Abstract
Efforts to trap early intermediates of the gold-catalyzed phenol synthesis failed. Neither inter- nor intramolecularly offered vinyl groups, ketones or alcohols were able to intercept the gold carbenoid species. This indicates that the competing steps of the gold-catalyzed phenol synthesis are much faster than the steps of the interception reaction. In the latter the barrier of activation is higher. At the same time this explains the high tolerance of this very efficient and general reaction towards functional groups.

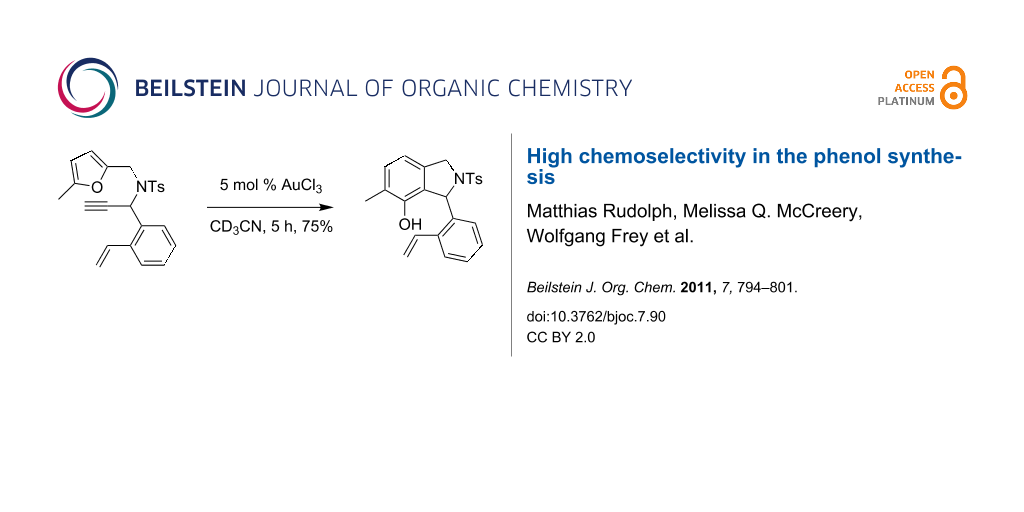
Graphical Abstract
Introduction
As documented in numerous reviews [1-10], over the last eleven years homogeneous gold catalysis has emerged from early examples [11,12] which documented its potential for organic synthesis of even complex molecules to an established tool in preparative organic chemistry [13,14]. One of these early examples is the gold-catalyzed phenol synthesis [12] in which the furan-ynes 1 used as substrates represent the first ene–yne-type compounds ever used in gold catalysis. While many investigations in the field focused on methodology, mechanistic research was much less widespread [2,3,15]. The gold-catalyzed ene–yne cycloisomerization reactions are, mechanistically, very complex reactions [16-18], and the furan–yne cycloisomerization is no exception. For the latter reaction arene oxides D [19] and oxepines C [20] could be detected as intermediates, and these could even be trapped by Diels–Alder reactions. In addition, labelling studies were carried out and the electronic influence of substituents was investigated [21]. Computational studies as well as side-products produced in the reaction pointed towards intermediates A and B (Scheme 1) [22-25]. Moreover, interesting new pathways were opened when ynamides and alkynyl ether substrates were employed: Here A is also a possible intermediate along these pathways [25].
![[1860-5397-7-90-i1]](/bjoc/content/inline/1860-5397-7-90-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanism of the furan–yne reaction.
Scheme 1: Mechanism of the furan–yne reaction.
Since direct experimental evidence existed only for C and D, we intended to intercept the postulated carbenoid intermediates A or B. Apart from intermolecular trapping [26-33], intramolecular trapping of such carbenoids has also been reported [34]. One option would be to offer a competing carbonyl group, to produce a carbonyl ylide, which could then undergo a 1,3-dipolar cycloaddition [35]. The second option would be a classical cyclopropanation of an olefin. A third option would be trapping of intermediate A with an intramolecular hydroxy nucleophile [36]. Here we report our observations when trying to apply these principles to intermediates of type A or B.
