Abstract
A class of novel, easily accessible and air-stable 1-[bis(trifluoromethyl)phosphine]-1’-oxazolinylferrocene ligands has been synthesized from ferrocene. It became apparent that these ligands can be used in the regio- and enantioselective Pd-catalyzed allylic alkylation of monosubstituted allyl substrates in a highly efficient manner. Excellent regio- and enantioselectivity could be obtained for a wide range of substrates.

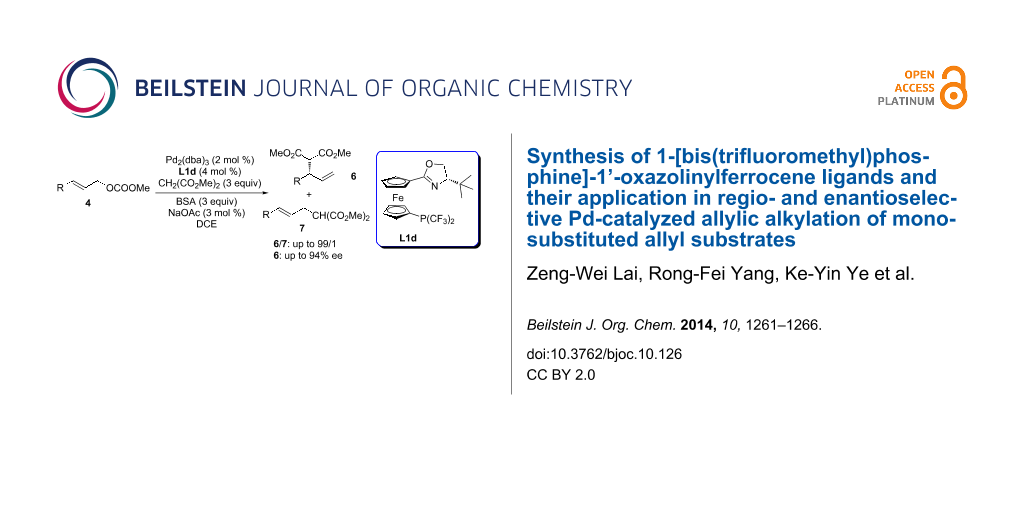
Graphical Abstract
Introduction
The palladium-catalyzed asymmetric allylic alkylation (AAA) reaction is now becoming an efficient method for the construction of carbon–carbon bonds [1-5]. Despite extensive investigation and noteworthy advances in this field, several challenges remain to be solved. For instance, with monosubstituted allyl substrates, the palladium-catalyzed allylic substitution reaction prefers to give linear products rather than the branched ones [6-9] (Scheme 1). Accordingly, the regio- and enantioselective allylic substitution reaction of monosubstituted allylic substrates to preferably obtain the branched products is one of the continuing challenges. To our knowledge, there are several cases in which high levels of both regio- and enantioselectivity have been realized by introducing special ligands [10-34] (Figure 1). Hayashi and coworkers reported a sterically bulky chiral monophosphine ligand (MeO-MOP) could be used for the Pd-catalyzed alkylation of branched monosubstituted allyl acetate favoring the branched products. However, linear products were favored when the linear allyl substrates were employed [23,24]. The chiral oxazoline–phosphite ligands introduced by Pfaltz and coworkers proved to be highly efficient for regio- and enantiocontrol in the Pd-catalyzed allylic alkylation reaction. Excellent results were obtained for the bulky and electron-rich aryl allyl substrates [25-27]. In 2001, Dai, Hou and their coworkers synthesized a new class of 1,1’-ferrocene-based P,N-ligands, namely SiocPhox. The application of these SiocPhox ligands in the Pd-catalyzed allylic substitution led to excellent regio- and enantioselectivities for a wide range of substrates in both allylic alkylation and amination reactions despite of the electronic properties of the allylic substrates [28-33]. Recently, Shen and co-workers reported an elegant synthesis of bis(perfluoroalkyl)phosphine-oxazoline ligands where small but strongly electron-withdrawing substituents were introduced at the phosphorus [34]. 1,2-Ferrocene based P,N-ligands were synthesized and gave excellent regio- and enantioselectivities in the Pd-catalyzed allylic alkylation reactions of monosubstituted allylic substrates. Inspired by these pioneering studies above and as our continuing interests in the transition metal-catalyzed asymmetric allylic alkylation reaction [35-38], we envisaged that the 1-[bis(trifluoromethyl)phosphine]-1’-oxazolinylferrocene ligands, a straightforward combination of the features of SiocPhox and Shen’s ligand, should be highly efficient for the Pd-catalyzed allylic alkylation reactions of monosubstituted allyl substrates. Herein, we report the synthesis of 1-[bis(trifluoromethyl)phosphine]-1’-oxazolinylferrocene ligands and their application in Pd-catalyzed allylic alkylation reactions of monosubstituted allyl substrates with excellent regio- and enantioselectivity.
