au]+ catalyst not only promoted the propargyl ester 3,3-rearrangement, but also influenced the allene reactivity, probably through gold catalyzed allene activation. The fact that TA–Au gave the dominant (E)-isomers strongly suggests that these complexes may be applied as the chemoselective catalyst in alkyne activation over allene. The reactions of propargyl ester 1a with TA–Au catalysts were then investigated as shown in Figure 1.
![[1860-5397-7-115-1]](/bjoc/content/figures/1860-5397-7-115-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemoselective activation of alkyne over allene by the TA–Au catalysts.
Figure 1: Chemoselective activation of alkyne over allene by the TA–Au catalysts.
As expected, with the cationic catalyst TA–Au-2 or TA–Au-3, the allene ester 2a was formed in excellent yields (1% loading, 91% yield). It is important to note here, that indene 3a was not observed even after 48 h reaction time, thus indicating excellent chemoselectivity of the triazole coordinated gold complexes. Various propargyl esters were synthesized to test the reaction substrate scope. The results are summarized in Table 1.
Table 1: The reaction substrate scope.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-115-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | Yield | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-115-i6.svg?max-width=637&scale=1.0)
|
1a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-115-i7.svg?max-width=637&scale=1.0)
|
2a | 91% |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-115-i8.svg?max-width=637&scale=1.0)
|
1b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-115-i9.svg?max-width=637&scale=1.0)
|
2b | 90% |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-115-i10.svg?max-width=637&scale=1.0)
|
1c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-115-i11.svg?max-width=637&scale=1.0)
|
2c | 87% |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-115-i12.svg?max-width=637&scale=1.0)
|
1d |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-115-i13.svg?max-width=637&scale=1.0)
|
2d | 89% |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-115-i14.svg?max-width=637&scale=1.0)
|
1e |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-115-i15.svg?max-width=637&scale=1.0)
|
2e | 89% |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-115-i16.svg?max-width=637&scale=1.0)
|
1f |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-115-i17.svg?max-width=637&scale=1.0)
|
2f | 85% |
| Substrates that did not form the desired allenesb | |||||
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-115-i18.svg?max-width=637&scale=1.0)
|
|||||
aGeneral reaction conditions: 1 (0.25 mmol) and TA–Au-2 (1.0 mol %) in dry DCM (2.5 mL), the reactions were monitored by TLC (2–10 h), rt. bTA–Au-1, TA–Au-2 and TA–Au-3 did not catalyze the reaction under the standard conditions.
As shown in Table 1, the transformation proceeded smoothly with substrates having both an aromatic group on the propargyl side and an aliphatic group on the alkyne side (entries 1–6). The desired allene products were formed in excellent yields, with 1% catalyst loading. The electronic density on the aromatic ring did not have a strong impact on the transformation: Both electron donating and electron-withdrawing groups were suitable for the reaction. Again, no indene by-products were observed in any of the tested cases, even with the electron-enriched p-OMe substituted alkyne 1d. These results highlighted the excellent chemoselective nature of the TA–Au catalyst.
The terminal alkyne 1i did not give any product when treated with TA–Au catalyst, even after an extended reaction time (24 h). This was probably caused by the preferred 1,2-rearrangement with the formation of a vinyl–Au intermediate. The aliphatic propargyl esters (1g, 1h) also did not give any desired allene products (enones from hydrations were produced after a long reaction time, 24–48 h; the crude NMR of the reaction mixtures did not show any allene products). This may be caused by the high reactivity of the corresponding aliphatic allenes under the reaction conditions (activated by TA–Au) and the overall better stability of the propargyl ester compared to the aliphatic substituted allenes (equilibrium favored the starting material). The reaction of cyclopropyl substituted propargyl ester 1j with the TA–Au catalyst gave a complex reaction mixture, which suggests possible ring opening and sequential cyclization as reported previously [47]. Overall, this study suggests that the propargyl ester rearrangement to form allene is highly substrate dependent. This could either be due to the similar reactivity of the alkyne and the allene (giving an equilibrium state favoring the alkyne over the allene) or it be could be due to a preferred alternative migration path (2,3-migration versus 3,3-migration). In any case, the TA–Au catalyst clearly displayed the interesting chemoselectivity, if the reaction could occur. To study the feasibility of this migration, we then investigated migrating groups other than esters. The results are summarized in Table 2.