Results and Discussion
Intermolecular olefinic trapping reagents
We started with the simplest experiments, namely the intermolecular trapping of the gold carbenoid intermediates. When 3 was reacted in the presence of an activated olefin, such as norbornene or styrene, phenol 4 was formed exclusively in essentially quantitative yield, no other products could be detected (Scheme 2).
![[1860-5397-7-90-i2]](/bjoc/content/inline/1860-5397-7-90-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Efforts for intermolecular trapping with olefins failed.
Scheme 2: Efforts for intermolecular trapping with olefins failed.
Experiments with a competing carbonyl group (competing with the carbonyl group in intermediate B) were also unsuccessful. Ketone 5 [37], prepared by the addition of methyllithium to commercially available hex-5-enoic acid, was used as an external carbonyl group. Reaction with both tosylamide 3 and ether 6 always delivered the phenolic products 4 or 7, respectively (Scheme 3). The same result was obtained when PtCl2 was used as the catalyst for the conversion of 3.
![[1860-5397-7-90-i3]](/bjoc/content/inline/1860-5397-7-90-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Efforts for intermolecular trapping with ketones failed.
Scheme 3: Efforts for intermolecular trapping with ketones failed.
Intramolecular olefinic trapping reagents
The next step was to offer the styrene unit in an intramolecular manner. Substrate 8 could potentially undergo three different modes of reaction (Scheme 4). After the initial step, the intermediate E would be produced (analogous to A). Cyclopropanation of the styrene subunit by the cyclopropyl carbenoid would deliver 9. If E rearranged to the vinylcarbenoid F, the two competing reactions would be the formation of the phenol 10 and cyclopropanation to form 11.
![[1860-5397-7-90-i4]](/bjoc/content/inline/1860-5397-7-90-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Potential products of an intramolecular trapping experiment with substrate 8.
Scheme 4: Potential products of an intramolecular trapping experiment with substrate 8.
The synthesis of 8 was possible by a short route (Scheme 5). Starting from the commercially available 2-bromostyrene (12), a halogen–metal exchange and subsequent formylation according to a procedure of Fukumoto et al. [38] gave 13. Addition of ethynylmagnesium bromide to 13 led to 14, which reacted with furan 15 [40] under Mitsunobu conditions [39] to afford 8. While the yields were good for the first two steps of the reaction sequence, the yield of the last step was only 32%.
![[1860-5397-7-90-i5]](/bjoc/content/inline/1860-5397-7-90-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
With AuCl3 the phenol 10 was formed exclusively (Scheme 6). The structure was unambiguously confirmed by X-ray crystal structure analysis (Figure 1). It shows an interesting hydrogen bond-like interaction of the phenolic hydroxy group and the alkene unit. After changing the solvent from acetonitrile to CDCl3, and the gold(I) catalyst to [Mes3PAu]NTf2 [41], only 10 was again observed. Thus, neither of the two oxidation states of the gold catalyst gave any product derived from the intercepted intermediate (the solvent was changed to CDCl3 since the activity of gold(I) is significantly reduced by MeCN).
![[1860-5397-7-90-i6]](/bjoc/content/inline/1860-5397-7-90-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: With substrate 8 the product of the phenol synthesis was exclusively obtained.
Scheme 6: With substrate 8 the product of the phenol synthesis was exclusively obtained.
![[1860-5397-7-90-1]](/bjoc/content/figures/1860-5397-7-90-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Solid-state molecular structure of 10.
Figure 1: Solid-state molecular structure of 10.
Intramolecular ketone as potential trapping reagent
Next we decided to use a carbonyl group as the competing unit. The intermediate G, formed from substrate 16, would offer the option of competition of the phenol synthesis (Scheme 7, pathway a) to yield 18, and reaction with the second carbonyl group (Scheme 7, pathway b). The latter would form intermediate H, which could then either afford product 17 via intramolecular 1,3-dipolar cycloaddition with the olefin, or could form the diene 19 by proton migration.
![[1860-5397-7-90-i7]](/bjoc/content/inline/1860-5397-7-90-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Potential products of an intramolecular trapping experiment with substrate 16.
Scheme 7: Potential products of an intramolecular trapping experiment with substrate 16.
