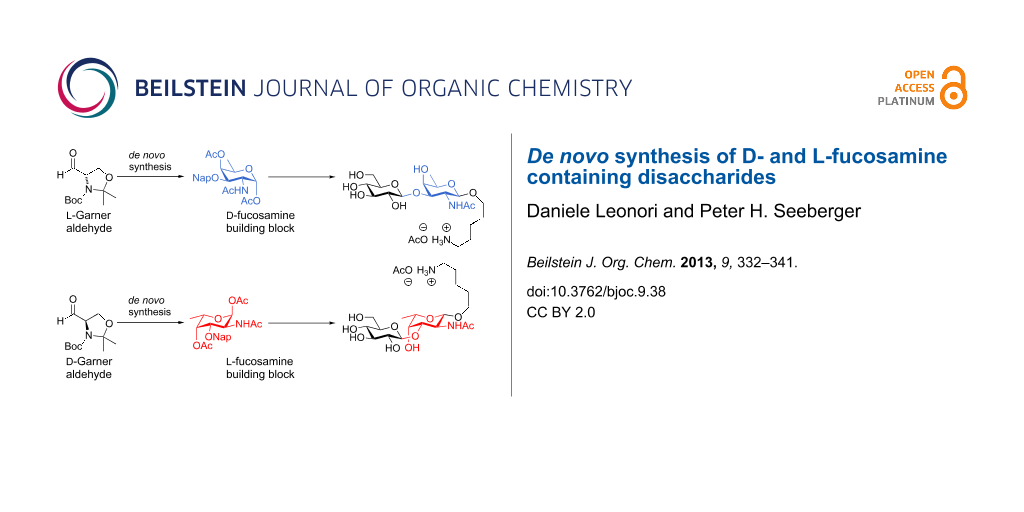
Abstract
The availability of rare monosaccharides that cannot be isolated from natural sources is currently limiting the access to the synthesis and the biological evaluation of complex bacterial cell-surface glycans. Here, we report the synthesis of D- and L-fucosamine building blocks by a de novo approach from L- and D-Garner aldehydes. These differentially protected monosaccharide building blocks were utilized to prepare disaccharides present on the surface of Pseudomonas aeruginosa bacteria.

Graphical Abstract
Introduction
Protein functions are directly influenced by their glycosylation patterns [1,2]. Therefore, an understanding of protein glycosylation is of utmost importance in order to develop new therapeutics [3-6]. The ability of bacteria to colonize human hosts and cause diseases is directly influenced by their capacity to synthesize glycoproteins and express them on cell surfaces. This evidence makes it particularly relevant especially for the identification of novel antibacterial agents as well as vaccines [7-9]. Those bacterial glycans often contain unusual monosaccharides that are not present in the human body. An immune response against these cell-surface glycans is the basis for the development of new vaccine candidates against bacterial infections [10-13].
Our efforts were directed to the development of new vaccine candidates [14-16] to prevent bacterial infections, including glycans of the highly pathogenic bacteria Pseudomonas aeruginosa. P. aeruginosa is a nosocomial pathogen that is involved in ventilator-associated pneumonia and has become resistant to many antimicrobials. The somatic pili of P. aeruginosa are a major virulence factor playing a pivotal role in the adherence and invasiveness of the bacterium. In 2001, the P. aeruginosa pilin O-linked glycans were found to be linear trisaccharides that are covalently attached to serine (Figure 1). The O-glycans contain a D-fucosamine residue at the protein-binding site. This unusual monosaccharide is not present in eukaryotes, and therefore may be used to stimulate an antibacterial response in the host organism [17-23]. Access to differentially protected D- and L-fucosamine building blocks, which can be used in preparing the corresponding glycans, is instrumental for the evaluation of oligosaccharide-based vaccine candidates against this bacterium [24-28].
![[1860-5397-9-38-1]](/bjoc/content/figures/1860-5397-9-38-1.svg?scale=1.4&max-width=1024&background=FFFFFF)
Figure 1: Structure of some O-linked glycans found on the cell surface of P. aeruginosa.
Figure 1: Structure of some O-linked glycans found on the cell surface of P. aeruginosa.
The synthesis of fucosamine building blocks has been reported in the literature, but it is highly affected by long synthetic sequences, extensive protecting group manipulations and expensive starting materials [29-33]. The de novo synthesis of rare sugars [34-43] provides an attractive alternative for rapid access to the required building blocks, but this approach has not been reported for D- or L-fucosamine [29-33,44-46].
Here, a full account of the de novo synthesis of differentially protected D- and L-fucosamine building blocks is described following a recent preliminary communication [47]. The building blocks prepared by de novo synthesis were used in the assembly of two disaccharides that are found on P. aeruginosa.
Results and Discussion
De novo synthesis of D- and L-fucosamine building blocks
Our retrosynthetic analysis of D-fucosamine envisioned the installation of the syn-1,2-diol unit by osmium-catalysed dihydroxylation of allylic ether A. It was anticipated that the conformation adopted by the molecule would allow for the formation of the required anti relationship between C3 and C4 hydroxy groups. A in turn would be accessed by the addition of a carbon nucleophile to L-Garner aldehyde L-1 (Scheme 1) [48]. The fact that D- and L-Garner aldehydes are commercially available greatly facilitates the synthesis of both D- and L-fucosamine building blocks. The fucosamine residues are usually further elongated at the C3 position in P. aeruginosa requiring two orthogonal protecting groups (PGs) at C3 and C4 of the building block in order to differentiate the two hydroxy groups at a later stage.
![[1860-5397-9-38-i1]](/bjoc/content/inline/1860-5397-9-38-i1.svg?scale=1.4&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of D- and L-fucosamine building blocks.
Scheme 1: Retrosynthetic analysis of D- and L-fucosamine building blocks.
The three carbon C4–C6 fragment of D-fucosamine was introduced by chelation-controlled addition [49,50] of commercially available propynylmagnesium bromide to L-1 (Scheme 2A) [51,52] The following E-selective alkyne reduction was accomplished by using diluted RedAl in Et2O [53]. This reaction proved to be highly dependent on the quality of the aluminium reagent obtained from commercial sources. Inspired by the work from the Trost group [54,55], an alternative reduction protocol based on a Ru-catalyzed hydrosilylation–protodesilylation sequence was pursued (Scheme 2B). Thus, exposure of alkyne 2 to Cp*Ru(CH3CN)3PF6 catalyst in the presence of the appropriate trialkylsilane gave the desired silylated products 7a–c (Table 1). The alkene geometry was confirmed as Z by nuclear Overauser effect (nOe). Treatment of 7a–c with TBAF and CuI delivered 3 in high yields. Generally, BnMe2SiH has been found to be optimal for these transformations [54,55]. In our hands however, the best results were achieved using the cheaper Et3SiH (Table 1). With a gram amount of 3 in hand, the C3 hydroxy group was protected and the acetonide removed by treatment with p-TSA. Oxidation of primary aminoalcohols 5a–b with the Dess–Martin reagent [56] yielded the desired aldehydes 6a–b, in five steps from commercially available starting materials. The two different O-protecting groups, namely naphthyl ether (Nap) [57-59] and benzoate ester (Bz), were introduced to gain access to two sets of electronically different and orthogonally protected derivatives (Scheme 2A).
