Abstract
The number of well-defined molybdenum and tungsten alkylidyne complexes that are able to catalyze alkyne metathesis reactions efficiently has been significantly expanded in recent years.The latest developments in this field featuring highly active imidazolin-2-iminato- and silanolate–alkylidyne complexes are outlined in this review.

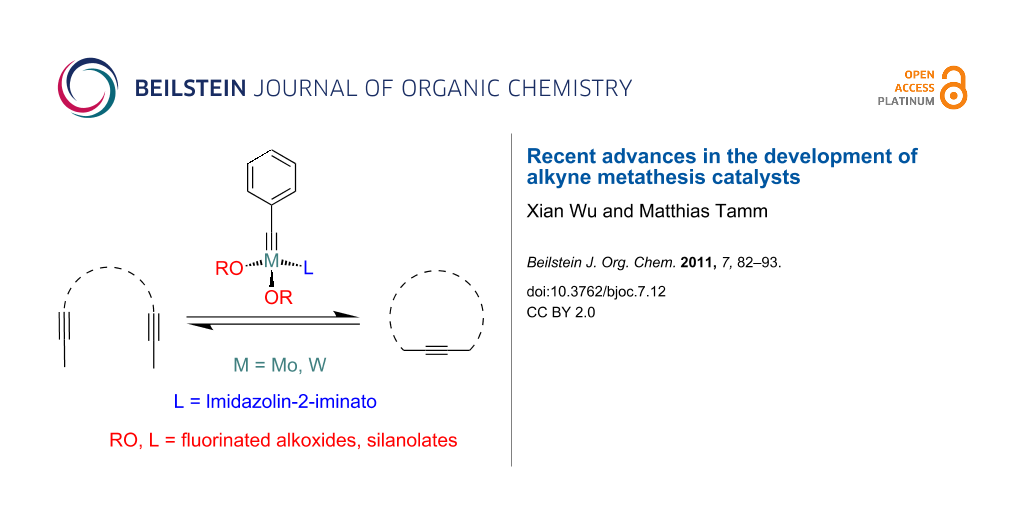
Graphical Abstract
Review
Introduction
C–C bond formation is one of the most important types of reaction in organic synthesis. Transformations employing organometallic compounds as catalysts have achieved a significant role because of their advantages such as simplicity (fewer reaction steps) and efficiency (higher yields) in comparison with traditional synthetic strategies. Nowadays, a plethora of methods is known, which can be used for the formation of C–C single and double bonds, whereas simple ways to create C–C triple bonds are less common, despite the importance and ubiquity of C–C triple bonds in research areas such as natural product synthesis and advanced material science [1].
Alkyne metathesis, which deals with the breaking and making of C–C triple bonds, has only relatively recently become part of the tool box of organic and polymer chemists for the preparation of their target molecules [2-11]. Catalyzed by organotransition metal complexes, this reaction type creates new C–C triple bonds very simply via the Katz mechanism (Scheme 1) [12], based on which a series of different reaction types such as alkyne cross metathesis (ACM), ring-closing alkyne metathesis (RCAM), ring-opening alkyne metathesis polymerization (ROAMP) and acyclic diyne metathesis polymerization (ADIMET) are known (Scheme 2).
![[1860-5397-7-12-i1]](/bjoc/content/inline/1860-5397-7-12-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Alkyne metathesis based on the Katz mechanism.
Scheme 1: Alkyne metathesis based on the Katz mechanism.
![[1860-5397-7-12-i2]](/bjoc/content/inline/1860-5397-7-12-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction patterns of alkyne metathesis.
Scheme 2: Reaction patterns of alkyne metathesis.
In contrast to olefin metathesis, the number of catalysts for alkyne metathesis is far more limited. The first catalyst for alkyne metathesis was a heterogeneous system based on WO3/silica, which was first reported by Pennella, Banks and Bailey in 1968 [13], while the first homogeneous system, which consisted of [Mo(CO)6] and resorcinol [14], was discovered by Mortreux and Blanchard in 1974. Since then, great efforts have been made to develop highly efficient alkyne metathesis catalysts and this has led to three major systems which have dominated this area, i.e., the Mortreux system, the Schrock system and the Cummins–Fürstner–Moore system. Only recently, two novel systems, which exhibit highly promising catalytic performance in alkyne metathesis, were successfully introduced: 1. A modified Schrock system containing imidazolin-2-iminato ligands that was developed by our group; 2. silanolate-supported complexes such as molybdenum nitride and alkylidyne complexes with Ph3SiO ligands developed by Fürstner and tungsten alkylidyne complexes with (t-BuO)3SiO ligands introduced by us. Since there are already several reviews available that cover research progress up to 2006 [2-11], this article will focus on the two novel catalyst systems, which were established over the last four years (2007–10), commencing with a brief introduction to the established systems that have already been widely used by synthetic chemists.
Traditional catalyst systems
Mortreux system
First reported in 1974, the Mortreux system consists of two components: [Mo(CO)6] and phenol or derivatives thereof [14-19]. During the last decades, this system was intensively studied and its performance was significantly improved. However, some drawbacks including the requirement of high reaction temperatures and low functional group tolerance greatly limit its applicability. Moreover, the catalytic mechanism and the active species involved remain unknown, preventing a further rational catalyst design. Nevertheless, because of the commercial availability and high stability of the pre-catalysts as well as the simplicity of operation, this classical system is still widely used by chemists [20-28].
Schrock system
Schrock-type catalysts are high oxidation state molybdenum or tungsten alkylidyne complexes which form metallacyclobutadienes (the key intermediate in the Katz mechanism) upon treatment with internal alkynes. Among these, the tungsten neopentylidyne complex [Me3C≡CW(OCMe3)3] is the most widely used species and is reliably synthesized in several steps from commercially available WCl6. Accordingly, numerous applications of this catalyst have been reported, which usually requires elevated reaction temperatures and relatively high catalyst loadings [29-35].
Cummins–Fürstner–Moore system
Cummins introduced triamido molybdenum(III) complexes of the type [Mo{NR(Ar)}3] in the mid 1990s, which are able to cleave the N–N triple bond in the dinitrogen molecule [36-38]. Based on this discovery, Fürstner developed a catalyst system that is formed upon treatment of [Mo{N(t-Bu)Ar}3] with dichloromethane to give the methylidyne complex [HC≡Mo{N(t-Bu)Ar}3] and the chloro complex [ClMo{N(t-Bu)Ar}3] [39]. Although the detailed reaction mechanism has not been fully uncovered, the latter complex is, somewhat counterintuitively, considered to be the active species. Similarly, Moore was able to isolate molybdenum alkylidyne complexes such as [EtC≡Mo{N(t-Bu)Ar}3], which are able to catalyze alkyne metathesis reactions efficiently, albeit only after treatment with phenol derivatives or by capture on silica [40-46]. The reaction with phenolic compounds presumably leads to partial or complete cleavage of the Mo–N bonds to produce catalytically active phenolate complexes. In agreement with this assumption, Cummins was able to report the synthesis of well-defined molybdenum benzylidyne complexes from the molybdaziridine [Mo(H)(η2-Me2CNAr){N(i-Pr}Ar)] and could demonstrate that these systems are efficient initiators for alkyne metathesis even at ambient temperature and low catalyst loadings [47]. Scheme 3 shows some typical examples of the three traditional catalyst systems.
![[1860-5397-7-12-i3]](/bjoc/content/inline/1860-5397-7-12-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Typical examples from traditional catalyst systems.
Scheme 3: Typical examples from traditional catalyst systems.
Novel catalyst systems
Imidazolin-2-iminato tungsten and molybdenum alkylidyne complexes
Imidazolin-2-iminato ligands, which are isolobal to phosphoraneimides (R3PN−) and cyclopentadienides (C5R5−) [48-52], can be described by the resonance structures shown in Scheme 4, indicating that the ability of the imidazolium ring to stabilize a positive charge affords highly basic ligands with a strong electron-donating capacity towards early transition metals or metals in a higher oxidation state [53-55]. In recent years, our group has significantly expanded the use of these 2σ,4π-electron donor ligands in organometallic chemistry and homogeneous catalysis [56-67]. Their synthesis starts from N-heterocyclic carbenes 1 which react with trimethylsilyl azide to afford 2-trimethylsilyliminoimidazolines 2. After treatment with methanol, the corresponding imidazolin-2-imines 3 can be conveniently isolated [60]. Deprotonation by alkyl lithium reagents leads to imidazolin-2-iminato lithium compounds 4, which serve as ligand transfer reagents during the catalyst preparation (Scheme 4).
![[1860-5397-7-12-i4]](/bjoc/content/inline/1860-5397-7-12-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Ligand synthesis and catalyst design.
Scheme 4: Ligand synthesis and catalyst design.
The idea to use imidazolin-2-iminato ligands for the modification of Schrock-type alkylidyne complexes is based on the consideration that they can be regarded as monoanionic analogues of dinegative imido ligands, which are present in some of the most active olefin metathesis catalysts, i. e., Schrock–Hoveyda-type tungsten and molybdenum imido-alkylidene complexes [10]. We presumed that substitution of the imido ligands by imidazolin-2-iminato ligands and concurrent conversion of the metal–carbon double bond into a triple bond would afford metal alkylidyne species with a well-preserved structural and electronic integrity, and therefore with potentially undiminished catalytic activity (Scheme 4). Thus, the resulting new complexes should then be highly active alkyne metathesis catalysts.