The synthesis of 16 was only possible by a 9-step sequence (Scheme 8). The starting point was a Claisen condensation of ester 20 and tert-butyl acetate (21) in the presence of lithium hexamethyldisilazide as the base. Ketoester 22 was obtained in 56% yield, however, the two-fold addition of 21 could not be suppressed completely and 14% of the corresponding tertiary alcohol 30 was also obtained. Reduction of the ketone 22 with sodium borohydride and protection of the alcohol 23 with tert-butyldimethylsilylchloride delivered 24 in excellent yield. Reduction of the ester group with diisobutylaluminiumhydride gave aldehyde 25. The addition of lithiated trimethylsilylacetylene provided the propargylic alcohol 26 and reaction with 15 under Mitsunobu conditions yielded 27. Deprotection of the alkyne 27 and the silyl ether 28, followed by the oxidation of the resulting alcohol 29 finally led to 16. It was not possible to remove both silyl groups simultaneously with TBAF, longer reaction times which would be necessary for the deprotection of the hydroxy group led to decomposition of the substrate. At 0 °C and with a very short reaction time, the alkyne was deprotected selectively. Selective deprotection of the alcohol was then possible with a mixture of acetic acid/water/THF. Another route, in which the alcohol function was deprotected first, then oxidized, followed by removal of the trimethylsilyl group from the alkyne also failed. Thus treatment of 27 with acetic acid in aqueous THF gave the desired alcohol 31in quantitative yield. However, whilst Ley oxidation [42] on the small-scale delivered ketone 32 in yields of up to 80%, on a larger scale the yield of 32 dropped dramatically to 28% and was accompanied by two side-products, 33 and 5. The latter are formed by an elimination reaction of the amide in 32. Furthermore, it was not possible to deprotect ketone 32 due to rapid decomposition.
![[1860-5397-7-90-i8]](/bjoc/content/inline/1860-5397-7-90-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
One of the diastereoisomers of 28 was identified as the anti-product 28a by an X-ray crystal structure analysis (Figure 2).
![[1860-5397-7-90-2]](/bjoc/content/figures/1860-5397-7-90-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Solid-state molecular structure of 28a.
Figure 2: Solid-state molecular structure of 28a.
The conversion of 16 with 5 mol % AuCl3 proceeded fast and gave exclusively phenol 18. No other products could be detected (Scheme 9).
![[1860-5397-7-90-i9]](/bjoc/content/inline/1860-5397-7-90-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: With substrate 16 the product of the phenol synthesis is obtained exclusively.
Scheme 9: With substrate 16 the product of the phenol synthesis is obtained exclusively.
The two gold(III) complexes 34 [43] and 35 [37] as well as the dinuclear gold(I) complex 36 [44] gave the same result (Figure 3). When the catalyst was changed to platinum(II) chloride in acetone, a complex mixture of inseparable products was obtained.
![[1860-5397-7-90-3]](/bjoc/content/figures/1860-5397-7-90-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Since the two diastereoisomers 28a and 28b with the propargylic stereocenters were separable, we investigated the gold-catalyzed conversion of the pure isomers. From the NMR spectra taken during the conversion (Figure 4), it could be clearly seen that no epimerization of the propargylic position occurred. In addition to the selective transformation to the phenols 37a and 37b as the main reaction products, partial removal of the TBS group was observed (38, Figure 5).
![[1860-5397-7-90-4]](/bjoc/content/figures/1860-5397-7-90-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR spectra of the separated diastereoisomers of the substrates for catalysis 28 (left) and of the products 37 (right, the small signals are due to the deprotected compounds 38).
Figure 4: 1H NMR spectra of the separated diastereoisomers of the substrates for catalysis 28 (left) and of t...
![[1860-5397-7-90-5]](/bjoc/content/figures/1860-5397-7-90-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Structure of the desilylation product 38.
Figure 5: Structure of the desilylation product 38.
Intramolecular alcohol as potential trapping reagent
For the interception of intermediate A we also considered the option of an intramolecular hydroxy nucleophile, compound 39 (Scheme 10) would represent this type of substrate. The intermediate I would be an analogue of A. Instead of the phenol synthesis to yield 40, an intramolecular nucleophilic attack at the activated three-membered ring could form intermediate J, which, after protodeauration, would provide ketal 41.