![[1860-5397-9-38-i2]](/bjoc/content/inline/1860-5397-9-38-i2.svg?scale=1.4&max-width=1024&background=FFFFFF)
Scheme 2: (A) Synthesis of aldehydes 6a and 6b. (B) Alkyne reduction by hydrosilylation–protodesilylation sequence (see Table 1).
Scheme 2: (A) Synthesis of aldehydes 6a and 6b. (B) Alkyne reduction by hydrosilylation–protodesilylation seq...
Dihydroxylation of aldehyde 6a under standard Upjohn conditions gave, after peracetylation with Ac2O, D-fucosamine building block D-8a in 81% yield and 5:1 dr (anti/syn, diastereomers separable by column chromatography) (Scheme 3) [60-62]. The formation of the desired C3–C4–C5 syn,syn cyclic product was confirmed based on observation of a 3JH3–H4 coupling of 3.5 Hz. When the same sequence of dihydroxylation–peracetylation was performed on Bz-substituted aldehyde 6b, compound D-8b was formed in 71% as a single diastereomer (Scheme 3). This product was crystallized from n-hexane/EtOAc solvent mixture, and the stereochemical assignment was confirmed by X-ray analysis (data shown in Supporting Information File 1).
![[1860-5397-9-38-i3]](/bjoc/content/inline/1860-5397-9-38-i3.svg?scale=1.4&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of D-fucosamine building blocks 8a and 8b.
Scheme 3: Synthesis of D-fucosamine building blocks 8a and 8b.
During the optimization of the synthetic sequence we explored potentially more economic oxidation protocols to convert the primary alcohol 5a to the D-fucosamine building block. Since α-amino protected aldehydes easily undergo α-epimerization, oxidation had to proceed under mild conditions in order to avoid the formation of the undesired D-talosamine building block D-9 (Scheme 4).
![[1860-5397-9-38-i4]](/bjoc/content/inline/1860-5397-9-38-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Several oxidation methods were tested (Table 2). All the reactions were performed sequentially without column-chromatographic purification of the aldehyde. Classic Swern oxidation [63] (Table 2, entry 2) gave a 2:1 mixture of C2 epimers demonstrating the acid lability of aldehyde 6a. This ratio in favour of the desired product was improved to 8:1 by switching the base from Et3N to iPr2EtN as reported by Dondoni and co-workers (Table 2, entry 3) [64]. Parrikh–Doering oxidation [65] and TCCA–TEMPO mediated oxidation [66] (Table 2, entries 4 and 6, respectively) were not suitable as considerable amounts of D-talosamine building block were formed. DMP emerged as the reagent of choice for the oxidation of 5a. Interestingly, the dihydroxylation of aldehyde epi-6a resulted in the formation of talosamine D-9 as a single diastereomer while the dihydroxylation of 6a gave D-8a in 5:1 dr (Scheme 3).
Table 2: Oxidation of 5a to D-fucosamine D-8a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-38-i7.svg?max-width=637&scale=1.0)
|
||
| entry | reaction conditions | D-Fuc/D-Tala |
|---|---|---|
| 1b | DMP, CH2Cl2, rt then 1 M Na2S2O3 in NaHCO3, sat. | >20:1 |
| 2 | DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C | 2:1 |
| 3 | DMSO, (COCl)2, iPr2NEt, CH2Cl2, −78 °C | 8:1 |
| 4 | SO3·pyr, DMSO, CH2Cl2, 0 °C | 2:1 |
| 5 | TCCA, TEMPO, CH2Cl2, 0 °C | 3:1 |
aDetermined by 1H NMR analysis of the crude product after dihydroxylation and peracetylation. bReaction run on 1 g scale.
Carrying out the synthetic sequence optimized for the synthesis of D-8a, on D-Garner aldehyde (D-1, commercially available) gave alcohol ent-5a (four steps, two chromatographic purifications) that after the direct oxidation–dihydroxylation–peracetylation protocol yielded the desired L-fucosamine building block L-8a (Scheme 5).
![[1860-5397-9-38-i5]](/bjoc/content/inline/1860-5397-9-38-i5.svg?scale=1.4&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of L-fucosamine building block L-8a from D-Garner aldehyde.
Scheme 5: Synthesis of L-fucosamine building block L-8a from D-Garner aldehyde.
Synthesis of fucosamine-containing disaccharides
To facilitate the use of the bacterial monosaccharides in the glycan microarray platform and their conjugation to carrier proteins, an appropriate linker was required at the anomeric position [67]. Placement of a C3 naphthyl ether anticipated the site for further glycosylation at this position, which typically serves as the connection to the next sugar. Therefore, building block 8a is ideal in terms of orthogonality and chemical synthesis. Hence, we tested the ability of D-8a to undergo anomeric functionalization and to effect glycosylation at the C3 hydroxy group. Due to the presence of N-acetylated D- and L-fucosamine residues in P. aeruginosa O-linked glycans, the strategic N-protecting group was evaluated. Direct glycosylation by using the building block D-8a was not possible. Hence, a direct N-Boc deprotection/N-acetylation sequence afforded D-10 in 84% yield (Scheme 6A). Direct glycosylation of D-10 by using glycosylating agent 11 [67] and BF3·Et2O as the activating agent, yielded the linker-functionalized monosaccharide D-12 as the β-anomer (3JH1–H2 = 8.3 Hz, Scheme 6A). At this point, the C3 naphthyl ether was cleaved under oxidative conditions and the corresponding alcohol was revealed by using a two-step deprotection protocol consisting of ester hydrolysis and hydrogenation. Monosaccharide D-13 was obtained in 85% yield (the β-linkage further confirmed by 1JC1–H1 164.1 Hz, Scheme 6A). Direct glycosylation after DDQ-deprotection was possible and the use of differentially protected glucose building block 15 [68] yielded the desired β-disaccharide 16 in 61% yield over two steps (β anomer, 3JH1–H2 of 7.8 Hz, Scheme 6A). Global deprotection employed saponification, and hydrogenation gave the fully deprotected D-fucosamine containing disaccharide 17 (Scheme 6A).
![[1860-5397-9-38-i6]](/bjoc/content/inline/1860-5397-9-38-i6.svg?scale=1.4&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of D- and L-fucosamine-containing mono- and disaccharides carrying the pentanolamine linker.
Scheme 6: Synthesis of D- and L-fucosamine-containing mono- and disaccharides carrying the pentanolamine link...
When the same synthetic sequence was performed on L-FucNAc building block L-10 the fully deprotected monomer L-13 and disaccharide 19 were obtained in similar yields and selectivities (Scheme 6B). Diagnostic 1JC–H analysis also corroborated the formation of the β-glycosydic linkages (Scheme 6B). Linker-terminated disaccharide 19 represents the terminal unit of the P. aeruginosa serotype O11 O-linked glycan (Figure 1).
Conclusion
The de novo synthesis of differentially protected D- and L-fucosamine building blocks from D- and L-Garner aldehyde is reported. Placement of a naphthyl ether protecting group at the C3 position allows for further elongation by glycosylation. The key oxidation step was optimized to minimize the formation of the unwanted D-talosamine building block D-9. The fucosamine building blocks prepared by de novo synthesis enabled the preparation of monosaccharides and disaccharides for attachment to microarray surfaces. The terminal disaccharide of P. aeruginosa O11 O-linked glycan has been prepared and will be the basis for biological studies involving this pathogen.