In order to verify this design strategy, high oxidation state tungsten and molybdenum alkylidyne complexes bearing imidazolin-2-iminato ligands (5 and 6) were synthesized by two different routes. The low-oxidation-state route (on the right-hand side in Scheme 5) starting from metal hexacarbonyl has advantages such as higher atom economy, easier operation and suitability for both tungsten and molybdenum [68-70] in comparison with the high-oxidation-state route (on the left- hand side in Scheme 5) starting from tungsten hexachloride [71-73]. The use of partially fluorinated alkoxides such as hexafluoro-tert-butoxide, OCCH3(CF3)2, proved to be essential for creating active catalysts [74], indicating that successful catalyst design in this system relies on establishing a push-pull situation in a similar fashion present in Schrock–Hoveyda olefin metathesis catalysts (Scheme 4) [10] and also in an isolobal rhenium(VII) imido-alkylidyne complex [Re(NAr)(Ct-Bu)(ORF)] (Ar = 2,6-diisopropylphenyl, RF = CCH3(CF3)2), which is able to metathesize aliphatic alkynes [75]. In contrast, however, anionic molybdenum imido-alkylidyne complexes such as [Mo(NAr)(Ct-Bu)(ORF)]− do not promote alkyne metathesis, since the more electron-rich nature of the alkylidyne anion may disfavor alkyne binding [76].
![[1860-5397-7-12-i5]](/bjoc/content/inline/1860-5397-7-12-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Catalysts synthesis using high- and low-oxidation-state routes (for 6a, X = Li or K; for 6b, X = K).
Scheme 5: Catalysts synthesis using high- and low-oxidation-state routes (for 6a, X = Li or K; for 6b, X = K)....
The catalysts 5 and 6 were proved to catalyze various alkyne metathesis reactions including ACM, RCAM and ROAMP. In addition, the isolation and structural characterization of a metallacyclobutadiene complex from the reaction of 5 with an excess of 3-hexyne confirmed that the [2 + 2]-cycloaddition (Katz) mechanism is operative [73,74]. The prototype 5 of our new catalyst system was used for the ACM of 1-phenylpropyne (7) and was shown to be significantly more active than the classic Schrock alkylidyne complex [Me3CC≡W(Ot-Bu)3] at both ambient and elevated temperatures [73,74]. Its performance was also compared with those of two other catalysts 9 and 10 bearing ImDippN and N(t-Bu)Ar ligands, respectively (Table 1, Figure 1). The results show that 5 is significantly more active than 9 and 10, whereas 10 is more active than 9. This is supported by DFT calculations for the metathesis of 2-butyne as the model reaction, which reveal that the activation barrier for the three catalysts follows the order 9 > 10 > 5.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-12-i10.svg?max-width=637&scale=1.0)
![[1860-5397-7-12-1]](/bjoc/content/figures/1860-5397-7-12-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
ACM reactions with more complex substrates bearing different functional groups were studied in the presence of 6a and 6b as catalysts [70]. In the ACM of the 3-pentynyl ether 11, tungsten and molybdenum benzylidyne complexes 6a and 6b were used as catalysts, both showing excellent activities under the same vacuum-driven reaction conditions (Table 2). In our hands, however, the tungsten system appeared to be a superior and more reliable catalyst system than its molybdenum congener, which was also supported by DFT calculations. Similar results were also found for the ACM of the 3-pentynyl benzoic esters 13 bearing a selection of functional groups in the 4-position of the phenyl ring (Table 2). With the tungsten catalyst 6a, excellent yields were obtained for X = Cl, OMe and SMe, whereas only 17% of 14d could be obtained for X = NO2. Increasing the catalyst loading to 2 mol % gave a higher conversion (33%), and we have obtained similar results for other substrates. For instance, ACM of 13e (X = NMe2) was hardly successful in the presence of 2 mol % of the catalyst, whereas 14e was isolated in 90% yield with a catalyst loading of 5 mol %. Further detailed studies are required to fully explain this ostensibly odd behavior.
Table 2: ACM of 11 and 13 using 6a and 6b as catalysts.
| substrate | product | catalyst | yield (%)a | |
|---|---|---|---|---|
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-12-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-12-i12.svg?max-width=637&scale=1.0)
|
6a
6b |
98
97 |
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-12-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-12-i14.svg?max-width=637&scale=1.0)
|
6a
6b |
a X = H
a X = H |
98
97 |
|
6a
6a 6a 6a 6a |
a X = H
b X = Cl c X = OMe d X = NO2 e X = NMe2 |
98
98 94 17 90b |
||
a1 mol % catalyst, toluene, rt, 1 h, 200 mbar. b5 mol % catalyst, toluene, rt, 2 h, 200 mbar, unpublished results.
Catalyst 5 was used in the RCAM of 6,15-dioxaeicosa-2,18-diyne (15) and o-, m- and p-bis(3-pentynyloxymethyl)benzenes 17 (Table 3). While the cyclic product 16 was obtained from 15 in high yield (95%), different selectivities toward the formation of monomeric [10]cyclophanes 18 and [10.10]cyclophanes 19 depending on the substitution pattern were observed [77]: The monomeric cycloalkyne 18b and the dimeric cyclodiyne 19c were exclusively formed from the m- and p-isomer 17b and 17c, respectively, whereas ring-closure of the o-isomer 17a gave a mixture of both 18a and 19a. This observation is in agreement with DFT calculations suggesting that reversible ring-opening and ring-closing metathesis (RORCM) leads to an equilibrium between monomeric and dimeric products and their ratios are determined by their relative stabilities [77].
Table 3: RCAM of 15 and 17 using 5 as catalyst.
| substrate | product | yield (%)a | |
|---|---|---|---|
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-12-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-12-i16.svg?max-width=637&scale=1.0)
|
95 | |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-12-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-12-i18.svg?max-width=637&scale=1.0)
|
a ortho
b meta c para |
24
100 0 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-12-i19.svg?max-width=637&scale=1.0)
|
a ortho
b meta c para |
76
0 100 |
|
a2 mol % catalyst 5, hexane, rt, 2 h, 350 mbar.
The catalytic performance of 6a and 6b in RCAM was demonstrated for the substrates m-bis(3-pentynyloxymethyl)benzene (17b) and bis(3-pentynyl)phthalate (20) (Table 4). The results showed that the tungsten benzylidyne complex can catalyze both reactions with high efficiency, whereas the molybdenum counterpart had a significantly lower activity, in agreement with a theoretically predicted higher activation barrier for the Mo system [70].
Table 4: RCAM of 17b and 20 using 6a and 6b as catalysts.
| substrate | product | catalyst | yield (%)a |
|---|---|---|---|
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-12-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-12-i21.svg?max-width=637&scale=1.0)
|
6a
6b |
86
47 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-12-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-12-i23.svg?max-width=637&scale=1.0)
|
6a
6b |
98
20 |
a2 mol % catalyst, 80 mL toluene, rt, 2 h, 300 mbar.
The ROAMP of cyclooctyne (22) was performed using 5 and 6a as catalysts (Table 5) [78]. According to gel permeation chromatography (GPC) analysis, polymer parameters such as the molecular weight (Mn and Mw) and the polydispersity index (PDI) depend on the catalyst and substrate concentration, and the reaction medium. Besides polymer formation, cyclooligomers were also detected by GPC and mass spectrometry. As shown in Table 5, both catalysts 5 and 6a catalyzed the ring-opening metathesis polymerization efficiently. It is also found that high yields of polymer were obtained when the reactions were performed on neat substrate, whereas lower substrate concentration increases the formation of cyclooligomers. This observation can be well explained by the Jacobson–Stockmayer theory of ring-chain equilibria [79].
Table 5: Ring-opening alkyne metathesis polymerization of cyclooctyne using 5 and 6a as catalysts.
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-12-i24.svg?max-width=637&scale=1.0)
|
|||||||
| cat | mol % | solvent |
csub
(mol/L) |
Mn
(g/mol) |
Mw
(g/mol) |
PDI | polymer yield (%) |
|---|---|---|---|---|---|---|---|
| 5 | 1 | - | neat | 33000 | 46800 | 1.4 | 70 |
| 6a | 1 | - | neat | 26400 | 41300 | 1.6 | 80 |
| 6a | 5 | - | neat | 9960 | 23200 | 2.3 | 95 |
| 6a | 5 | toluene | 0.03 | 82000 | 100000 | 1.2 | 7 |
| 6a | 5 | toluene | 0.02 | -a | - a | - a | 0 |
| 6a | 5 | n-hexane | 0.02 | - a | - a | - a | 0 |
aonly cyclic oligomers were obtained.
Molybdenum nitride and alkylidyne complexes with silanolate ligands
Fürstner recently established a different design strategy for the development of novel alkyne metathesis catalysts. Inspired both by a report of Johnson and co-workers, who found that molybdenum and tungsten nitride complexes 24 with fluorinated alkoxide ligands react with alkynes to generate the corresponding metal alkylidynes 25 in situ (Scheme 6) [80,81], and by the work of Chiu et al. on the preparation of a silanolate-supported molybdenum-nitride complex [82], Fürstner’s group introduced a novel user-friendly catalyst system for alkyne metathesis by employing triphenylsilanol (Ph3SiOH) [83,84]. Two synthetic routes were developed, which are shown in Scheme 7. The one on the left-hand side starting from Na2MoO4 leads to molybdenum nitride pre-catalysts, while the one on the right-hand side starting from [Mo(CO)6] directly affords molybdenum alkylidyne complexes. This procedure resembles the low-oxidation-state route presented in Scheme 5.