![[1860-5397-7-90-i10]](/bjoc/content/inline/1860-5397-7-90-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Potential products of an intramolecular trapping experiment with substrate 39.
Scheme 10: Potential products of an intramolecular trapping experiment with substrate 39.
The synthesis of 39 was readily accomplished by the addition of lithiated sylvan 42 to the PMB-protected aldehyde 43 (Scheme 11) [45]. The resulting furfuryl alcohol 44 was then propargylated to give 45. The deprotection was however, problematic. Treatment of the latter with cerium ammonium nitrate led to decomposition. Only with DDQ was the desired alcohol 39 obtained in moderate yield.
![[1860-5397-7-90-i11]](/bjoc/content/inline/1860-5397-7-90-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of the substrate 39.
Scheme 11: Synthesis of the substrate 39.
The conversion of 39, catalyzed by AuCl3 in CDCl3, again only produced the expected phenol 40 (Scheme 12). Not unexpectedly, the PMB-protected alcohol 45 was similarly converted to 46. PtCl2 did not lead to a change in selectivity.
![[1860-5397-7-90-i12]](/bjoc/content/inline/1860-5397-7-90-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: With substrate 39 and 45 exclusively the product of the phenol synthesis is obtained.
Scheme 12: With substrate 39 and 45 exclusively the product of the phenol synthesis is obtained.
Conclusion
The complete failure of both the inter- and the intramolecular trapping experiments shows that the gold-catalyzed phenol synthesis follows a reaction pathway low in energy. These observations also nicely explain the high functional group tolerance, for example, towards olefins and alcohols.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of synthesized compounds. | ||
| Format: PDF | Size: 503.3 KB | Download |
References
-
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem. 2006, 118, 8064–8105. doi:10.1002/ange.200602454
Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454
Return to citation in text: [1] -
Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592
Return to citation in text: [1] [2] -
Fürstner, A.; Davies, P. W. Angew. Chem. 2007, 119, 3478–3519. doi:10.1002/ange.200604335
Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335
Return to citation in text: [1] [2] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Muzart, J. Tetrahedron 2008, 64, 5815–5849. doi:10.1016/j.tet.2008.04.018
Return to citation in text: [1] -
Shen, H. C. Tetrahedron 2008, 64, 7847–7870. doi:10.1016/j.tet.2008.05.082
Return to citation in text: [1] -
Hashmi, A. S. K.; Bührle, M. Aldrichimica Acta 2010, 43, 27–33.
Return to citation in text: [1] -
Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088
Return to citation in text: [1] -
Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem. 2000, 112, 2382–2385. doi:10.1002/1521-3757(20000703)112:13<2382::AID-ANGE2382>3.0.CO;2-R
Angew. Chem., Int. Ed. 2000, 39, 2285–2288. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F
Return to citation in text: [1] -
Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d
Return to citation in text: [1] [2] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j
Return to citation in text: [1] -
Hashmi, A. S. K. Angew. Chem. 2010, 122, 5360–5369. doi:10.1002/ange.200907078
Angew. Chem., Int. Ed. 2010, 49, 5232–5241. doi:10.1002/anie.200907078
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Jiménez-Núñez, E.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 5916–5923. doi:10.1002/chem.200600174
Return to citation in text: [1] -
Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Aldrichimica Acta 2010, 43, 37–46.