Experimental
General experimental details
All reagents were obtained from commercial suppliers and were used without further purification unless otherwise specified. All reactions were conducted under an Ar atmosphere by using standard Schlenk techniques. THF and Et2O were distilled from purple Na/benzophenone diketyl; CH2Cl2, pyridine and BF3·Et2O were distilled from CaH2. Deionized water was obtained from an in-house purification system. The compounds purified by flash chromatography were further concentrated by the removal of residual solvent under high vacuum (<0.2 mbar). 1H NMR and 13C NMR spectra were measured with a Varian 400-MR or Varian 600 spectrometer. The proton signal of residual, nondeuterated solvent (δ 7.26 ppm for CHCl3 or δ 4.79 ppm for HDO) was used as an internal reference for 1H spectra. For 13C spectra, the chemical shifts are reported relative to the δ 77.36 ppm resonance of CDCl3. Coupling constants (J values) are quoted to one decimal place with values in hertz (Hz) and were corrected. Infrared (IR) spectra were recorded as thin films on a Perkin Elmer Spectrum 100 FTIR spectrophotometer. Optical rotations (OR) were measured with a Schmidt & Haensch UniPol L 1000 spectrometer at a concentration (c) expressed in grams per hundred millilitres (g/100 mL). High-resolution mass spectra (HRMS) were recorded with an Agilent 6210 ESI–TOF mass spectrometer at the Freie Universität Berlin, Mass Spectrometry Core Facility. Analytical thin-layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. The TLC plates were visualized with UV light and by staining with Hanessian solution (ceric sulfate and ammonium molybdate in aqueous sulfuric acid) or potassium permanganate solution (potassium permanganate in basic aqueous solution). Column chromatography was performed by using Kieselgel 60 (230–400 mesh) silica gel with a typical 50–100:1 weight ratio of silica gel to crude product. For the preparation and characterization of compounds 2–10, D-12 and D-16 see [14].
L-Fucosamine monosaccharide L-12
Under the same reaction conditions reported in [14], L-10 (75 mg, 0.17 mmol, 1.0 equiv), 11 (86 mg, 0.26 mmol, 1.5 equiv) and BF3·Et2O (33 mL, 0.26 mmol, 1.5 equiv) gave L-12 (74 mg, 61%) as an oil; [α]D20 −63.2 (c 2.0, CHCl3), other data as above.
D-Fucosamine monosaccharide D-13
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-38-i8.svg?max-width=637&scale=0.827274)
A solution of D-12 (56 mg, 80.0 mmol, 1.0 equiv) in CH2Cl2 (0.9 mL) and H2O (90 mL) was treated with DDQ (21 mg, 94.0 mmol, 1.2 equiv) and stirred in the dark at rt for 2 h. Saturated NaHCO3 (2 mL) and CH2Cl2 (5 mL) were added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 3 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. The crude was purified by filtration over a short plug of silica gel eluting with CH2Cl2/MeOH 9:1 (34 mg, 76%) and immediately used for the next step. The monodeprotected compound (18 mg, 32.0 mmol, 1.0 equiv) in MeOH (0.4 mL) and THF (0.4 mL) was treated with KOH (1.8 mg, 32.0 mmol, 1.0 equiv) and stirred at rt for 30 min. H2O (2 mL) was added and the solvents were removed under vacuum. CH2Cl2 (5 mL) was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. The crude product was solubilised in MeOH/H2O/AcOH (2.0:1.0:0.05 mL) and Pd/C (10 mg) was added. The heterogeneous mixture was stirred under an atmosphere of H2 for 24 h. The mixture was filtered over celite to give D-13 (8 mg, 85%) as an amorphous solid; [α]D20 +112.9 (c 1.1, H2O); 1H NMR (400 MHz, D2O) δ 4.38 (d, J = 8.4 Hz, 1H, CHF), 3.83 (dt, J = 10.1, 6.3 Hz, 1H, CHG), 3.79 (dd, J = 10.7, 8.4 Hz, 1H, CHE), 3.75–3.69 (m, 2H, CHC and NH), 3.68 (dd, J = 10.7, 3.4 Hz, 1H, CHD), 3.66 (dt, J = 10.1, 6.3 Hz, 1H, CHG), 3.41 (q, J = 6.4 Hz, 1H, CHB), 2.95 (t, J = 7.6 Hz, 2H, CH2K), 1.99 (s, 3H, CH3), 1.87 (AcOH), 1.71–1.62 (m, 2H, CH2J), 1.59–1.51 (m, 2H, CH2H), 1.39–1.29 (m, 2H, CH2I), 1.23 (d, J = 6.4 Hz, 3H, CH3A); 13C NMR (100 MHz, D2O) δ 101.4 (CHF), 71.0 (CHD), 70.4 (CHC), 70.3 (CHB), 69.8 (CH2G), 52.0 (CHE), 39.2 (CH2K), 28.0 (CH2J), 26.3 (CH2H), 23.1 (CH3), 22.1 (CH3), 21.9 (CH2I), 15.3 (CH3A); LRMS–ESI (m/z): 291.2 [M + H+]; HRMS–ESI (m/z): [M + H+] calcd for C13H27N2O5, 291.1914; found, 291.1919.
L-Fucosamine monosaccharide L-13
Using the same reaction conditions reported for D-13, D-12 (24 mg, 34.0 mmol, 1.0 equiv), DDQ (21 mg, 41.0 mmol, 1.2 equiv), KOH (1.8 mg, 34.0 mmol, 1.0 equiv) and Pd/C (10 mg), gave L-13 (9 mg, 75%) as an amorphous solid; [α]D20 −111.1 (c 1.0, H2O), other data as above.
D-Fucosamine disaccharide 17
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-38-i9.svg?max-width=637&scale=0.827274)
A solution of 16 (21 mg, 19.0 mmol, 1.0 equiv) in MeOH (0.4 mL) and THF (0.4 mL) was treated with KOH (1.0 mg, 19.0 mmol, 1.0 equiv) and stirred at rt for 30 min. H2O (2 mL) was added and the solvents were removed under vacuum. CH2Cl2 (5 mL) was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. The crude product was solubilised in MeOH/H2O/AcOH (2.0:1.0:0.05 mL) and Pd/C (20 mg) was added. The heterogeneous mixture was stirred under an atmosphere of H2 for 24 h. The mixture was filtered over celite to give 17 (9 mg, 91%) as an amorphous solid; [α]D20 +13.5 (c 0.9, H2O); 1H NMR (400 MHz, D2O) δ 4.47 (d, J = 7.9 Hz, 1H, CHG), 4.41 (d, J = 7.9 Hz, 1H, CHF), 3.94 (d, J = 2.7 Hz, 1H, CHC), 3.90 (dd, J = 10.3, 7.9 Hz, 1H, CHE), 3.87–3.77 (m, 3H, CHL & CHM & CHD), 3.70 (q, J = 6.5 Hz, 1H, CHB), 3.68 (dd, J = 12.1, 3.4 Hz, 1H, CHL), 3.54 (dt, J = 10.0, 6.2 Hz, 1H, CHM), 3.44–3.32 (m, 3H, CHI, CHJ and CHK), 3.25 (t, J = 7.9 Hz, 1H, CHH), 2.94 (t, J = 7.6 Hz, 2H, CH2Q), 1.97 (s, 3H, CH3), 1.90 (s, 3H, AcOH), 1.68–1.59 (m, 2H, CH2P), 1.58–1.50 (m, 2H, CH2N), 1.41–1.32 (m, 2H, CH2O), 1.22 (d, J = 6.5 Hz, 3H, CH3A); 13C NMR (100 MHz, D2O) δ 104.2 (CHG), 101.1 (CHF), 80.2 (CHD), 75.5 (CH), 75.4 (CH), 72.7 (CHH), 70.4 (2 × CHB&C), 69.8 (CH2L), 69.2 (CH), 60.3 (CH2M), 50.8 (CHE), 39.1 (CH2Q), 28.0 (CH2N), 26.2 (CH2P), 23.1 (CH3), 22.1 (CH3), 22.0 (CH2O), 15.3 (CH3A); LRMS–ESI (m/z): 453.2 [M + Na+]; HRMS–ESI (m/z): [M + H+] calcd for C19H37N2O10, 453.2443; found, 453.2468.