Table 2: Different migrating groups.a
| Entry | Substrate | Product | Yield | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-115-i19.svg?max-width=637&scale=1.0)
|
4a |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-115-i20.svg?max-width=637&scale=1.0)
|
5a | 92% |
| 2 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-115-i21.svg?max-width=637&scale=1.0)
|
4b |
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-115-i22.svg?max-width=637&scale=1.0)
|
5b | 91% |
| 3 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-115-i23.svg?max-width=637&scale=1.0)
|
4c |
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-115-i24.svg?max-width=637&scale=1.0)
|
5c | 88% |
| 4 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-7-115-i25.svg?max-width=637&scale=1.0)
|
4d |
![[Graphic 22]](/bjoc/content/inline/1860-5397-7-115-i26.svg?max-width=637&scale=1.0)
|
5d | 92% |
| 5 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-7-115-i27.svg?max-width=637&scale=1.0)
|
4e |
![[Graphic 24]](/bjoc/content/inline/1860-5397-7-115-i28.svg?max-width=637&scale=1.0)
|
5e | 89% |
| 6 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-7-115-i29.svg?max-width=637&scale=1.0)
|
4f |
![[Graphic 26]](/bjoc/content/inline/1860-5397-7-115-i30.svg?max-width=637&scale=1.0)
|
5f | 85% |
aGeneral reaction condition: 4 (0.25 mmol) and TA–Au-2 (1.0 mol %) in dry DCM (2.5 mL), the reactions were monitored by TLC (2–10 h), rt.
As indicated in Table 2, carbonates (entries 1–5) and carbamate (entry 6) were also suitable for this transformation. Compared to the allene-acetates, the allene-carbonates and allene-carbamates were more stable in water. Notably, although the alkene was considered as a readily reactive functional group in gold catalysis, the substrate 4c was suitable for this transformation, giving the desired allene-ene 5c in excellent yield.
Conclusion
In this letter, we reported the application of triazole-coordinated gold(I) complexes as the effective catalysts for the promotion of the propargyl ester, carbonate and carbamate 3,3-rearrangement for the synthesis of the corresponding substituted allene derivatives. The chemoselective nature of the TA–Au catalysts was clearly demonstrated, which makes them an interesting class of new catalysts for promoting organic transformations. The application of the allene-carbonates and allene-carbamates as building blocks for development of new synthetic methodologies is currently underway in our group.