Return to citation in text: [1] -
Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075–6089. doi:10.1002/ejoc.200900790
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M.; Weyrauch, J. P.; Wölfle, M.; Frey, W.; Bats, J. W. Angew. Chem. 2005, 117, 2858–2861. doi:10.1002/ange.200462672
Angew. Chem., Int. Ed. 2005, 44, 2798–2801. doi:10.1002/anie.200462672
Return to citation in text: [1] -
Hashmi, A. S. K.; Kurpejović, E.; Wölfle, M.; Frey, W.; Bats, J. W. Adv. Synth. Catal. 2007, 349, 1743–1750. doi:10.1002/adsc.200600653
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M.; Siehl, H.-U.; Tanaka, M.; Bats, J. W.; Frey, W. Chem.–Eur. J. 2008, 14, 3703–3708. doi:10.1002/chem.200701795
Return to citation in text: [1] -
Martín-Matute, B.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem. 2001, 113, 4890–4893. doi:10.1002/1521-3757(20011217)113:24<4890::AID-ANGE4890>3.0.CO;2-V
Angew. Chem., Int. Ed. 2001, 40, 4754–4757. doi:10.1002/1521-3773(20011217)40:24<4754::AID-ANIE4754>3.0.CO;2-9
Return to citation in text: [1] -
Martín-Matute, B.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2003, 125, 5757–5766. doi:10.1021/ja029125p
Return to citation in text: [1] -
Hashmi, A. S. K.; Wölfle, M.; Ata, F.; Hamzic, M.; Salathé, R.; Frey, W. Adv. Synth. Catal. 2006, 348, 2501–2508. doi:10.1002/adsc.200600367
Return to citation in text: [1] -
Hashmi, A. S. K. Pure Appl. Chem. 2010, 82, 1517–1528. doi:10.1351/PAC-CON-09-10-34
Return to citation in text: [1] [2] -
Miki, K.; Ohe, K.; Uemura, S. Tetrahedron Lett. 2003, 44, 2019–2022. doi:10.1016/S0040-4039(03)00219-3
Return to citation in text: [1] -
Miki, K.; Ohe, K.; Uemura, S. J. Org. Chem. 2003, 68, 8505–8513. doi:10.1021/jo034841a
Return to citation in text: [1] -
Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 18002–18003. doi:10.1021/ja0552500
Return to citation in text: [1] -
Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. J. Am. Chem. Soc. 2004, 126, 8654–8655. doi:10.1021/ja048094q
Return to citation in text: [1] -
Fürstner, A.; Hannen, P. Chem. Commun. 2004, 2546–2547. doi:10.1039/b412354a
Return to citation in text: [1] -
Fürstner, A.; Hannen, P. Chem.–Eur. J. 2006, 12, 3006–3019. doi:10.1002/chem.200501299
Return to citation in text: [1] -
Fehr, C.; Galindo, J. Angew. Chem. 2006, 118, 2967–2970. doi:10.1002/ange.200504543
Angew. Chem., Int. Ed. 2006, 45, 2901–2904. doi:10.1002/anie.200504543
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Muňoz, M. P.; Jiménez-Núñez, E.; Buñuel, E.; Cárdenas, D. J.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 1694–1702. doi:10.1002/chem.200501089
Return to citation in text: [1] -
López, S.; Herrero-Gómez, E.; Pérez-Galán, P.; Nieto-Oberhuber, C.; Echavarren, A. M. Angew. Chem. 2006, 118, 6175–6178. doi:10.1002/ange.200602448
Angew. Chem., Int. Ed. 2006, 45, 6029–6032. doi:10.1002/anie.200602448
Return to citation in text: [1] -
Padwa, A.; Curtis, E. A.; Sandanayaka, V. P. J. Org. Chem. 1996, 61, 73–81. doi:10.1021/jo951371e
Return to citation in text: [1] -
Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2005, 127, 6962–6963. doi:10.1021/ja051110e
Return to citation in text: [1] -
Rubottom, G. M.; Kim, C. J. Org. Chem. 1983, 48, 1550–1552. doi:10.1021/jo00157a038
Return to citation in text: [1] [2] -
Hibino, S.; Sugino, E.; Adachi, Y.; Nomi, K.; Sato, K.; Fukumoto, K. Heterocycles 1989, 28, 275–282. doi:10.3987/COM-88-S12
Return to citation in text: [1] -
Mitsunobu, O.; Kato, K.; Tomari, M. Tetrahedron 1970, 26, 5731–5736. doi:10.1016/0040-4020(70)80009-6
Return to citation in text: [1] -
Carrettin, S.; Blanco, M. C.; Corma, A.; Hashmi, A. S. K. Adv. Synth. Catal. 2006, 348, 1283–1288. doi:10.1002/adsc.200606099
Return to citation in text: [1] -
Blanco, M. C. AURICAT EU-RTN final report, Universität Stuttgart, 2006.