![[1860-5397-7-12-i6]](/bjoc/content/inline/1860-5397-7-12-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Design strategy of Fürstner’s new system.
Scheme 6: Design strategy of Fürstner’s new system.
![[1860-5397-7-12-i7]](/bjoc/content/inline/1860-5397-7-12-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthetic routes of Fürstner’s new catalysts.
Scheme 7: Synthetic routes of Fürstner’s new catalysts.
The catalytic activities of the complexes 26, 28, 29, 31, 32 in ACM and RCAM were studied for a variety of substrates. In their initial publication [83], catalytic reactions were performed using 26/Ph3SiOH and 28 as catalysts. Although satisfactory to good yields were achieved, all reactions required elevated reaction temperatures (≥ 80 °C) and, in most cases, high catalyst loadings (up to 20%). However, the results were greatly improved for the 1,10-phenanthroline (phen) systems 29/MnCl2 and 32/MnCl2 – MnCl2 is added to remove the phen-ligand by precipitation of MnCl2•phen – and for the diethyl ether (Et2O) complex 31 by addition of molecular sieves (MS 5 Å) to adsorb the 2-butyne formed during the metathesis reaction [84]. This method constitutes a significant advance, since it allows all reactions to be run in a closed system at ambient pressure. Accordingly, only the latter, improved results will be presented here, and Table 6 and Table 7 summarize the results for ACM and RCAM with the pre-catalysts 29/MnCl2, 31 and 32/MnCl2 [84].
Table 6: ACM of 33 using 29/MnCl2, 31 and 32/MnCl2 as catalysts.
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-12-i25.svg?max-width=637&scale=1.0)
|
||||
| 33 | R- | catalyst and yield (%) | ||
|---|---|---|---|---|
| 29/MnCl2a | 31b | 32/MnCl2c | ||
| af |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-12-i26.svg?max-width=637&scale=1.0)
|
99 | 99 | 99 |
| b |
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-12-i27.svg?max-width=637&scale=1.0)
|
96 | 97 | 97 |
| c |
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-12-i28.svg?max-width=637&scale=1.0)
|
87 | 98d | 96d |
| d |
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-12-i29.svg?max-width=637&scale=1.0)
|
72e | 95 | 97 |
| e |
![[Graphic 21]](/bjoc/content/inline/1860-5397-7-12-i30.svg?max-width=637&scale=1.0)
|
94 | 93 | 95 |
| f |
![[Graphic 22]](/bjoc/content/inline/1860-5397-7-12-i31.svg?max-width=637&scale=1.0)
|
no reaction | no reaction | no reaction |
| g |
![[Graphic 23]](/bjoc/content/inline/1860-5397-7-12-i32.svg?max-width=637&scale=1.0)
|
<40e | 84 | 84 |
| h |
![[Graphic 24]](/bjoc/content/inline/1860-5397-7-12-i33.svg?max-width=637&scale=1.0)
|
76e | 90d | 88d |
| i |
![[Graphic 25]](/bjoc/content/inline/1860-5397-7-12-i34.svg?max-width=637&scale=1.0)
|
86 | 88 | 87 |
| j |
![[Graphic 26]](/bjoc/content/inline/1860-5397-7-12-i35.svg?max-width=637&scale=1.0)
|
95 | 92 | 92 |
| k |
![[Graphic 27]](/bjoc/content/inline/1860-5397-7-12-i36.svg?max-width=637&scale=1.0)
|
85 | 89 | 81 |
| l |
![[Graphic 28]](/bjoc/content/inline/1860-5397-7-12-i37.svg?max-width=637&scale=1.0)
|
92 | 88 | |
| m |
![[Graphic 29]](/bjoc/content/inline/1860-5397-7-12-i38.svg?max-width=637&scale=1.0)
|
81 | 87 | 89 |
a29 (10 mol %), activated by MnCl2 (10 mol %) at 80 °C, molecular sieve, 80 °C. b31 (2 mol %), molecular sieve, ambient temperature. c32 (5 mol %), activated by MnCl2 (5 mol %) at 80 °C, molecular sieve, ambient temperature. d50 °C. e100 °C. f 33a and 34a are the same as 7 and 8, respectively.
Table 7: RCAM of 35 using 29/MnCl2, 31 and 32/MnCl2 as catalysts.
![[Graphic 30]](/bjoc/content/inline/1860-5397-7-12-i39.svg?max-width=637&scale=1.0)
|
||||
| 35 | catalyst and yield (%) | |||
|---|---|---|---|---|
| 29/MnCl2a | 31b | 32/MnCl2c | ||
| a |
![[Graphic 31]](/bjoc/content/inline/1860-5397-7-12-i40.svg?max-width=637&scale=1.0)
|
70 | 97 | 94 |
| b |
![[Graphic 32]](/bjoc/content/inline/1860-5397-7-12-i41.svg?max-width=637&scale=1.0)
|
85 | ||
| c |
![[Graphic 33]](/bjoc/content/inline/1860-5397-7-12-i42.svg?max-width=637&scale=1.0)
|
67 | 72 | |
| d |
![[Graphic 34]](/bjoc/content/inline/1860-5397-7-12-i43.svg?max-width=637&scale=1.0)
|
91 | 73 | 78 |
| e |
![[Graphic 35]](/bjoc/content/inline/1860-5397-7-12-i44.svg?max-width=637&scale=1.0)
|
85 | 92 | 90 |
a29 (10 mol %), activated by MnCl2 (10 mol %) at 80 °C, without molecular sieve, 80 °C. b31 (2 mol %), molecular sieve, ambient temperature. c32 (5 mol %), activated by MnCl2 (5 mol %) at 80 °C, molecular sieve, ambient temperature.
The air-stable nitride complex 29 performs satisfactorily in the presence of MnCl2 and MS 5 Å, however, its stability comes at the expense of higher catalyst loadings (10 mol %) and elevated reaction temperatures. In contrast, the phenanthroline–alkylidyne system 32 requires higher temperatures (80 °C) only for the activation with MnCl2, whereas the metathesis reaction can be carried out at ambient temperature. Noteworthy, it is the Et2O complex 31 that sets a new standard in alkyne metathesis, despite its reduced robustness in comparison with 32. Like 29 and 32, 31 – in combination with MS 5 Å – shows an excellent functional group tolerance together with a significantly enhanced catalytic performance even at lower catalyst concentrations and temperatures than indicated in Table 6 and Table 7 [84]. In addition, this catalyst was employed for the synthesis of various bioactive natural products and also for the total synthesis of natural occurring macrolactides [85,86], confirming and highlighting the strong potential of alkyne metathesis as a tool in organic synthetic methodology [9].
In a very recent report, Finke and Moore reported on the Lewis acid activation of the molybdenum nitrides 26 and 28, which afforded the pre-catalysts 37 and 38 upon addition of one or two equivalents of B(C6F5)3, respectively (Scheme 8) [87]. While the adduct 38 is found to be active in alkyne metathesis, the complex 37 requires additional activation by treatment with the electron-poor phenol 2-(F3C)C6H4OH to facilitate the formation of a catalytically active molybdenum alkylidyne species. The latter system was tested for the metathesis of several phenylalkynes, and yields up to 64% were obtained by application of relatively forcing reaction conditions (10 mol % nitride, 20 mol % borane, 30 mol % phenol, T = 90 °C). Nevertheless, the rate of metathesis is enhanced in comparison with the performance of the borane-free complexes, and these results might therefore pave the way for the development of alkyne metathesis catalysts based on transition metal nitrides.
![[1860-5397-7-12-i8]](/bjoc/content/inline/1860-5397-7-12-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Lewis acid addition of 26 and 28.
Scheme 8: Lewis acid addition of 26 and 28.
Silanolate-supported tungsten alkylidyne complexes
The suitability of silanolates as suitable ancillary ligands for the development of alkyne metathesis catalysts is further confirmed by our independent synthesis of the tungsten benzylidyne complex 39 (Scheme 9), which can be isolated in high yield as a yellow crystalline solid from the reaction of the tribromide [PhC≡WBr3(dme)] (dme = 1,2-dimethoxyethane) with the lithium salt of the silanol (t-BuO)3SiOH [88]. Since this silanol can be regarded as a mimic for silica surfaces [89-94], 39 might be regarded as a homogeneous model for silica-supported alkylidyne complexes [45,46,91-94]. Compound 39 exhibits excellent catalytic behavior in a number of ACM and RCAM reactions [88] (Table 8 and Table 9), and in analogy to Fürstner’s report [84], our studies also indicate that the addition of MS 5 Å does further improve the activity and the ease of applicability of this catalyst system.
![[1860-5397-7-12-i9]](/bjoc/content/inline/1860-5397-7-12-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Preparation of the silanolate–alkylidyne tungsten complex 39.
Scheme 9: Preparation of the silanolate–alkylidyne tungsten complex 39.