L-Fucosamine disaccharide 18
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-38-i10.svg?max-width=637&scale=0.827274)
A solution of L-12 (60 mg, 86 mmol, 1.0 equiv) in CH2Cl2 (1 mL) and H2O (0.1 mL) was treated with DDQ (21 mg, 95 mmol, 1.2 equiv) and stirred in the dark at rt for 2 h. Saturated NaHCO3 (2 mL) and CH2Cl2 (5 mL) were added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 3 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. The crude was purified by filtration over a short plug of silica gel eluting with CH2Cl2/MeOH (9:1) and immediately used for the next step. An oven-dried flask was immediately charged with the product and 15 (77 mg, 0.12 mmol, 1.5 equiv) was added. The flask was left under high vacuum for 4 h. After refilling the flask with argon, freshly distilled CH2Cl2 (1.5 mL) was added, and the mixture was cooled to −30 °C. NIS (29 mg, 0.13 mmol, 1.5 equiv) and TfOH (4 mL, 43 mmol, 0.5 equiv) were added. The mixture was stirred at −30 °C for 4 h and then it was allowed to warm to rt overnight. Saturated Na2SO3 (3 mL) and CH2Cl2 (3 mL) were added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica gel, eluting with n-hexane/EtOAc (6:4), gave 18 (45 mg, 48%) as an oil; Rf 0.22 [n-hexane/EtOAc (4:1)]; [α]D20 −5.5 (c 0.3, CHCl3); FTIR (film) νmax (cm−1): 3321, 2925, 2857, 1743, 1716, 1705, 1662, 1368, 1271, 1240, 1071; 1H NMR (400 MHz, CDCl3, rotamers) δ 8.04 (d, 2H, J = 7.3 Hz, 2 × CH), 7.57 (tt, J = 7.4, 1H, 1.2 CH), 7.44 (t, J = 7.7 Hz, 2H, 2 × CH), 7.35–7.22 (m, 17H, 17 × CH), 7.17–7.04 (m, 8H, 8 × CH), 5.88 (br d, J = 6.5 Hz, 0.5H, NH), 5.73 (br d, J = 6.0 Hz, 0.5H, NH), 5.21 (dd, J = 9.1, 8.0 Hz, 1H, CHH), 5.17 (br s, 1H, CHQ), 5.14 (br s, 1H, CHQ), 5.13 (d, J = 3.5 Hz, 1H, CHC), 5.07 (d, J = 8.0 Hz, 1H, CHF), 4.79 (d, J = 10.8 Hz, 1H, CHM), 4.70 (d, J = 11.0 Hz, 1H, CHM), 4.65–4.53 (m, 6H, CHD & CHG & 4 × CHM), 4.52–4.46 (m, 2H, CH2R), 3.86 (t, J = 9.3 Hz, 1H, CHI), 3.82–3.74 (m, 4H, CHJ, CH2L, CHN), 3.67 (q, J = 6.4 Hz, 1H, CHB), 3.50 (br dt, J = 9.6, 2.3 Hz, 1H, CHK), 3.44–3.40 (m, 1H, CHN), 3.23–3.17 (m, 3H, CHE and CH2P), 1.85 and 1.81 (s, 3H, CH3), 1.53–1.50 (m, 3H, 3 × CHO), 1.45 (s, 3H, CH3), 1.29–1.21 (m, 3H, 3 × CHO), 1.08 (d, J = 6.4 Hz, 3H, CH3A); 13C NMR (100 MHz, CDCl3, rotamers) δ 171.2 (C=O), 170.5 (C=O), 165.1 (C=O), 137.9 (2 × C), 137.7 (2 × C), 137.6 (C), 133.2 (2 × CH), 130.1 (2 × CH), 129.5 (C), 128.5 (2 × CH), 128.4 (4 × CH), 128.3 (4 × CH), 128.2 (4 × CH), 127.9 (2 × CH), 127.8 (4 × CH), 127.7 (CH), 127.6 (CH), 127.3 (CH), 127.2 (CH), 127.1 (CH), 99.6 (CH), 98.4 (CH), 82.9 (CH), 77.7 (CH), 75.2 (CH2), 75.0 (CH2), 74.7 (CH), 73.5 (CH), 73.4 (CH2), 72.5 (CH), 69.7 and 69.5 (CH2), 69.1 (CH), 68.9 (CH), 68.7 (CH2), 67.1 (CH2), 54.4 (CH), 50.4 and 50.1 (CH2), 47.1 and 46.1 (CH2), 29.7 and 29.1 (CH2), 27.8 and 27.4 (CH2), 23.5 (CH3), 23.1 (CH2), 19.9 (CH3), 16.4 (CH3); LRMS–ESI (m/z): 1115.4 [M + Na+].