Supporting Information
| Supporting Information File 1: General methods, characterization data and NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 912.5 KB | Download |
References
-
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] [2] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335
Return to citation in text: [1] [2] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/B612008C
Return to citation in text: [1] -
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368
Return to citation in text: [1] -
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M.; Huck, J.; Frey, W.; Bats, J. W.; Hamzić, M. Angew. Chem., Int. Ed. 2009, 48, 5848–5852. doi:10.1002/anie.200900887
Return to citation in text: [1] -
Hashmi, A. S. K.; Loos, A.; Littmann, A.; Braun, I.; Knight, J.; Doherty, S.; Rominger, F. Adv. Synth. Catal. 2009, 351, 576–582. doi:10.1002/adsc.200800681
Return to citation in text: [1] -
Hashmi, A. S. K.; Salathé, R.; Frey, W. Chem.–Eur. J. 2006, 12, 6991–6996. doi:10.1002/chem.200600533
Return to citation in text: [1] -
Hashmi, A. S. K.; Blanco, M. C.; Kurpejović, E.; Frey, W.; Bats, J. W. Adv. Synth. Catal. 2006, 348, 709–713. doi:10.1002/adsc.200606012
Return to citation in text: [1] -
Hashmi, A. S. K.; Weyrauch, J. P.; Kurpejović, E.; Frost, T. M.; Miehlich, B.; Frey, W.; Bats, J. W. Chem.–Eur. J. 2006, 12, 5806–5814. doi:10.1002/chem.200501268
Return to citation in text: [1] -
Carrettin, S.; Blanco, M. C.; Corma, A.; Hashmi, A. S. K. Adv. Synth. Catal. 2006, 348, 1283–1288. doi:10.1002/adsc.200606099
Return to citation in text: [1] -
Shapiro, N. D.; Toste, F. D. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2779–2782. doi:10.1073/pnas.0710500105
Return to citation in text: [1] -
de Frémont, P.; Scott, N. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, 2411–2418. doi:10.1021/om050111c
Return to citation in text: [1] -
Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/cr900074m
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Acc. Chem. Res. 2008, 41, 1440–1449. doi:10.1021/ar800020y
Return to citation in text: [1] -
Díez-González, S.; Nolan, S. P. Acc. Chem. Res. 2008, 41, 349–358. doi:10.1021/ar7001655
Return to citation in text: [1] -
Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804–16805. doi:10.1021/ja056419c
Return to citation in text: [1] -
Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q
Return to citation in text: [1] -
Buzas, A.; Istrate, F.; Gagosz, F. Org. Lett. 2006, 8, 1957–1959. doi:10.1021/ol0606839
Return to citation in text: [1] -
Wang, S.; Zhang, L. Org. Lett. 2006, 8, 4585–4587. doi:10.1021/ol0618151
Return to citation in text: [1] -
Wang, S.; Zhang, L. J. Am. Chem. Soc. 2006, 128, 8414–8415. doi:10.1021/ja062777j
Return to citation in text: [1] -
Buzas, A.; Gagosz, F. J. Am. Chem. Soc. 2006, 128, 12614–12615. doi:10.1021/ja064223m
Return to citation in text: [1] -
Wang, S.; Zhang, L. J. Am. Chem. Soc. 2006, 128, 14274–14275. doi:10.1021/ja066220f
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773
Return to citation in text: [1] -
Marco-Contelles, J.; Soriano, E. Chem.–Eur. J. 2007, 13, 1350–1357. doi:10.1002/chem.200601522
Return to citation in text: [1] -
Barluenga, J.; Riesgo, L.; Vicente, R.; Lopéz, L. A.; Tomás, M. J. Am. Chem. Soc. 2007, 129, 7772–7773. doi:10.1021/ja072864r
Return to citation in text: [1] -
Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503–15512. doi:10.1021/ja071070+
Return to citation in text: [1] -
Saucy, R.; Marbet, R.; Lindlar, H.; Isler, O. Helv. Chim. Acta 1959, 42, 1945–1955. doi:10.1002/hlca.19590420624