Table 8: ACM using 39 as catalyst.
| substrate | product | yield (%)a | yield (%)b | ||
|---|---|---|---|---|---|
![[Graphic 36]](/bjoc/content/inline/1860-5397-7-12-i45.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-7-12-i46.svg?max-width=637&scale=1.0)
|
85 | 95 | ||
![[Graphic 38]](/bjoc/content/inline/1860-5397-7-12-i47.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-7-12-i48.svg?max-width=637&scale=1.0)
|
a X = H
b X = Cl c X = OMe d X = SMe |
97
92 96 94c |
a X = H
b X = Cl c X = OMe d X = SMe |
99
97 97 99 |
a0.5 mmol substrate, 2 mol % catalyst 39, 8 mL toluene, rt, 1 h, 200 mbar. b0.5 mmol substrate, 1 mol % catalyst 39, 2 mL toluene, rt, 1 h, 500 mg, molecular sieve. c0.5 mmol, substrate, 5 mol % catalyst 39, 8 mL toluene, rt, 1 h, 200 mbar.
![[Graphic 40]](/bjoc/content/inline/1860-5397-7-12-i49.svg?max-width=637&scale=1.0)
![[Graphic 41]](/bjoc/content/inline/1860-5397-7-12-i50.svg?max-width=637&scale=1.0)
![[Graphic 42]](/bjoc/content/inline/1860-5397-7-12-i51.svg?max-width=637&scale=1.0)
![[Graphic 43]](/bjoc/content/inline/1860-5397-7-12-i52.svg?max-width=637&scale=1.0)
![[Graphic 44]](/bjoc/content/inline/1860-5397-7-12-i53.svg?max-width=637&scale=1.0)
![[Graphic 45]](/bjoc/content/inline/1860-5397-7-12-i54.svg?max-width=637&scale=1.0)
![[Graphic 46]](/bjoc/content/inline/1860-5397-7-12-i55.svg?max-width=637&scale=1.0)
![[Graphic 47]](/bjoc/content/inline/1860-5397-7-12-i56.svg?max-width=637&scale=1.0)
Conclusion
“Although alkyne metathesis may never reach the breadth of alkene metathesis because of a smaller substrate base” [84], the recent additions to the comparatively small family of alkyne metathesis catalysts – imidazolin-2-iminato- and silanolate-supported molybdenum and tungsten alkylidyne complexes – should certainly help to boost the recognition of alkyne metathesis and to overcome the prevalence of olefin metathesis. The synthetic protocols developed for the synthesis of these new (pre-) catalysts allow for fine-tuning of their steric and electronic properties in order to further optimize their stability and catalytic performance and to modulate their structure according to the requirements of specific applications and substrate classes. However, the development in alkyne metathesis has yet to overcome one major obstacle, and that is the impracticability of employing terminal alkynes as substrates, since these tend to form polymers [95] and were also shown to degrade Schrock alkylidynes by formation of deprotonated, inactive metallacyclobutadienes [96]. Hence, future efforts should also re-address this issue, e. g., by adjusting the properties of the metallacyclobutadiene key intermediates [97] in order to prevent their degeneration and therefore ineffectiveness in undergoing the Katz [2 + 2]cycloaddition/cycloreversion mechanism (Scheme 1).
References
-
Diederich, F.; Stang, P. J.; Tykwinski, R. R., Eds. Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Zhang, W.; Moore, J. S. Adv. Synth. Catal. 2007, 349, 93–120. doi:10.1002/adsc.200600476
Return to citation in text: [1] [2] -
Schrock, R. R.; Czekelius, C. Adv. Synth. Catal. 2007, 349, 55–77. doi:10.1002/adsc.200600459
Return to citation in text: [1] [2] -
Van de Weghe, P.; Bisseret, P.; Blanchard, N.; Eustache, J. J. Organomet. Chem. 2006, 691, 5078–5108. doi:10.1016/j.jorganchem.2006.07.022
Return to citation in text: [1] [2] -
Mortreux, A.; Coutelier, O. J. Mol. Catal. A: Chem. 2006, 254, 96–104. doi:10.1016/j.molcata.2006.03.054
Return to citation in text: [1] [2] -
Schrock, R. R. Chem. Commun. 2005, 22, 2773–2777. doi:10.1039/b504541j
Return to citation in text: [1] [2] -
Bunz, U. H. F. Science 2005, 308, 216–217. doi:10.1126/science.1111098
Return to citation in text: [1] [2] -
Schrock, R. R. Chem. Rev. 2002, 102, 145–180. doi:10.1021/cr0103726
Return to citation in text: [1] [2] -
Fürstner, A.; Davis, P. W. Chem. Commun. 2005, 2307–2320. doi:10.1039/b419143a
Return to citation in text: [1] [2] [3] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 115, 4740–4782. doi:10.1002/anie.200300576
Return to citation in text: [1] [2] [3] [4] -
Fürstner, A. Alkyne Metathesis. In Handbook of Metathesis; Grubbs, R., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Vol. 2, pp 432–462. doi:10.1002/9783527619481.ch27
Return to citation in text: [1] [2] -
Katz, T. J.; McGinnis, J. J. Am. Chem. Soc. 1975, 97, 1592–1594. doi:10.1021/ja00839a063
Return to citation in text: [1] -
Pennella, F.; Banks, R. L.; Bailey, G. C. Chem. Commun. (London) 1968, 1548–1549. doi:10.1039/c19680001548
Return to citation in text: [1] -
Mortreux, A.; Blanchard, M. J. Chem. Soc., Chem. Commun. 1974, 786–787. doi:10.1039/C39740000786
Return to citation in text: [1] [2] -
Mortreux, A.; Dy, N.; Blanchard, M. J. Mol. Catal. 1975, 76, 101–109. doi:10.1016/0304-5102(76)80004-1
Return to citation in text: [1] -
Mortreux, A.; Petit, F.; Blanchard, M. Tetrahedron Lett. 1978, 19, 4967–4968. doi:10.1016/S0040-4039(01)85783-X
Return to citation in text: [1] -
Bencheick, A.; Petit, M.; Mortreux, A.; Petit, F. J. Mol. Catal. 1982, 15, 93–101. doi:10.1016/0304-5102(82)80008-4
Return to citation in text: [1] -
Mortreux, A.; Delgrange, J. C.; Blanchard, M.; Lubochinsky, B. J. Mol. Catal. 1977, 2, 73–82. doi:10.1016/0304-5102(77)85018-9
Return to citation in text: [1] -
Mortreux, A.; Petit, F.; Blanchard, M. J. Mol. Catal. 1980, 8, 97–106. doi:10.1016/0304-5102(80)87009-X
Return to citation in text: [1] -
Kaneta, N.; Hikichi, K.; Asaka, S.-i.; Uemura, M.; Mori, M. Chem. Lett. 1995, 1055–1066. doi:10.1246/cl.1995.1055
Return to citation in text: [1] -
Zhang, W.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 118, 4524–4548. doi:10.1002/anie.200503988
Return to citation in text: [1] -
Zhao, D.; Moore, J. S. Chem. Commun. 2003, 807–818. doi:10.1039/b207442g
Return to citation in text: [1] -
Brizius, G.; Pschirer, N. G.; Steffen, W.; Stitzer, K.; zur Loye, H. C.; Bunz, U. H. F. J. Am. Chem. Soc. 2000, 122, 12435–12440. doi:10.1021/ja0010251
Return to citation in text: [1] -