L-Fucosamine disaccharide 19
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-38-i11.svg?max-width=637&scale=0.827274)
A solution of 18 (29 mg, 27.0 mmol, 1.0 equiv) in MeOH (0.6 mL) and THF (0.6 mL) was treated with KOH (0.8 mg, 13.5 mmol, 0.5 equiv) and stirred at rt for 30 min. H2O (2 mL) was added and the solvents were removed under vacuum. CH2Cl2 (5 mL) was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. The crude product was solubilised in MeOH/H2O/AcOH (3.0:1.5:0.06 mL) and Pd/C (30 mg) was added. The heterogeneous mixture was stirred under an atmosphere of H2 for 24 h. The mixture was filtered over celite to give 19 (10 mg, 74%) as an amorphous solid; [α]D20 +59.2 (c 0.9, H2O); 1H NMR (400 MHz, D2O) δ 4.48 (d, J = 7.9 Hz, 1H, CHG), 4.43 (d, J = 7.9 Hz, 1H, CHF), 3.96–3.89 (m, 3H, CHC, CHD and CHL), 3.88–3.80 (m, 2H, CHE and CHM), 3.72 (q, J = 6.5 Hz, 1H, CHB), 3.67 (dd, J = 12.1, 6.5 Hz, 1H, CHL), 3.56 (dt, J = 10.2, 6.2 Hz, 1H, CHM), 3.45 (t, J = 9.5 Hz, 1H, CHI), 3.42–3.46 (m, 1H, CHJ), 3.36–3.31 (m, 1H, CHK), 3.27 (dd, J = 9.5, 7.9 Hz, 1H, CHH), 2.95 (t, J = 7.6 Hz, 2H, CH2Q), 1.99 (s, 3H, CH3), 1.97 (s, 3H, AcOH), 1.69–1.60 (m, 2H, CH2P), 1.59–1.50 (m, 2H, CH2N), 1.42–1.32 (m, 2H, CH2O), 1.26 (d, J = 6.5 Hz, 3H, CH3A); 13C NMR (100 MHz, D2O) δ 177.0 (C=O), 103.9 (CHF), 102.7 (CHG), 80.1 (CHD), 79.5 (CHI), 77.9 (CHJ), 75.3 (CHB), 73.0 (CHH), 72.4 (CH2L), 72.2 (CHK), 70.4 (CHC), 63.5 (CH2M), 52.8 (CHE), 41.7 (CH2Q), 30.5 (CH2N), 28.8 (CH2P), 24.8 (CH3), 24.5 (CH2O), 24.4 (CH3), 17.8 (CH3); LRMS–ESI (m/z): 453.2 [M + H+]; HRMS–ESI (m/z): [M + H+] calcd for C19H37N2O10, 453.244; found, 453.2468.
Supporting Information
| Supporting Information File 1: 1H NMR, COSY, 13C NMR and HSQC spectra and the crystallographic data file for D-8b. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Mariño, K.; Bones, J.; Kattla, J. J.; Rudd, P. M. Nat. Chem. Biol. 2010, 6, 713. doi:10.1038/nchembio.437
Return to citation in text: [1] -
Arnold, J. N.; Wormald, M. R.; Sim, R. B.; Rudd, P. M.; Dwek, R. A. Annu. Rev. Immunol. 2007, 25, 21. doi:10.1146/annurev.immunol.25.022106.141702
Return to citation in text: [1] -
Dube, D. H.; Bertozzi, C. R. Nat. Rev. Drug Discovery 2005, 4, 477. doi:10.1038/nrd1751
Return to citation in text: [1] -
Stallforth, P.; Lepenies, B.; Adibekian, A.; Seeberger, P. H. J. Med. Chem. 2009, 52, 5561. doi:10.1021/jm900819p
Return to citation in text: [1] -
Dube, D. H.; Champasa, K.; Wang, B. Chem. Commun. 2011, 47, 87. doi:10.1039/c0cc01557a
Return to citation in text: [1] -
Boltje, T. J.; Buskas, T.; Boons, G.-J. Nat. Chem. 2009, 1, 611. doi:10.1038/nchem.399
Return to citation in text: [1] -
Bieber, D.; Ramer, S. W.; Wu, C.-Y.; Murray, W. J.; Tobe, T.; Fernandez, R.; Schoolnik, G. K. Science 1998, 280, 2114. doi:10.1126/science.280.5372.2114
Return to citation in text: [1] -
Banerjee, A.; Wang, R.; Supernavage, S. L.; Ghosh, S. K.; Parker, J.; Ganesh, N. F.; Wang, P. G.; Gulati, S.; Rice, P. A. J. Exp. Med. 2002, 196, 147. doi:10.1084/jem.20012022
Return to citation in text: [1] -
Power, P. M.; Ku, S. C.; Rutter, K.; Warren, M. J.; Limnios, E. A.; Tapsall, J. W.; Jennings, M. P. Infect. Immun. 2007, 75, 3202. doi:10.1128/IAI.01501-06
Return to citation in text: [1] -
Takeuchi, K.; Taguchi, F.; Inagaki, Y.; Toyoda, K.; Shiraishi, T.; Ichinose, Y. J. Bacteriol. 2003, 185, 6658. doi:10.1128/JB.185.22.6658-6665.2003
Return to citation in text: [1] -
Horzempa, J.; Carlson, P. E., Jr.; O’Dee, D. M.; Shanks, R. M. Q.; Nau, G. J. BMC Microbiol. 2008, 8, No. 172. doi:10.1186/1471-2180-8-172
Return to citation in text: [1] -
van Sorge, N. M.; Bleumink, N. M. C.; van Vliet, S. J.; Saeland, E.; van der Pol, W.-L.; van Kooyk, Y.; van Putten, J. P. M. Cell. Microbiol. 2009, 11, 1768. doi:10.1111/j.1462-5822.2009.01370.x
Return to citation in text: [1] -
Verma, A.; Arora, S. K.; Kuravi, S. K.; Ramphal, R. Infect. Immun. 2005, 73, 8237. doi:10.1128/IAI.73.12.8237-8246.2005
Return to citation in text: [1] -
Yang, Y.; Martin, C. E.; Seeberger, P. H. Chem. Sci. 2012, 3, 896. doi:10.1039/c1sc00804h
Return to citation in text: [1] [2] [3] -
Martin, C. E.; Weishaupt, M. W.; Seeberger, P. H. Chem. Commun. 2011, 47, 10260. doi:10.1039/c1cc13614c
Return to citation in text: [1] -
Ohara, T.; Adibekian, A.; Esposito, D.; Stallforth, P.; Seeberger, P. H. Chem. Commun. 2010, 46, 4106. doi:10.1039/c000784f
Return to citation in text: [1] -
Smedley, J. G., III; Jewell, E.; Roguskie, J.; Horzempa, J.; Syboldt, A.; Stolz, D. B.; Castric, P. Infect. Immun. 2005, 73, 7922. doi:10.1128/IAI.73.12.7922-7931.2005
Return to citation in text: [1] -
Castric, P.; Cassels, F. J.; Carlson, R. W. J. Biol. Chem. 2001, 276, 26479. doi:10.1074/jbc.M102685200
Return to citation in text: [1] -
Chamot-Rooke, J.; Rousseau, B.; Lanternier, F.; Mikaty, G.; Mairey, E.; Malosse, C.; Bouchoux, G.; Pelicic, V.; Camoin, L.; Nassif, X.; Duménil, G. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14783. doi:10.1073/pnas.0705335104
Return to citation in text: [1] -
Kus, J. V.; Kelly, J.; Tessier, L.; Harvey, H.; Cvitkovitch, D. G.; Burrows, L. L. J. Bacteriol. 2008, 190, 7464. doi:10.1128/JB.01075-08
Return to citation in text: [1] -
Castric, P. Microbiology (Reading, U. K.) 1995, 141, 1247. doi:10.1099/13500872-141-5-1247
Return to citation in text: [1] -
Sugawara, K.; Tsunakawa, M.; Konishi, M.; Kawaguchi, J.; Krishnan, B.; He, C. H.; Clardy, J. J. Org. Chem. 1987, 52, 996. doi:10.1021/jo00382a005