Return to citation in text: [1] -
Cherbuliez, E.; Moll, H.; Baehler, B. R.; Rabinowitz, J. Helv. Chim. Acta 1967, 50, 1154–1158. doi:10.1002/hlca.19670500422
Return to citation in text: [1] -
Wang, D.; Gautam, L. N. S.; Bollinger, C.; Harris, A.; Li, M.; Shi, X. Org. Lett. 2011, 13, 2618–2621. doi:10.1021/ol200714h
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; de Fremont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571
Return to citation in text: [1] -
Sengupta, S.; Duan, H.; Lu, W.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 1493–1496. doi:10.1021/ol8002783
Return to citation in text: [1] -
Chen, Y.; Liu, Y.; Petersen, J. L.; Shi, X. Chem. Commun. 2008, 3254–3256. doi:10.1039/b805328f
Return to citation in text: [1] -
Liu, Y.; Yan, W.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 5389–5392. doi:10.1021/ol802246q
Return to citation in text: [1] -
Duan, H.; Yan, W.; Sengupta, S.; Shi, X. Bioorg. Med. Chem. Lett. 2009, 19, 3899–3902. doi:10.1016/j.bmcl.2009.03.096
Return to citation in text: [1] -
Yan, W.; Wang, Q.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2010, 12, 3308–3311. doi:10.1021/ol101082v
Return to citation in text: [1] -
Duan, H.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2009, 131, 12100–12102. doi:10.1021/ja9041093
Return to citation in text: [1] -
Chen, Y.; Yan, W.; Akhmedov, N. G.; Shi, X. Org. Lett. 2010, 12, 344–347. doi:10.1021/ol902680k
Return to citation in text: [1] -
Wang, D.; Ye, X.; Shi, X. Org. Lett. 2010, 12, 2088–2091. doi:10.1021/ol100576m
Return to citation in text: [1] -
Yu, M.; Zhang, G.; Zhang, L. Org. Lett. 2007, 9, 2147–2150. doi:10.1021/ol070637o
Return to citation in text: [1] -
Ye, L.; Zhang, L. Org. Lett. 2009, 11, 3646–3649. doi:10.1021/ol901346k
Return to citation in text: [1] -
Yu, M.; Zhang, G.; Zhang, L. Tetrahedron 2009, 65, 1846–1855. doi:10.1016/j.tet.2008.11.107
Return to citation in text: [1] -
Mauleón, P.; Krinsky, J. L.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 4513–4520. doi:10.1021/ja900456m
Return to citation in text: [1]
| 1. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 2. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 3. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 4. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 5. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 6. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 7. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/B612008C |
| 8. | Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368 |
| 9. | Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454 |
| 17. | de Frémont, P.; Scott, N. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, 2411–2418. doi:10.1021/om050111c |
| 18. | Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/cr900074m |
| 19. | Marion, N.; Nolan, S. P. Acc. Chem. Res. 2008, 41, 1440–1449. doi:10.1021/ar800020y |
| 20. | Díez-González, S.; Nolan, S. P. Acc. Chem. Res. 2008, 41, 349–358. doi:10.1021/ar7001655 |
| 47. | Mauleón, P.; Krinsky, J. L.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 4513–4520. doi:10.1021/ja900456m |
| 16. | Shapiro, N. D.; Toste, F. D. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2779–2782. doi:10.1073/pnas.0710500105 |
| 5. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 6. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 43. | Wang, D.; Ye, X.; Shi, X. Org. Lett. 2010, 12, 2088–2091. doi:10.1021/ol100576m |