Ge, P. H.; Fu, W.; Herrmann, W. A.; Herdtweck, E.; Campana, C.; Adams, R. D.; Bunz, U. H. F. Angew. Chem., Int. Ed. 2000, 112, 3753–3756. doi:10.1002/1521-3773(20001016)39:20<3607::AID-ANIE3607>3.0.CO;2-S
Return to citation in text: [1] -
Höger, S. Angew. Chem., Int. Ed. 2005, 117, 3872–3875. doi:10.1002/anie.200500681
Return to citation in text: [1] -
Miljanic, O. S.; Vollhardt, K. P. C.; Whitener, G. D. Synlett 2003, 29–34. doi:10.1055/s-2003-36233
Return to citation in text: [1] -
Johnson, C. A., II; Lu, Y.; Haley, M. M. Org. Lett. 2007, 9, 3725–3728. doi:10.1021/ol7014253
Return to citation in text: [1] -
Fürstner, A.; Guth, O.; Rumbo, A.; Seidel, G. J. Am. Chem. Soc. 1999, 121, 11108–11113. doi:10.1021/ja992074k
Return to citation in text: [1] -
Sancho, J.; Schrock, R. R. J. Mol. Catal. 1982, 15, 75–79. doi:10.1016/0304-5102(82)80006-0
Return to citation in text: [1] -
Schrock, R. R.; Clark, D. N.; Sancho, J.; Wengrovius, J. H.; Rocklage, S. M.; Pederson, S. F. Organometallics 1982, 1, 1645–1651. doi:10.1021/om00072a018
Return to citation in text: [1] -
Wengrovius, J. H.; Sancho, J.; Schrock, R. R. J. Am. Chem. Soc. 1981, 103, 3932–3934. doi:10.1021/ja00403a058
Return to citation in text: [1] -
Fürstner, A.; Seidel, G. Angew. Chem., Int. Ed. 1998, 110, 1758–1760. doi:10.1002/(SICI)1521-3773(19980703)37:12<1734::AID-ANIE1734>3.0.CO;2-6
Return to citation in text: [1] -
Grela, K.; Ignatowska, J. Org. Lett. 2002, 4, 3747–3749. doi:10.1021/ol026690o
Return to citation in text: [1] -
Song, D.; Blond, G.; Fürstner, A. Tetrahedron 2003, 59, 6899–6904. doi:10.1016/S0040-4020(03)00815-9
Return to citation in text: [1] -
Fürstner, A.; Müller, G. J. Organomet. Chem. 2000, 606, 75–78. doi:10.1016/S0022-328X(00)00096-6
Return to citation in text: [1] -
Laplaza, C. E.; Odom, A. L.; Davis, M. W.; Cummins, C. C.; Protasiewicz, J. D. J. Am. Chem. Soc. 1995, 117, 4999–5000. doi:10.1021/ja00122a033
Return to citation in text: [1] -
Laplaza, C. E.; Cummins, C. C. Science 1995, 268, 861–863. doi:10.1126/science.268.5212.861
Return to citation in text: [1] -
Laplaza, C. E.; Johnson, A. R.; Cummins, C. C. J. Am. Chem. Soc. 1996, 118, 709–710. doi:10.1021/ja953573y
Return to citation in text: [1] -
Fürstner, A.; Mathes, C.; Lehmann, C. W. J. Am. Chem. Soc. 1999, 121, 9453–9454. doi:10.1021/ja991340r
Return to citation in text: [1] -
Zhang, W.; Kraft, S.; Moore, J. S. Chem. Commun. 2003, 832–833. doi:10.1039/b212405j
Return to citation in text: [1] -
Zhang, W.; Kraft, S.; Moore, J. S. J. Am. Chem. Soc. 2004, 126, 329–335. doi:10.1021/ja0379868
Return to citation in text: [1] -
Zhang, W.; Moore, J. S. J. Am. Chem. Soc. 2004, 126, 12796. doi:10.1021/ja046531v
Return to citation in text: [1] -
Zhang, W.; Moore, J. S. J. Am. Chem. Soc. 2005, 127, 11863–11870. doi:10.1021/ja053466w
Return to citation in text: [1] -
Zhang, W.; Brombosz, S. M.; Mendoza, J. L.; Moore, J. S. J. Org. Chem. 2005, 70, 10198–10201. doi:10.1021/jo0517803
Return to citation in text: [1] -
Weissmann, H.; Plunkett, K. N.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 118, 599–602. doi:10.1002/anie.200502840
Return to citation in text: [1] [2] -
Cho, H. M.; Weissmann, H.; Moore, J. S. J. Org. Chem. 2008, 73, 4256–4258. doi:10.1021/jo8003919
Return to citation in text: [1] [2] -
Blackwell, J. M.; Figueroa, J. S.; Stephens, F. H.; Cummins, C. C. Organometallics 2003, 22, 3351–3353. doi:10.1021/om0301482
Return to citation in text: [1] -
Dehnicke, K.; Greiner, A. Angew. Chem., Int. Ed. 2003, 115, 1378–1392. doi:10.1002/anie.200390346
Return to citation in text: [1] -
Dehnicke, K.; Krieger, M.; Massa, W. Coord. Chem. Rev. 1999, 182, 19–65. doi:10.1016/S0010-8545(98)00191-X
Return to citation in text: [1] -
Dehnicke, K.; Weller, F. Coord. Chem. Rev. 1997, 158, 103–169. doi:10.1016/S0010-8545(97)90055-2
Return to citation in text: [1] -
Dehnicke, K.; Strähle, J. Polyhedron 1989, 8, 707–726. doi:10.1016/S0277-5387(00)83838-3
Return to citation in text: [1] -
Diefenbach, A.; Bickelhaupt, F. M. Z. Anorg. Allg. Chem. 1999, 625, 892–900. doi:10.1002/(SICI)1521-3749(199906)625:6<892::AID-ZAAC892>3.0.CO;2-7
Return to citation in text: [1] -
Kuhn, N.; Göhner, M.; Grathwohl, M.; Wiethoff, J.; Frenking, G.; Chen, Y. Z. Anorg. Allg. Chem. 2003, 629, 793–802. doi:10.1002/zaac.200390141
Return to citation in text: [1] -
Kuhn, N.; Fawzi, R.; Steinmann, M.; Wiethoff, J. Z. Anorg. Allg. Chem. 1997, 623, 769–774. doi:10.1002/zaac.199762301121
Return to citation in text: [1] -
Kuhn, N.; Fawzi, R.; Steinmann, M.; Wiethoff, J.; Bläser, D.; Boese, R. Z. Naturforsch. 1995, 50b, 1779–1784.
Return to citation in text: [1] -
Tamm, M.; Randoll, S.; Bannenberg, T.; Herdtweck, E. Chem. Commun. 2004, 876–877. doi:10.1039/b401041h
Return to citation in text: [1] -
Tamm, M.; Beer, S.; Herdtweck, E. Z. Naturforsch. 2004, 59b, 1497–1504.
Return to citation in text: [1] -
Tamm, M.; Randoll, S.; Herdtweck, E.; Kleigrewe, N.; Kehr, G.; Erker, G.; Rieger, B. Dalton Trans. 2006, 459–467. doi:10.1039/b511752f
Return to citation in text: [1] -
Petrovic, D.; Tamm, M.; Herdtweck, E. Acta Crystallogr. 2006, C62, 217–219. doi:10.1107/S0108270106008778
Return to citation in text: [1] -
Tamm, M.; Petrovic, D.; Randoll, S.; Beer, S.; Bannenberg, T.; Jones, P. G.; Grunenberg, J. Org. Biomol. Chem. 2007, 5, 523–530. doi:10.1039/b615418b
Return to citation in text: [1] [2] -
Panda, T. K.; Randoll, S.; Hrib, C. G.; Jones, P. G.; Bannenberg, T.; Tamm, M. Chem. Commun. 2007, 5007–5009. doi:10.1039/b711669a
Return to citation in text: [1] -
Stelzig, S. H.; Tamm, M.; Waymouth, R. M. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 6064–6070. doi:10.1002/pola.22918
Return to citation in text: [1] -
Panda, T. K.; Trambitas, A. G.; Bannenberg, T.; Hrib, C. G.; Randoll, S.; Jones, P. G.; Tamm, M. Inorg. Chem. 2009, 48, 5462–5472. doi:10.1021/ic900503q
Return to citation in text: [1] -
Trambitas, A. G.; Panda, T. K.; Jenter, J.; Roesky, P. W.; Daniliuc, C.; Hrib, C. G.; Jones, P. G.; Tamm, M. Inorg. Chem. 2010, 49, 2435–2446. doi:10.1021/ic9024052
Return to citation in text: [1] -
Tamm, M.; Trambitas, A. G.; Hrib, C. G.; Jones, P. G. Terrae Rarae 2010, 7, 1–4.