Return to citation in text: [1] -
Myers, A. G.; Liang, J.; Hammond, M.; Harrington, P. M.; Wu, Y.; Kuo, E. Y. J. Am. Chem. Soc. 1998, 120, 5319. doi:10.1021/ja980588y
Return to citation in text: [1] -
Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900. doi:10.1002/anie.200802036
Return to citation in text: [1] -
Bongat, A. F. G.; Demchenko, A. V. Carbohydr. Res. 2007, 342, 374. doi:10.1016/j.carres.2006.10.021
Return to citation in text: [1] -
Buskas, T.; Ingale, S.; Boons, G.-J. Glycobiology 2006, 16, 113R. doi:10.1093/glycob/cwj125
Return to citation in text: [1] -
Codée, J. D. C.; Litjens, R. E. J. N.; van den Bos, L. J.; Overkleeft, H. S.; van der Marel, G. A. Chem. Soc. Rev. 2005, 34, 769. doi:10.1039/b417138c
Return to citation in text: [1] -
Boons, G.-J.; Demchenko, A. V. Chem. Rev. 2000, 100, 4539. doi:10.1021/cr990313g
Return to citation in text: [1] -
Liav, A.; Hildesheim, J.; Zehavi, U.; Sharon, N. Carbohydr. Res. 1974, 33, 217. doi:10.1016/S0008-6215(00)82799-8
Return to citation in text: [1] [2] -
Busca, P.; Martin, O. R. Tetrahedron Lett. 2004, 45, 4433. doi:10.1016/j.tetlet.2004.04.074
Return to citation in text: [1] [2] -
Jones, G. B.; Lin, Y.; Xiao, Z.; Kappen, L.; Goldberg, I. H. Bioorg. Med. Chem. 2007, 15, 784. doi:10.1016/j.bmc.2006.10.052
Return to citation in text: [1] [2] -
Weerapana, E.; Glover, K. J.; Chen, M. M.; Imperiali, B. J. Am. Chem. Soc. 2005, 127, 13766. doi:10.1021/ja054265v
Return to citation in text: [1] [2] -
Amin, M. N.; Ishiwata, A.; Ito, Y. Carbohydr. Res. 2006, 341, 1922. doi:10.1016/j.carres.2006.04.031
Return to citation in text: [1] [2] -
Schmidt, R. R. Pure Appl. Chem. 1987, 59, 415. doi:10.1351/pac198759030415
Return to citation in text: [1] -
Kirschning, A.; Jesberger, M.; Schöning, K.-U. Synthesis 2001, 507. doi:10.1055/s-2001-12342
Return to citation in text: [1] -
Hemeon, I.; Bennet, A. J. Synthesis 2007, 1899. doi:10.1055/s-2007-983723
Return to citation in text: [1] -
Vogel, P. In The Organic Chemistry of Sugars; Levy, D. E.; Fugedi, P., Eds.; Taylor and Francis Group/CRC Press: Boca Raton, FL, 2006; pp 629 ff.
Return to citation in text: [1] -
Voigt, B.; Scheffler, U.; Mahrwald, R. Chem. Commun. 2012, 48, 5304. doi:10.1039/c2cc31541f
Return to citation in text: [1] -
Crich, D.; Navuluri, C. Org. Lett. 2011, 13, 6288. doi:10.1021/ol202773t
Return to citation in text: [1] -
Lorpitthaya, R.; Suryawanshi, S. B.; Wang, S.; Pasunooti, K. K.; Cai, S.; Ma, J.; Liu, X.-W. Angew. Chem., Int. Ed. 2011, 50, 12054. doi:10.1002/anie.201104516
Return to citation in text: [1] -
Northrup, A. B.; MacMillan, D. W. C. Science 2004, 305, 1752. doi:10.1126/science.1101710
Return to citation in text: [1] -
Babu, R. S.; Zhou, M.; O’Doherty, G. A. J. Am. Chem. Soc. 2004, 126, 3428. doi:10.1021/ja039400n
Return to citation in text: [1] -
Enders, D.; Grondal, C. Angew. Chem., Int. Ed. 2005, 44, 1210. doi:10.1002/anie.200462428
Return to citation in text: [1] -
Calin, O.; Pragani, R.; Seeberger, P. H. J. Org. Chem. 2012, 77, 870. doi:10.1021/jo201883k
Return to citation in text: [1] -
Pragani, R.; Seeberger, P. H. J. Am. Chem. Soc. 2011, 133, 102. doi:10.1021/ja1087375
Return to citation in text: [1] -
Pragani, R.; Stallforth, P.; Seeberger, P. H. Org. Lett. 2010, 12, 1624. doi:10.1021/ol1003912
Return to citation in text: [1] -
Leonori, D.; Seeberger, P. H. Org. Lett. 2012, 14, 4954. doi:10.1021/ol3023227
Return to citation in text: [1] -
Garner, P. Tetrahedron Lett. 1984, 25, 5855. doi:10.1016/S0040-4039(01)81703-2
Return to citation in text: [1] -
Cram, D. J.; Kopecky, K. R. J. Am. Chem. Soc. 1959, 81, 2748. doi:10.1021/ja01520a036
Return to citation in text: [1] -
O’Brien, A. G. Tetrahedron 2011, 67, 9639. doi:10.1016/j.tet.2011.10.002
Return to citation in text: [1] -
Zhang, X.; van der Donk, W. A. J. Am. Chem. Soc. 2007, 129, 2212. doi:10.1021/ja067672v
Return to citation in text: [1] -
Garner, P.; Park, J. M.; Malecki, E. J. Org. Chem. 1988, 53, 4395. doi:10.1021/jo00253a039
Return to citation in text: [1] -
Blot, V.; Jacquemard, U.; Reissig, H.-U.; Kleuser, B. Synthesis 2009, 759. doi:10.1055/s-0028-1083373
Return to citation in text: [1] -
Trost, B. M.; Ball, Z. T.; Jöge, T. J. Am. Chem. Soc. 2002, 124, 7922. doi:10.1021/ja026457l
Return to citation in text: [1] [2] -
Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2005, 127, 17644. doi:10.1021/ja0528580
Return to citation in text: [1] [2] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155. doi:10.1021/jo00170a070
Return to citation in text: [1] -
Gaunt, M. J.; Yu, J.; Spencer, J. B. J. Org. Chem. 1998, 63, 4172. doi:10.1021/jo980823v
Return to citation in text: [1] -
Liao, W.; Locke, R. D.; Matta, K. L. Chem. Commun. 2000, 369. doi:10.1039/a908511d
Return to citation in text: [1] -
Lipták, A.; Borbás, A.; Jánossy, L.; Szilágyi, L. Tetrahedron Lett. 2000, 41, 4949. doi:10.1016/S0040-4039(00)00735-8
Return to citation in text: [1] -
Cha, J. K.; Kim, N.-S. Chem. Rev. 1995, 95, 1761. doi:10.1021/cr00038a003
Return to citation in text: [1] -
Cha, J. K.; Christ, W. J.; Kishi, Y. Tetrahedron Lett. 1983, 24, 3943. doi:10.1016/S0040-4039(00)88231-3
Return to citation in text: [1] -
Karjalainen, O. K.; Koskinen, A. M. P. Org. Biomol. Chem. 2011, 9, 1231. doi:10.1039/c0ob00747a
Return to citation in text: [1] -
Mancuso, A. J.; Swern, D. Synthesis 1981, 165. doi:10.1055/s-1981-29377
Return to citation in text: [1] -
Dondoni, A.; Perrone, D. Org. Synth. 2004, Coll. Vol. 10, 320.