Return to citation in text: [1] -
Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Synthesis 1994, 639–666. doi:10.1055/s-1994-25538
Return to citation in text: [1] -
Canovese, L.; Cattalini, L.; Marangoni, G.; Tobe, M. L. J. Chem. Soc., Dalton Trans. 1985, 731–735. doi:10.1039/DT9850000731
Return to citation in text: [1] -
Hashmi, A. S. K.; Blanco, M. C.; Kurpejovic, E.; Frey, W.; Bats, J. W. Adv. Synth. Catal. 2006, 348, 709–713. doi:10.1002/adsc.200606012
Return to citation in text: [1] -
Herb, C.; Maier, M. E. J. Org. Chem. 2003, 68, 8129–8135. doi:10.1021/jo035054g
Return to citation in text: [1]
| 1. |
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem. 2006, 118, 8064–8105. doi:10.1002/ange.200602454
Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454 |
| 2. | Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592 |
| 3. |
Fürstner, A.; Davies, P. W. Angew. Chem. 2007, 119, 3478–3519. doi:10.1002/ange.200604335
Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 4. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c |
| 5. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 6. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 7. | Muzart, J. Tetrahedron 2008, 64, 5815–5849. doi:10.1016/j.tet.2008.04.018 |
| 8. | Shen, H. C. Tetrahedron 2008, 64, 7847–7870. doi:10.1016/j.tet.2008.05.082 |
| 9. | Hashmi, A. S. K.; Bührle, M. Aldrichimica Acta 2010, 43, 27–33. |
| 10. | Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088 |
| 2. | Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592 |
| 3. |
Fürstner, A.; Davies, P. W. Angew. Chem. 2007, 119, 3478–3519. doi:10.1002/ange.200604335
Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 15. |
Hashmi, A. S. K. Angew. Chem. 2010, 122, 5360–5369. doi:10.1002/ange.200907078
Angew. Chem., Int. Ed. 2010, 49, 5232–5241. doi:10.1002/anie.200907078 |
| 36. | Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2005, 127, 6962–6963. doi:10.1021/ja051110e |
| 12. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d |
| 37. | Rubottom, G. M.; Kim, C. J. Org. Chem. 1983, 48, 1550–1552. doi:10.1021/jo00157a038 |
| 13. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 14. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j |
| 34. |
López, S.; Herrero-Gómez, E.; Pérez-Galán, P.; Nieto-Oberhuber, C.; Echavarren, A. M. Angew. Chem. 2006, 118, 6175–6178. doi:10.1002/ange.200602448
Angew. Chem., Int. Ed. 2006, 45, 6029–6032. doi:10.1002/anie.200602448 |
| 11. |
Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem. 2000, 112, 2382–2385. doi:10.1002/1521-3757(20000703)112:13<2382::AID-ANGE2382>3.0.CO;2-R
Angew. Chem., Int. Ed. 2000, 39, 2285–2288. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F |
| 12. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d |
| 35. | Padwa, A.; Curtis, E. A.; Sandanayaka, V. P. J. Org. Chem. 1996, 61, 73–81. doi:10.1021/jo951371e |
| 21. | Hashmi, A. S. K.; Rudolph, M.; Siehl, H.-U.; Tanaka, M.; Bats, J. W.; Frey, W. Chem.–Eur. J. 2008, 14, 3703–3708. doi:10.1002/chem.200701795 |
| 25. | Hashmi, A. S. K. Pure Appl. Chem. 2010, 82, 1517–1528. doi:10.1351/PAC-CON-09-10-34 |
| 20. | Hashmi, A. S. K.; Kurpejović, E.; Wölfle, M.; Frey, W.; Bats, J. W. Adv. Synth. Catal. 2007, 349, 1743–1750. doi:10.1002/adsc.200600653 |