Return to citation in text: [1] -
Panda, T. K.; Hrib, C. G.; Jones, P. G.; Tamm, M. J. Organomet. Chem. 2010, 695, 2768–2773. doi:10.1016/j.jorganchem.2010.06.028
Return to citation in text: [1] -
Trambitas, A. G.; Panda, T. K.; Tamm, M. Z. Anorg. Allg. Chem. 2010, 636, 2156–2171. doi:10.1002/zaac.201000224
Return to citation in text: [1] -
Mayr, A.; McDermott, G. A. J. Am. Chem. Soc. 1986, 108, 548–549. doi:10.1021/ja00263a054
Return to citation in text: [1] -
McDermott, G. A.; Dorries, A. M.; Mayr, A. Organometallics 1987, 6, 925–931. doi:10.1021/om00148a005
Return to citation in text: [1] -
Haberlag, B.; Wu, X.; Brandhorst, K.; Grunenberg, J.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. Chem.–Eur. J. 2010, 16, 8868–8877. doi:10.1002/chem.201000597
Return to citation in text: [1] [2] [3] -
Schrock, R. R.; Sancho, J.; Pederson, S. F. Inorg. Synth. 1989, 26, 44–51. doi:10.1002/9780470132579.ch10
Return to citation in text: [1] -
Freudenberger, J. H.; Schrock, R. R.; Churchill, M. R.; Rheingold, A. L.; Ziller, J. W. Organometallics 1984, 3, 1563–1573. doi:10.1021/om00088a019
Return to citation in text: [1] -
Beer, S.; Hrib, C. G.; Jones, P. G.; Brandhorst, K.; Grunenberg, J.; Tamm, M. Angew. Chem., Int. Ed. 2007, 119, 9047–9051. doi:10.1002/anie.200703184
Return to citation in text: [1] [2] [3] -
Beer, S.; Brandhorst, K.; Hrib, C. G.; Wu, X.; Haberlag, B.; Grunenberg, J.; Jones, P. G.; Tamm, M. Organometallics 2009, 28, 1534–1545. doi:10.1021/om801119t
Return to citation in text: [1] [2] [3] -
Schrock, R. R.; Weinstock, I. A.; Horton, A. D.; Liu, A. H.; Schofield, M. H. J. Am. Chem. Soc. 1988, 110, 2686–2687. doi:10.1021/ja00216a071
Return to citation in text: [1] -
Tonzetich, Z. J.; Schrock, R. R.; Müller, P. Organometallics 2006, 25, 4301–4306. doi:10.1021/om060501e
Return to citation in text: [1] -
Beer, S.; Brandhorst, K.; Grunenberg, J.; Hrib, C. G.; Jones, P. G.; Tamm, M. Org. Lett. 2008, 10, 981–984. doi:10.1021/ol800154y
Return to citation in text: [1] [2] -
Lysenko, S.; Haberlag, B.; Wu, X.; Tamm, M. Macromol. Symp. 2010, 293, 20–23. doi:10.1002/masy.200900046
Return to citation in text: [1] -
Monfette, S.; Fogg, D. E. Chem. Rev. 2009, 109, 3783–3816. doi:10.1021/cr800541y
Return to citation in text: [1] -
Gdula, R. L.; Johnson, M. J. A. J. Am. Chem. Soc. 2006, 128, 9614–9615. doi:10.1021/ja058036k
Return to citation in text: [1] -
Geyer, A. M.; Wiedner, E. S.; Gary, J. B.; Gdula, R. L.; Kuhlmann, N. C.; Johnson, M. J. A.; Dunietz, B. D.; Kampf, J. W. J. Am. Chem. Soc. 2008, 130, 8984–8999. doi:10.1021/ja800020w
Return to citation in text: [1] -
Chiu, H.-T.; Chen, Y.-P.; Chuang, S.-H.; Jen, J.-S.; Lee, G.-H.; Peng, S.-M. Chem. Commun. 1996, 139–140. doi:10.1039/cc9960000139
Return to citation in text: [1] -
Bindl, M.; Stade, R.; Heilmann, E. K.; Picot, A.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2009, 131, 9468–9470. doi:10.1021/ja903259g
Return to citation in text: [1] [2] -
Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Hickmann, V.; Alcarazo, M.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11042–11044. doi:10.1021/ja104796a
Return to citation in text: [1] -
Micoine, K.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 14064–14066. doi:10.1021/ja107141p
Return to citation in text: [1] -
Finke, A. D.; Moore, J. S. Chem. Commun. 2010, 46, 7939–7941. doi:10.1039/c0cc03113e
Return to citation in text: [1] -
Lysenko, S.; Haberlag, B.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. ChemCatChem 2011, 3, 115–118. doi:10.1002/cctc.201000355
Return to citation in text: [1] [2] -
Fischbach, A.; Klimpel, M. G.; Widenmeyer, M.; Herdtweck, E.; Scherer, W.; Anwander, R. Angew. Chem., Int. Ed. 2004, 116, 2284–2289. doi:10.1002/anie.200352730
Return to citation in text: [1] -
Duchateau, R. Chem. Rev. 2002, 102, 3525–3542. doi:10.1021/cr010386b
Return to citation in text: [1] -
Chandrasekhar, V.; Boomishankar, R.; Nagendran, S. Chem. Rev. 2004, 104, 5847–5910. doi:10.1021/cr0306135
Return to citation in text: [1] [2] -
Cho, H. M.; Weissman, H.; Wilson, S. R.; Moore, J. S. J. Am. Chem. Soc. 2006, 128, 14742–14743. doi:10.1021/ja065101x
Return to citation in text: [1] [2] -
Chabanas, M.; Baudouin, A.; Copéret, C.; Basset, J. M. J. Am. Chem. Soc. 2001, 123, 2062–2063. doi:10.1021/ja000900f
Return to citation in text: [1] [2] -
Merle, N.; Taoufik, M.; Nayer, M.; Baudouin, A.; Le Boux, E.; Gauvin, R. M.; Lefebvre, F.; Thivolle-Gazat, J.; Basset, J. M. J. Organomet. Chem. 2008, 693, 1733–1737. doi:10.1016/j.jorganchem.2008.02.020
Return to citation in text: [1] [2] -
Bray, A.; Mortreux, A.; Petit, F.; Petit, M.; Szymanska-Buzar, T. J. Chem. Soc., Chem. Commun. 1993, 197–199. doi:10.1039/C39930000197
Return to citation in text: [1] -
McCullough, L. G.; Listemann, M. L.; Schrock, R. R.; Churchill, M. R.; Ziller, J. W. J. Am. Chem. Soc. 1983, 105, 6729–6730. doi:10.1021/ja00360a040
Return to citation in text: [1] -
Suresh, C. H.; Frenking, G. Organometallics 2010, 29, 4766–4769. doi:10.1021/om100260p
Return to citation in text: [1]
| 77. | Beer, S.; Brandhorst, K.; Grunenberg, J.; Hrib, C. G.; Jones, P. G.; Tamm, M. Org. Lett. 2008, 10, 981–984. doi:10.1021/ol800154y |
| 77. | Beer, S.; Brandhorst, K.; Grunenberg, J.; Hrib, C. G.; Jones, P. G.; Tamm, M. Org. Lett. 2008, 10, 981–984. doi:10.1021/ol800154y |
| 70. | Haberlag, B.; Wu, X.; Brandhorst, K.; Grunenberg, J.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. Chem.–Eur. J. 2010, 16, 8868–8877. doi:10.1002/chem.201000597 |
| 84. | Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w |
| 84. | Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w |
| 83. | Bindl, M.; Stade, R.; Heilmann, E. K.; Picot, A.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2009, 131, 9468–9470. doi:10.1021/ja903259g |
| 84. | Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w |
| 83. | Bindl, M.; Stade, R.; Heilmann, E. K.; Picot, A.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2009, 131, 9468–9470. doi:10.1021/ja903259g |
| 80. | Gdula, R. L.; Johnson, M. J. A. J. Am. Chem. Soc. 2006, 128, 9614–9615. doi:10.1021/ja058036k |
| 81. | Geyer, A. M.; Wiedner, E. S.; Gary, J. B.; Gdula, R. L.; Kuhlmann, N. C.; Johnson, M. J. A.; Dunietz, B. D.; Kampf, J. W. J. Am. Chem. Soc. 2008, 130, 8984–8999. doi:10.1021/ja800020w |
| 82. | Chiu, H.-T.; Chen, Y.-P.; Chuang, S.-H.; Jen, J.-S.; Lee, G.-H.; Peng, S.-M. Chem. Commun. 1996, 139–140. doi:10.1039/cc9960000139 |
| 78. | Lysenko, S.; Haberlag, B.; Wu, X.; Tamm, M. Macromol. Symp. 2010, 293, 20–23. doi:10.1002/masy.200900046 |
| 79. | Monfette, S.; Fogg, D. E. Chem. Rev. 2009, 109, 3783–3816. doi:10.1021/cr800541y |
| 84. | Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w |
| 85. | Hickmann, V.; Alcarazo, M.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11042–11044. doi:10.1021/ja104796a |
| 86. | Micoine, K.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 14064–14066. doi:10.1021/ja107141p |
| 84. | Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w |
| 95. | Bray, A.; Mortreux, A.; Petit, F.; Petit, M.; Szymanska-Buzar, T. J. Chem. Soc., Chem. Commun. 1993, 197–199. doi:10.1039/C39930000197 |