Return to citation in text: [1] -
Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505. doi:10.1021/ja00997a067
Return to citation in text: [1] -
De Luca, L.; Giacomelli, G.; Masala, S.; Porcheddu, A. J. Org. Chem. 2003, 68, 4999. doi:10.1021/jo034276b
Return to citation in text: [1] -
Snapper, C. M.; Mond, J. J. J. Immunol. 1996, 157, 2229.
Return to citation in text: [1] [2] -
Plé, K.; Chwalek, M.; Voutquenne-Nazabadioko, L. Tetrahedron 2005, 61, 4347. doi:10.1016/j.tet.2005.03.003
Return to citation in text: [1]
| 14. | Yang, Y.; Martin, C. E.; Seeberger, P. H. Chem. Sci. 2012, 3, 896. doi:10.1039/c1sc00804h |
| 14. | Yang, Y.; Martin, C. E.; Seeberger, P. H. Chem. Sci. 2012, 3, 896. doi:10.1039/c1sc00804h |
| 1. | Mariño, K.; Bones, J.; Kattla, J. J.; Rudd, P. M. Nat. Chem. Biol. 2010, 6, 713. doi:10.1038/nchembio.437 |
| 2. | Arnold, J. N.; Wormald, M. R.; Sim, R. B.; Rudd, P. M.; Dwek, R. A. Annu. Rev. Immunol. 2007, 25, 21. doi:10.1146/annurev.immunol.25.022106.141702 |
| 14. | Yang, Y.; Martin, C. E.; Seeberger, P. H. Chem. Sci. 2012, 3, 896. doi:10.1039/c1sc00804h |
| 15. | Martin, C. E.; Weishaupt, M. W.; Seeberger, P. H. Chem. Commun. 2011, 47, 10260. doi:10.1039/c1cc13614c |
| 16. | Ohara, T.; Adibekian, A.; Esposito, D.; Stallforth, P.; Seeberger, P. H. Chem. Commun. 2010, 46, 4106. doi:10.1039/c000784f |
| 53. | Blot, V.; Jacquemard, U.; Reissig, H.-U.; Kleuser, B. Synthesis 2009, 759. doi:10.1055/s-0028-1083373 |
| 10. | Takeuchi, K.; Taguchi, F.; Inagaki, Y.; Toyoda, K.; Shiraishi, T.; Ichinose, Y. J. Bacteriol. 2003, 185, 6658. doi:10.1128/JB.185.22.6658-6665.2003 |
| 11. | Horzempa, J.; Carlson, P. E., Jr.; O’Dee, D. M.; Shanks, R. M. Q.; Nau, G. J. BMC Microbiol. 2008, 8, No. 172. doi:10.1186/1471-2180-8-172 |
| 12. | van Sorge, N. M.; Bleumink, N. M. C.; van Vliet, S. J.; Saeland, E.; van der Pol, W.-L.; van Kooyk, Y.; van Putten, J. P. M. Cell. Microbiol. 2009, 11, 1768. doi:10.1111/j.1462-5822.2009.01370.x |
| 13. | Verma, A.; Arora, S. K.; Kuravi, S. K.; Ramphal, R. Infect. Immun. 2005, 73, 8237. doi:10.1128/IAI.73.12.8237-8246.2005 |
| 54. | Trost, B. M.; Ball, Z. T.; Jöge, T. J. Am. Chem. Soc. 2002, 124, 7922. doi:10.1021/ja026457l |
| 55. | Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2005, 127, 17644. doi:10.1021/ja0528580 |
| 7. | Bieber, D.; Ramer, S. W.; Wu, C.-Y.; Murray, W. J.; Tobe, T.; Fernandez, R.; Schoolnik, G. K. Science 1998, 280, 2114. doi:10.1126/science.280.5372.2114 |
| 8. | Banerjee, A.; Wang, R.; Supernavage, S. L.; Ghosh, S. K.; Parker, J.; Ganesh, N. F.; Wang, P. G.; Gulati, S.; Rice, P. A. J. Exp. Med. 2002, 196, 147. doi:10.1084/jem.20012022 |
| 9. | Power, P. M.; Ku, S. C.; Rutter, K.; Warren, M. J.; Limnios, E. A.; Tapsall, J. W.; Jennings, M. P. Infect. Immun. 2007, 75, 3202. doi:10.1128/IAI.01501-06 |
| 49. | Cram, D. J.; Kopecky, K. R. J. Am. Chem. Soc. 1959, 81, 2748. doi:10.1021/ja01520a036 |
| 50. | O’Brien, A. G. Tetrahedron 2011, 67, 9639. doi:10.1016/j.tet.2011.10.002 |
| 3. | Dube, D. H.; Bertozzi, C. R. Nat. Rev. Drug Discovery 2005, 4, 477. doi:10.1038/nrd1751 |
| 4. | Stallforth, P.; Lepenies, B.; Adibekian, A.; Seeberger, P. H. J. Med. Chem. 2009, 52, 5561. doi:10.1021/jm900819p |
| 5. | Dube, D. H.; Champasa, K.; Wang, B. Chem. Commun. 2011, 47, 87. doi:10.1039/c0cc01557a |
| 6. | Boltje, T. J.; Buskas, T.; Boons, G.-J. Nat. Chem. 2009, 1, 611. doi:10.1038/nchem.399 |
| 51. | Zhang, X.; van der Donk, W. A. J. Am. Chem. Soc. 2007, 129, 2212. doi:10.1021/ja067672v |
| 52. | Garner, P.; Park, J. M.; Malecki, E. J. Org. Chem. 1988, 53, 4395. doi:10.1021/jo00253a039 |
| 34. | Schmidt, R. R. Pure Appl. Chem. 1987, 59, 415. doi:10.1351/pac198759030415 |
| 35. | Kirschning, A.; Jesberger, M.; Schöning, K.-U. Synthesis 2001, 507. doi:10.1055/s-2001-12342 |
| 36. | Hemeon, I.; Bennet, A. J. Synthesis 2007, 1899. doi:10.1055/s-2007-983723 |
| 37. | Vogel, P. In The Organic Chemistry of Sugars; Levy, D. E.; Fugedi, P., Eds.; Taylor and Francis Group/CRC Press: Boca Raton, FL, 2006; pp 629 ff. |
| 38. | Voigt, B.; Scheffler, U.; Mahrwald, R. Chem. Commun. 2012, 48, 5304. doi:10.1039/c2cc31541f |
| 39. | Crich, D.; Navuluri, C. Org. Lett. 2011, 13, 6288. doi:10.1021/ol202773t |
| 40. | Lorpitthaya, R.; Suryawanshi, S. B.; Wang, S.; Pasunooti, K. K.; Cai, S.; Ma, J.; Liu, X.-W. Angew. Chem., Int. Ed. 2011, 50, 12054. doi:10.1002/anie.201104516 |
| 41. | Northrup, A. B.; MacMillan, D. W. C. Science 2004, 305, 1752. doi:10.1126/science.1101710 |
| 42. | Babu, R. S.; Zhou, M.; O’Doherty, G. A. J. Am. Chem. Soc. 2004, 126, 3428. doi:10.1021/ja039400n |
| 43. | Enders, D.; Grondal, C. Angew. Chem., Int. Ed. 2005, 44, 1210. doi:10.1002/anie.200462428 |