| 26. | Miki, K.; Ohe, K.; Uemura, S. Tetrahedron Lett. 2003, 44, 2019–2022. doi:10.1016/S0040-4039(03)00219-3 |
| 27. | Miki, K.; Ohe, K.; Uemura, S. J. Org. Chem. 2003, 68, 8505–8513. doi:10.1021/jo034841a |
| 28. | Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 18002–18003. doi:10.1021/ja0552500 |
| 29. | Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. J. Am. Chem. Soc. 2004, 126, 8654–8655. doi:10.1021/ja048094q |
| 30. | Fürstner, A.; Hannen, P. Chem. Commun. 2004, 2546–2547. doi:10.1039/b412354a |
| 31. | Fürstner, A.; Hannen, P. Chem.–Eur. J. 2006, 12, 3006–3019. doi:10.1002/chem.200501299 |
| 32. |
Fehr, C.; Galindo, J. Angew. Chem. 2006, 118, 2967–2970. doi:10.1002/ange.200504543
Angew. Chem., Int. Ed. 2006, 45, 2901–2904. doi:10.1002/anie.200504543 |
| 33. | Nieto-Oberhuber, C.; López, S.; Muňoz, M. P.; Jiménez-Núñez, E.; Buñuel, E.; Cárdenas, D. J.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 1694–1702. doi:10.1002/chem.200501089 |
| 19. |
Hashmi, A. S. K.; Rudolph, M.; Weyrauch, J. P.; Wölfle, M.; Frey, W.; Bats, J. W. Angew. Chem. 2005, 117, 2858–2861. doi:10.1002/ange.200462672
Angew. Chem., Int. Ed. 2005, 44, 2798–2801. doi:10.1002/anie.200462672 |
| 16. | Nieto-Oberhuber, C.; López, S.; Jiménez-Núñez, E.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 5916–5923. doi:10.1002/chem.200600174 |
| 17. | Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Aldrichimica Acta 2010, 43, 37–46. |
| 18. | Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075–6089. doi:10.1002/ejoc.200900790 |
| 22. |
Martín-Matute, B.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem. 2001, 113, 4890–4893. doi:10.1002/1521-3757(20011217)113:24<4890::AID-ANGE4890>3.0.CO;2-V
Angew. Chem., Int. Ed. 2001, 40, 4754–4757. doi:10.1002/1521-3773(20011217)40:24<4754::AID-ANIE4754>3.0.CO;2-9 |
| 23. | Martín-Matute, B.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2003, 125, 5757–5766. doi:10.1021/ja029125p |
| 24. | Hashmi, A. S. K.; Wölfle, M.; Ata, F.; Hamzic, M.; Salathé, R.; Frey, W. Adv. Synth. Catal. 2006, 348, 2501–2508. doi:10.1002/adsc.200600367 |
| 25. | Hashmi, A. S. K. Pure Appl. Chem. 2010, 82, 1517–1528. doi:10.1351/PAC-CON-09-10-34 |
| 39. | Mitsunobu, O.; Kato, K.; Tomari, M. Tetrahedron 1970, 26, 5731–5736. doi:10.1016/0040-4020(70)80009-6 |
| 38. | Hibino, S.; Sugino, E.; Adachi, Y.; Nomi, K.; Sato, K.; Fukumoto, K. Heterocycles 1989, 28, 275–282. doi:10.3987/COM-88-S12 |
| 40. | Carrettin, S.; Blanco, M. C.; Corma, A.; Hashmi, A. S. K. Adv. Synth. Catal. 2006, 348, 1283–1288. doi:10.1002/adsc.200606099 |
| 44. | Hashmi, A. S. K.; Blanco, M. C.; Kurpejovic, E.; Frey, W.; Bats, J. W. Adv. Synth. Catal. 2006, 348, 709–713. doi:10.1002/adsc.200606012 |
| 45. | Herb, C.; Maier, M. E. J. Org. Chem. 2003, 68, 8129–8135. doi:10.1021/jo035054g |
| 43. | Canovese, L.; Cattalini, L.; Marangoni, G.; Tobe, M. L. J. Chem. Soc., Dalton Trans. 1985, 731–735. doi:10.1039/DT9850000731 |
| 37. | Rubottom, G. M.; Kim, C. J. Org. Chem. 1983, 48, 1550–1552. doi:10.1021/jo00157a038 |
| 42. | Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Synthesis 1994, 639–666. doi:10.1055/s-1994-25538 |
© 2011 Rudolph et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)