| 88. | Lysenko, S.; Haberlag, B.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. ChemCatChem 2011, 3, 115–118. doi:10.1002/cctc.201000355 |
| 84. | Heppekausen, J.; Stade, R.; Goddard, R.; Fürstner, A. J. Am. Chem. Soc. 2010, 132, 11045–11057. doi:10.1021/ja104800w |
| 89. | Fischbach, A.; Klimpel, M. G.; Widenmeyer, M.; Herdtweck, E.; Scherer, W.; Anwander, R. Angew. Chem., Int. Ed. 2004, 116, 2284–2289. doi:10.1002/anie.200352730 |
| 90. | Duchateau, R. Chem. Rev. 2002, 102, 3525–3542. doi:10.1021/cr010386b |
| 91. | Chandrasekhar, V.; Boomishankar, R.; Nagendran, S. Chem. Rev. 2004, 104, 5847–5910. doi:10.1021/cr0306135 |
| 92. | Cho, H. M.; Weissman, H.; Wilson, S. R.; Moore, J. S. J. Am. Chem. Soc. 2006, 128, 14742–14743. doi:10.1021/ja065101x |
| 93. | Chabanas, M.; Baudouin, A.; Copéret, C.; Basset, J. M. J. Am. Chem. Soc. 2001, 123, 2062–2063. doi:10.1021/ja000900f |
| 94. | Merle, N.; Taoufik, M.; Nayer, M.; Baudouin, A.; Le Boux, E.; Gauvin, R. M.; Lefebvre, F.; Thivolle-Gazat, J.; Basset, J. M. J. Organomet. Chem. 2008, 693, 1733–1737. doi:10.1016/j.jorganchem.2008.02.020 |
| 45. | Weissmann, H.; Plunkett, K. N.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 118, 599–602. doi:10.1002/anie.200502840 |
| 46. | Cho, H. M.; Weissmann, H.; Moore, J. S. J. Org. Chem. 2008, 73, 4256–4258. doi:10.1021/jo8003919 |
| 91. | Chandrasekhar, V.; Boomishankar, R.; Nagendran, S. Chem. Rev. 2004, 104, 5847–5910. doi:10.1021/cr0306135 |
| 92. | Cho, H. M.; Weissman, H.; Wilson, S. R.; Moore, J. S. J. Am. Chem. Soc. 2006, 128, 14742–14743. doi:10.1021/ja065101x |
| 93. | Chabanas, M.; Baudouin, A.; Copéret, C.; Basset, J. M. J. Am. Chem. Soc. 2001, 123, 2062–2063. doi:10.1021/ja000900f |
| 94. | Merle, N.; Taoufik, M.; Nayer, M.; Baudouin, A.; Le Boux, E.; Gauvin, R. M.; Lefebvre, F.; Thivolle-Gazat, J.; Basset, J. M. J. Organomet. Chem. 2008, 693, 1733–1737. doi:10.1016/j.jorganchem.2008.02.020 |
| 87. | Finke, A. D.; Moore, J. S. Chem. Commun. 2010, 46, 7939–7941. doi:10.1039/c0cc03113e |
| 88. | Lysenko, S.; Haberlag, B.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. ChemCatChem 2011, 3, 115–118. doi:10.1002/cctc.201000355 |
| 97. | Suresh, C. H.; Frenking, G. Organometallics 2010, 29, 4766–4769. doi:10.1021/om100260p |
| 96. | McCullough, L. G.; Listemann, M. L.; Schrock, R. R.; Churchill, M. R.; Ziller, J. W. J. Am. Chem. Soc. 1983, 105, 6729–6730. doi:10.1021/ja00360a040 |
| 1. | Diederich, F.; Stang, P. J.; Tykwinski, R. R., Eds. Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005. |
| 14. | Mortreux, A.; Blanchard, M. J. Chem. Soc., Chem. Commun. 1974, 786–787. doi:10.1039/C39740000786 |
| 53. | Kuhn, N.; Göhner, M.; Grathwohl, M.; Wiethoff, J.; Frenking, G.; Chen, Y. Z. Anorg. Allg. Chem. 2003, 629, 793–802. doi:10.1002/zaac.200390141 |
| 54. | Kuhn, N.; Fawzi, R.; Steinmann, M.; Wiethoff, J. Z. Anorg. Allg. Chem. 1997, 623, 769–774. doi:10.1002/zaac.199762301121 |
| 55. | Kuhn, N.; Fawzi, R.; Steinmann, M.; Wiethoff, J.; Bläser, D.; Boese, R. Z. Naturforsch. 1995, 50b, 1779–1784. |
| 13. | Pennella, F.; Banks, R. L.; Bailey, G. C. Chem. Commun. (London) 1968, 1548–1549. doi:10.1039/c19680001548 |
| 56. | Tamm, M.; Randoll, S.; Bannenberg, T.; Herdtweck, E. Chem. Commun. 2004, 876–877. doi:10.1039/b401041h |
| 57. | Tamm, M.; Beer, S.; Herdtweck, E. Z. Naturforsch. 2004, 59b, 1497–1504. |
| 58. | Tamm, M.; Randoll, S.; Herdtweck, E.; Kleigrewe, N.; Kehr, G.; Erker, G.; Rieger, B. Dalton Trans. 2006, 459–467. doi:10.1039/b511752f |
| 59. | Petrovic, D.; Tamm, M.; Herdtweck, E. Acta Crystallogr. 2006, C62, 217–219. doi:10.1107/S0108270106008778 |
| 60. | Tamm, M.; Petrovic, D.; Randoll, S.; Beer, S.; Bannenberg, T.; Jones, P. G.; Grunenberg, J. Org. Biomol. Chem. 2007, 5, 523–530. doi:10.1039/b615418b |
| 61. | Panda, T. K.; Randoll, S.; Hrib, C. G.; Jones, P. G.; Bannenberg, T.; Tamm, M. Chem. Commun. 2007, 5007–5009. doi:10.1039/b711669a |
| 62. | Stelzig, S. H.; Tamm, M.; Waymouth, R. M. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 6064–6070. doi:10.1002/pola.22918 |
| 63. | Panda, T. K.; Trambitas, A. G.; Bannenberg, T.; Hrib, C. G.; Randoll, S.; Jones, P. G.; Tamm, M. Inorg. Chem. 2009, 48, 5462–5472. doi:10.1021/ic900503q |
| 64. | Trambitas, A. G.; Panda, T. K.; Jenter, J.; Roesky, P. W.; Daniliuc, C.; Hrib, C. G.; Jones, P. G.; Tamm, M. Inorg. Chem. 2010, 49, 2435–2446. doi:10.1021/ic9024052 |
| 65. | Tamm, M.; Trambitas, A. G.; Hrib, C. G.; Jones, P. G. Terrae Rarae 2010, 7, 1–4. |
| 66. | Panda, T. K.; Hrib, C. G.; Jones, P. G.; Tamm, M. J. Organomet. Chem. 2010, 695, 2768–2773. doi:10.1016/j.jorganchem.2010.06.028 |
| 67. | Trambitas, A. G.; Panda, T. K.; Tamm, M. Z. Anorg. Allg. Chem. 2010, 636, 2156–2171. doi:10.1002/zaac.201000224 |
| 12. | Katz, T. J.; McGinnis, J. J. Am. Chem. Soc. 1975, 97, 1592–1594. doi:10.1021/ja00839a063 |
| 47. | Blackwell, J. M.; Figueroa, J. S.; Stephens, F. H.; Cummins, C. C. Organometallics 2003, 22, 3351–3353. doi:10.1021/om0301482 |
| 2. | Zhang, W.; Moore, J. S. Adv. Synth. Catal. 2007, 349, 93–120. doi:10.1002/adsc.200600476 |
| 3. | Schrock, R. R.; Czekelius, C. Adv. Synth. Catal. 2007, 349, 55–77. doi:10.1002/adsc.200600459 |
| 4. | Van de Weghe, P.; Bisseret, P.; Blanchard, N.; Eustache, J. J. Organomet. Chem. 2006, 691, 5078–5108. doi:10.1016/j.jorganchem.2006.07.022 |
| 5. | Mortreux, A.; Coutelier, O. J. Mol. Catal. A: Chem. 2006, 254, 96–104. doi:10.1016/j.molcata.2006.03.054 |
| 6. | Schrock, R. R. Chem. Commun. 2005, 22, 2773–2777. doi:10.1039/b504541j |
| 7. | Bunz, U. H. F. Science 2005, 308, 216–217. doi:10.1126/science.1111098 |
| 8. | Schrock, R. R. Chem. Rev. 2002, 102, 145–180. doi:10.1021/cr0103726 |
| 9. | Fürstner, A.; Davis, P. W. Chem. Commun. 2005, 2307–2320. doi:10.1039/b419143a |
| 10. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 115, 4740–4782. doi:10.1002/anie.200300576 |
| 11. | Fürstner, A. Alkyne Metathesis. In Handbook of Metathesis; Grubbs, R., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Vol. 2, pp 432–462. doi:10.1002/9783527619481.ch27 |
| 48. | Dehnicke, K.; Greiner, A. Angew. Chem., Int. Ed. 2003, 115, 1378–1392. doi:10.1002/anie.200390346 |
| 49. | Dehnicke, K.; Krieger, M.; Massa, W. Coord. Chem. Rev. 1999, 182, 19–65. doi:10.1016/S0010-8545(98)00191-X |
| 50. | Dehnicke, K.; Weller, F. Coord. Chem. Rev. 1997, 158, 103–169. doi:10.1016/S0010-8545(97)90055-2 |
| 51. | Dehnicke, K.; Strähle, J. Polyhedron 1989, 8, 707–726. doi:10.1016/S0277-5387(00)83838-3 |
| 52. | Diefenbach, A.; Bickelhaupt, F. M. Z. Anorg. Allg. Chem. 1999, 625, 892–900. doi:10.1002/(SICI)1521-3749(199906)625:6<892::AID-ZAAC892>3.0.CO;2-7 |
| 29. | Sancho, J.; Schrock, R. R. J. Mol. Catal. 1982, 15, 75–79. doi:10.1016/0304-5102(82)80006-0 |
| 30. | Schrock, R. R.; Clark, D. N.; Sancho, J.; Wengrovius, J. H.; Rocklage, S. M.; Pederson, S. F. Organometallics 1982, 1, 1645–1651. doi:10.1021/om00072a018 |
| 31. | Wengrovius, J. H.; Sancho, J.; Schrock, R. R. J. Am. Chem. Soc. 1981, 103, 3932–3934. doi:10.1021/ja00403a058 |