| 47. | Leonori, D.; Seeberger, P. H. Org. Lett. 2012, 14, 4954. doi:10.1021/ol3023227 |
| 29. | Liav, A.; Hildesheim, J.; Zehavi, U.; Sharon, N. Carbohydr. Res. 1974, 33, 217. doi:10.1016/S0008-6215(00)82799-8 |
| 30. | Busca, P.; Martin, O. R. Tetrahedron Lett. 2004, 45, 4433. doi:10.1016/j.tetlet.2004.04.074 |
| 31. | Jones, G. B.; Lin, Y.; Xiao, Z.; Kappen, L.; Goldberg, I. H. Bioorg. Med. Chem. 2007, 15, 784. doi:10.1016/j.bmc.2006.10.052 |
| 32. | Weerapana, E.; Glover, K. J.; Chen, M. M.; Imperiali, B. J. Am. Chem. Soc. 2005, 127, 13766. doi:10.1021/ja054265v |
| 33. | Amin, M. N.; Ishiwata, A.; Ito, Y. Carbohydr. Res. 2006, 341, 1922. doi:10.1016/j.carres.2006.04.031 |
| 48. | Garner, P. Tetrahedron Lett. 1984, 25, 5855. doi:10.1016/S0040-4039(01)81703-2 |
| 24. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900. doi:10.1002/anie.200802036 |
| 25. | Bongat, A. F. G.; Demchenko, A. V. Carbohydr. Res. 2007, 342, 374. doi:10.1016/j.carres.2006.10.021 |
| 26. | Buskas, T.; Ingale, S.; Boons, G.-J. Glycobiology 2006, 16, 113R. doi:10.1093/glycob/cwj125 |
| 27. | Codée, J. D. C.; Litjens, R. E. J. N.; van den Bos, L. J.; Overkleeft, H. S.; van der Marel, G. A. Chem. Soc. Rev. 2005, 34, 769. doi:10.1039/b417138c |
| 28. | Boons, G.-J.; Demchenko, A. V. Chem. Rev. 2000, 100, 4539. doi:10.1021/cr990313g |
| 17. | Smedley, J. G., III; Jewell, E.; Roguskie, J.; Horzempa, J.; Syboldt, A.; Stolz, D. B.; Castric, P. Infect. Immun. 2005, 73, 7922. doi:10.1128/IAI.73.12.7922-7931.2005 |
| 18. | Castric, P.; Cassels, F. J.; Carlson, R. W. J. Biol. Chem. 2001, 276, 26479. doi:10.1074/jbc.M102685200 |
| 19. | Chamot-Rooke, J.; Rousseau, B.; Lanternier, F.; Mikaty, G.; Mairey, E.; Malosse, C.; Bouchoux, G.; Pelicic, V.; Camoin, L.; Nassif, X.; Duménil, G. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14783. doi:10.1073/pnas.0705335104 |
| 20. | Kus, J. V.; Kelly, J.; Tessier, L.; Harvey, H.; Cvitkovitch, D. G.; Burrows, L. L. J. Bacteriol. 2008, 190, 7464. doi:10.1128/JB.01075-08 |
| 21. | Castric, P. Microbiology (Reading, U. K.) 1995, 141, 1247. doi:10.1099/13500872-141-5-1247 |
| 22. | Sugawara, K.; Tsunakawa, M.; Konishi, M.; Kawaguchi, J.; Krishnan, B.; He, C. H.; Clardy, J. J. Org. Chem. 1987, 52, 996. doi:10.1021/jo00382a005 |
| 23. | Myers, A. G.; Liang, J.; Hammond, M.; Harrington, P. M.; Wu, Y.; Kuo, E. Y. J. Am. Chem. Soc. 1998, 120, 5319. doi:10.1021/ja980588y |
| 29. | Liav, A.; Hildesheim, J.; Zehavi, U.; Sharon, N. Carbohydr. Res. 1974, 33, 217. doi:10.1016/S0008-6215(00)82799-8 |
| 30. | Busca, P.; Martin, O. R. Tetrahedron Lett. 2004, 45, 4433. doi:10.1016/j.tetlet.2004.04.074 |
| 31. | Jones, G. B.; Lin, Y.; Xiao, Z.; Kappen, L.; Goldberg, I. H. Bioorg. Med. Chem. 2007, 15, 784. doi:10.1016/j.bmc.2006.10.052 |
| 32. | Weerapana, E.; Glover, K. J.; Chen, M. M.; Imperiali, B. J. Am. Chem. Soc. 2005, 127, 13766. doi:10.1021/ja054265v |
| 33. | Amin, M. N.; Ishiwata, A.; Ito, Y. Carbohydr. Res. 2006, 341, 1922. doi:10.1016/j.carres.2006.04.031 |
| 44. | Calin, O.; Pragani, R.; Seeberger, P. H. J. Org. Chem. 2012, 77, 870. doi:10.1021/jo201883k |
| 45. | Pragani, R.; Seeberger, P. H. J. Am. Chem. Soc. 2011, 133, 102. doi:10.1021/ja1087375 |
| 46. | Pragani, R.; Stallforth, P.; Seeberger, P. H. Org. Lett. 2010, 12, 1624. doi:10.1021/ol1003912 |
| 57. | Gaunt, M. J.; Yu, J.; Spencer, J. B. J. Org. Chem. 1998, 63, 4172. doi:10.1021/jo980823v |
| 58. | Liao, W.; Locke, R. D.; Matta, K. L. Chem. Commun. 2000, 369. doi:10.1039/a908511d |
| 59. | Lipták, A.; Borbás, A.; Jánossy, L.; Szilágyi, L. Tetrahedron Lett. 2000, 41, 4949. doi:10.1016/S0040-4039(00)00735-8 |
| 54. | Trost, B. M.; Ball, Z. T.; Jöge, T. J. Am. Chem. Soc. 2002, 124, 7922. doi:10.1021/ja026457l |
| 55. | Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2005, 127, 17644. doi:10.1021/ja0528580 |
| 56. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155. doi:10.1021/jo00170a070 |
| 68. | Plé, K.; Chwalek, M.; Voutquenne-Nazabadioko, L. Tetrahedron 2005, 61, 4347. doi:10.1016/j.tet.2005.03.003 |
| 66. | De Luca, L.; Giacomelli, G.; Masala, S.; Porcheddu, A. J. Org. Chem. 2003, 68, 4999. doi:10.1021/jo034276b |
| 65. | Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505. doi:10.1021/ja00997a067 |
| 60. | Cha, J. K.; Kim, N.-S. Chem. Rev. 1995, 95, 1761. doi:10.1021/cr00038a003 |
| 61. | Cha, J. K.; Christ, W. J.; Kishi, Y. Tetrahedron Lett. 1983, 24, 3943. doi:10.1016/S0040-4039(00)88231-3 |
| 62. | Karjalainen, O. K.; Koskinen, A. M. P. Org. Biomol. Chem. 2011, 9, 1231. doi:10.1039/c0ob00747a |
© 2013 Leonori and Seeberger; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)