| 10. | Hashmi, A. S. K.; Rudolph, M.; Huck, J.; Frey, W.; Bats, J. W.; Hamzić, M. Angew. Chem., Int. Ed. 2009, 48, 5848–5852. doi:10.1002/anie.200900887 |
| 11. | Hashmi, A. S. K.; Loos, A.; Littmann, A.; Braun, I.; Knight, J.; Doherty, S.; Rominger, F. Adv. Synth. Catal. 2009, 351, 576–582. doi:10.1002/adsc.200800681 |
| 12. | Hashmi, A. S. K.; Salathé, R.; Frey, W. Chem.–Eur. J. 2006, 12, 6991–6996. doi:10.1002/chem.200600533 |
| 13. | Hashmi, A. S. K.; Blanco, M. C.; Kurpejović, E.; Frey, W.; Bats, J. W. Adv. Synth. Catal. 2006, 348, 709–713. doi:10.1002/adsc.200606012 |
| 14. | Hashmi, A. S. K.; Weyrauch, J. P.; Kurpejović, E.; Frost, T. M.; Miehlich, B.; Frey, W.; Bats, J. W. Chem.–Eur. J. 2006, 12, 5806–5814. doi:10.1002/chem.200501268 |
| 15. | Carrettin, S.; Blanco, M. C.; Corma, A.; Hashmi, A. S. K. Adv. Synth. Catal. 2006, 348, 1283–1288. doi:10.1002/adsc.200606099 |
| 44. | Yu, M.; Zhang, G.; Zhang, L. Org. Lett. 2007, 9, 2147–2150. doi:10.1021/ol070637o |
| 45. | Ye, L.; Zhang, L. Org. Lett. 2009, 11, 3646–3649. doi:10.1021/ol901346k |
| 46. | Yu, M.; Zhang, G.; Zhang, L. Tetrahedron 2009, 65, 1846–1855. doi:10.1016/j.tet.2008.11.107 |
| 35. | Marion, N.; Díez-González, S.; de Fremont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571 |
| 41. | Duan, H.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2009, 131, 12100–12102. doi:10.1021/ja9041093 |
| 34. | Wang, D.; Gautam, L. N. S.; Bollinger, C.; Harris, A.; Li, M.; Shi, X. Org. Lett. 2011, 13, 2618–2621. doi:10.1021/ol200714h |
| 42. | Chen, Y.; Yan, W.; Akhmedov, N. G.; Shi, X. Org. Lett. 2010, 12, 344–347. doi:10.1021/ol902680k |
| 32. | Saucy, R.; Marbet, R.; Lindlar, H.; Isler, O. Helv. Chim. Acta 1959, 42, 1945–1955. doi:10.1002/hlca.19590420624 |
| 33. | Cherbuliez, E.; Moll, H.; Baehler, B. R.; Rabinowitz, J. Helv. Chim. Acta 1967, 50, 1154–1158. doi:10.1002/hlca.19670500422 |
| 21. | Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804–16805. doi:10.1021/ja056419c |
| 22. | Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q |
| 23. | Buzas, A.; Istrate, F.; Gagosz, F. Org. Lett. 2006, 8, 1957–1959. doi:10.1021/ol0606839 |
| 24. | Wang, S.; Zhang, L. Org. Lett. 2006, 8, 4585–4587. doi:10.1021/ol0618151 |
| 25. | Wang, S.; Zhang, L. J. Am. Chem. Soc. 2006, 128, 8414–8415. doi:10.1021/ja062777j |
| 26. | Buzas, A.; Gagosz, F. J. Am. Chem. Soc. 2006, 128, 12614–12615. doi:10.1021/ja064223m |
| 27. | Wang, S.; Zhang, L. J. Am. Chem. Soc. 2006, 128, 14274–14275. doi:10.1021/ja066220f |
| 28. | Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773 |
| 29. | Marco-Contelles, J.; Soriano, E. Chem.–Eur. J. 2007, 13, 1350–1357. doi:10.1002/chem.200601522 |
| 30. | Barluenga, J.; Riesgo, L.; Vicente, R.; Lopéz, L. A.; Tomás, M. J. Am. Chem. Soc. 2007, 129, 7772–7773. doi:10.1021/ja072864r |
| 31. | Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503–15512. doi:10.1021/ja071070+ |
| 36. | Sengupta, S.; Duan, H.; Lu, W.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 1493–1496. doi:10.1021/ol8002783 |
| 37. | Chen, Y.; Liu, Y.; Petersen, J. L.; Shi, X. Chem. Commun. 2008, 3254–3256. doi:10.1039/b805328f |
| 38. | Liu, Y.; Yan, W.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 5389–5392. doi:10.1021/ol802246q |
| 39. | Duan, H.; Yan, W.; Sengupta, S.; Shi, X. Bioorg. Med. Chem. Lett. 2009, 19, 3899–3902. doi:10.1016/j.bmcl.2009.03.096 |
| 40. | Yan, W.; Wang, Q.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2010, 12, 3308–3311. doi:10.1021/ol101082v |
© 2011 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)