| 32. | Fürstner, A.; Seidel, G. Angew. Chem., Int. Ed. 1998, 110, 1758–1760. doi:10.1002/(SICI)1521-3773(19980703)37:12<1734::AID-ANIE1734>3.0.CO;2-6 |
| 33. | Grela, K.; Ignatowska, J. Org. Lett. 2002, 4, 3747–3749. doi:10.1021/ol026690o |
| 34. | Song, D.; Blond, G.; Fürstner, A. Tetrahedron 2003, 59, 6899–6904. doi:10.1016/S0040-4020(03)00815-9 |
| 35. | Fürstner, A.; Müller, G. J. Organomet. Chem. 2000, 606, 75–78. doi:10.1016/S0022-328X(00)00096-6 |
| 39. | Fürstner, A.; Mathes, C.; Lehmann, C. W. J. Am. Chem. Soc. 1999, 121, 9453–9454. doi:10.1021/ja991340r |
| 20. | Kaneta, N.; Hikichi, K.; Asaka, S.-i.; Uemura, M.; Mori, M. Chem. Lett. 1995, 1055–1066. doi:10.1246/cl.1995.1055 |
| 21. | Zhang, W.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 118, 4524–4548. doi:10.1002/anie.200503988 |
| 22. | Zhao, D.; Moore, J. S. Chem. Commun. 2003, 807–818. doi:10.1039/b207442g |
| 23. | Brizius, G.; Pschirer, N. G.; Steffen, W.; Stitzer, K.; zur Loye, H. C.; Bunz, U. H. F. J. Am. Chem. Soc. 2000, 122, 12435–12440. doi:10.1021/ja0010251 |
| 24. | Ge, P. H.; Fu, W.; Herrmann, W. A.; Herdtweck, E.; Campana, C.; Adams, R. D.; Bunz, U. H. F. Angew. Chem., Int. Ed. 2000, 112, 3753–3756. doi:10.1002/1521-3773(20001016)39:20<3607::AID-ANIE3607>3.0.CO;2-S |
| 25. | Höger, S. Angew. Chem., Int. Ed. 2005, 117, 3872–3875. doi:10.1002/anie.200500681 |
| 26. | Miljanic, O. S.; Vollhardt, K. P. C.; Whitener, G. D. Synlett 2003, 29–34. doi:10.1055/s-2003-36233 |
| 27. | Johnson, C. A., II; Lu, Y.; Haley, M. M. Org. Lett. 2007, 9, 3725–3728. doi:10.1021/ol7014253 |
| 28. | Fürstner, A.; Guth, O.; Rumbo, A.; Seidel, G. J. Am. Chem. Soc. 1999, 121, 11108–11113. doi:10.1021/ja992074k |
| 40. | Zhang, W.; Kraft, S.; Moore, J. S. Chem. Commun. 2003, 832–833. doi:10.1039/b212405j |
| 41. | Zhang, W.; Kraft, S.; Moore, J. S. J. Am. Chem. Soc. 2004, 126, 329–335. doi:10.1021/ja0379868 |
| 42. | Zhang, W.; Moore, J. S. J. Am. Chem. Soc. 2004, 126, 12796. doi:10.1021/ja046531v |
| 43. | Zhang, W.; Moore, J. S. J. Am. Chem. Soc. 2005, 127, 11863–11870. doi:10.1021/ja053466w |
| 44. | Zhang, W.; Brombosz, S. M.; Mendoza, J. L.; Moore, J. S. J. Org. Chem. 2005, 70, 10198–10201. doi:10.1021/jo0517803 |
| 45. | Weissmann, H.; Plunkett, K. N.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 118, 599–602. doi:10.1002/anie.200502840 |
| 46. | Cho, H. M.; Weissmann, H.; Moore, J. S. J. Org. Chem. 2008, 73, 4256–4258. doi:10.1021/jo8003919 |
| 14. | Mortreux, A.; Blanchard, M. J. Chem. Soc., Chem. Commun. 1974, 786–787. doi:10.1039/C39740000786 |
| 15. | Mortreux, A.; Dy, N.; Blanchard, M. J. Mol. Catal. 1975, 76, 101–109. doi:10.1016/0304-5102(76)80004-1 |
| 16. | Mortreux, A.; Petit, F.; Blanchard, M. Tetrahedron Lett. 1978, 19, 4967–4968. doi:10.1016/S0040-4039(01)85783-X |
| 17. | Bencheick, A.; Petit, M.; Mortreux, A.; Petit, F. J. Mol. Catal. 1982, 15, 93–101. doi:10.1016/0304-5102(82)80008-4 |
| 18. | Mortreux, A.; Delgrange, J. C.; Blanchard, M.; Lubochinsky, B. J. Mol. Catal. 1977, 2, 73–82. doi:10.1016/0304-5102(77)85018-9 |
| 19. | Mortreux, A.; Petit, F.; Blanchard, M. J. Mol. Catal. 1980, 8, 97–106. doi:10.1016/0304-5102(80)87009-X |
| 2. | Zhang, W.; Moore, J. S. Adv. Synth. Catal. 2007, 349, 93–120. doi:10.1002/adsc.200600476 |
| 3. | Schrock, R. R.; Czekelius, C. Adv. Synth. Catal. 2007, 349, 55–77. doi:10.1002/adsc.200600459 |
| 4. | Van de Weghe, P.; Bisseret, P.; Blanchard, N.; Eustache, J. J. Organomet. Chem. 2006, 691, 5078–5108. doi:10.1016/j.jorganchem.2006.07.022 |
| 5. | Mortreux, A.; Coutelier, O. J. Mol. Catal. A: Chem. 2006, 254, 96–104. doi:10.1016/j.molcata.2006.03.054 |
| 6. | Schrock, R. R. Chem. Commun. 2005, 22, 2773–2777. doi:10.1039/b504541j |
| 7. | Bunz, U. H. F. Science 2005, 308, 216–217. doi:10.1126/science.1111098 |
| 8. | Schrock, R. R. Chem. Rev. 2002, 102, 145–180. doi:10.1021/cr0103726 |
| 9. | Fürstner, A.; Davis, P. W. Chem. Commun. 2005, 2307–2320. doi:10.1039/b419143a |
| 10. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 115, 4740–4782. doi:10.1002/anie.200300576 |
| 11. | Fürstner, A. Alkyne Metathesis. In Handbook of Metathesis; Grubbs, R., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Vol. 2, pp 432–462. doi:10.1002/9783527619481.ch27 |
| 36. | Laplaza, C. E.; Odom, A. L.; Davis, M. W.; Cummins, C. C.; Protasiewicz, J. D. J. Am. Chem. Soc. 1995, 117, 4999–5000. doi:10.1021/ja00122a033 |
| 37. | Laplaza, C. E.; Cummins, C. C. Science 1995, 268, 861–863. doi:10.1126/science.268.5212.861 |
| 38. | Laplaza, C. E.; Johnson, A. R.; Cummins, C. C. J. Am. Chem. Soc. 1996, 118, 709–710. doi:10.1021/ja953573y |
| 68. | Mayr, A.; McDermott, G. A. J. Am. Chem. Soc. 1986, 108, 548–549. doi:10.1021/ja00263a054 |
| 69. | McDermott, G. A.; Dorries, A. M.; Mayr, A. Organometallics 1987, 6, 925–931. doi:10.1021/om00148a005 |
| 70. | Haberlag, B.; Wu, X.; Brandhorst, K.; Grunenberg, J.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. Chem.–Eur. J. 2010, 16, 8868–8877. doi:10.1002/chem.201000597 |
| 60. | Tamm, M.; Petrovic, D.; Randoll, S.; Beer, S.; Bannenberg, T.; Jones, P. G.; Grunenberg, J. Org. Biomol. Chem. 2007, 5, 523–530. doi:10.1039/b615418b |
| 10. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 115, 4740–4782. doi:10.1002/anie.200300576 |
| 73. | Beer, S.; Hrib, C. G.; Jones, P. G.; Brandhorst, K.; Grunenberg, J.; Tamm, M. Angew. Chem., Int. Ed. 2007, 119, 9047–9051. doi:10.1002/anie.200703184 |
| 74. | Beer, S.; Brandhorst, K.; Hrib, C. G.; Wu, X.; Haberlag, B.; Grunenberg, J.; Jones, P. G.; Tamm, M. Organometallics 2009, 28, 1534–1545. doi:10.1021/om801119t |
| 70. | Haberlag, B.; Wu, X.; Brandhorst, K.; Grunenberg, J.; Daniliuc, C. G.; Jones, P. G.; Tamm, M. Chem.–Eur. J. 2010, 16, 8868–8877. doi:10.1002/chem.201000597 |
| 76. | Tonzetich, Z. J.; Schrock, R. R.; Müller, P. Organometallics 2006, 25, 4301–4306. doi:10.1021/om060501e |
| 73. | Beer, S.; Hrib, C. G.; Jones, P. G.; Brandhorst, K.; Grunenberg, J.; Tamm, M. Angew. Chem., Int. Ed. 2007, 119, 9047–9051. doi:10.1002/anie.200703184 |
| 74. | Beer, S.; Brandhorst, K.; Hrib, C. G.; Wu, X.; Haberlag, B.; Grunenberg, J.; Jones, P. G.; Tamm, M. Organometallics 2009, 28, 1534–1545. doi:10.1021/om801119t |
| 10. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 115, 4740–4782. doi:10.1002/anie.200300576 |
| 75. | Schrock, R. R.; Weinstock, I. A.; Horton, A. D.; Liu, A. H.; Schofield, M. H. J. Am. Chem. Soc. 1988, 110, 2686–2687. doi:10.1021/ja00216a071 |
| 71. | Schrock, R. R.; Sancho, J.; Pederson, S. F. Inorg. Synth. 1989, 26, 44–51. doi:10.1002/9780470132579.ch10 |
| 72. | Freudenberger, J. H.; Schrock, R. R.; Churchill, M. R.; Rheingold, A. L.; Ziller, J. W. Organometallics 1984, 3, 1563–1573. doi:10.1021/om00088a019 |
| 73. | Beer, S.; Hrib, C. G.; Jones, P. G.; Brandhorst, K.; Grunenberg, J.; Tamm, M. Angew. Chem., Int. Ed. 2007, 119, 9047–9051. doi:10.1002/anie.200703184 |
| 74. | Beer, S.; Brandhorst, K.; Hrib, C. G.; Wu, X.; Haberlag, B.; Grunenberg, J.; Jones, P. G.; Tamm, M. Organometallics 2009, 28, 1534–1545. doi:10.1021/om801119t |
© 2011 Wu and Tamm; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)