![[1860-5397-10-126-i1]](/bjoc/content/inline/1860-5397-10-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Transition metal-catalyzed allylic substitution reactions with monosubstituted allyl substrates.
Scheme 1: Transition metal-catalyzed allylic substitution reactions with monosubstituted allyl substrates.
![[1860-5397-10-126-1]](/bjoc/content/figures/1860-5397-10-126-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative ligands developed for the regio- and enantioselective Pd-catalyzed allylic alkylation.
Figure 1: Representative ligands developed for the regio- and enantioselective Pd-catalyzed allylic alkylatio...
Results and Discussion
As depicted in Scheme 2, ligands L1a–L1d were synthesized from known compounds 3, which were obtained from ferrocene in three steps according to the reported procedures [39-41]. The commercially available ferrocene was dilithiated with n-BuLi and then quenched with dibromoterafluoroethane to give dibromoferrocence 1. Treatment of 1 with n-BuLi at −20 °C followed by trapping with CO2 afforded compound 2. Treatment of compound 2 with (COCl)2 and then chiral amino alcohols yielded the amide intermediates which were transformed to their corresponding 1-bromo-1’-oxazolinylferrocenes 3. Eventually, lithium–bromide exchange of 3 with n-BuLi at −78 °C, followed by quenching with P(OPh)3, provided the phosphonite intermediates which were used without further purification. Subsequently, trifluoromethylation provided the ligands L1a–d in moderate yields, upon treatment with Ruppert’s reagent (TMSCF3) and CsF [42-45]. Notably, ligands L1a–d are moisture and air-stable, and their NMR spectra show no change even after being stored over six months under ambient atmosphere.
![[1860-5397-10-126-i2]](/bjoc/content/inline/1860-5397-10-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of 1-[bis(trifluoromethyl)phosphine]-1’-oxazolinylferrocene ligands. Reagents and conditions: (a) (i) n-BuLi, TMEDA, Et2O, rt; (ii) (BrCF2)2, −78 °C. (b) n-BuLi, CO2, THF, −20 °C. (c) (i) (COCl)2, DCM, rt; then TEA, amino alcohol, DCM, rt; (ii) Ph3P, CCl4, TEA, CH3CN, rt. (d) (i) n-BuLi, TMEDA, P(OPh)3, Et2O, −78 °C; (ii) TMSCF3, CsF, Et2O, rt.
Scheme 2: Preparation of 1-[bis(trifluoromethyl)phosphine]-1’-oxazolinylferrocene ligands. Reagents and condi...
To test the suitability of these 1-[bis(trifluoromethyl)phosphine]-1’-oxazolinylferrocene ligands in Pd-catalyzed allylic alkylation reactions, we began our study by choosing methyl cinnamyl carbonate and dimethyl malonate as the model substrates, along with the catalysts derived from Pd2(dba)3 and ligands 1a–d. The results are summarized in Table 1. Ligands L1a–d were screened in the reaction using bis(trimethylsilyl)acetamide (BSA) as the base and LiOAc as the additive. The results suggested that ligands L1a–d were effective for this reaction with full conversion and high selectivities (entries 1–4, Table 1). The catalyst derived from L1d gave the highest selectivities [b/l (branched/linear): 95/5, 82% ee; entry 4, Table 1]. With ligand L1d, different reaction parameters including the Pd precursor and solvent were further optimized. The utilization of [Pd(C3H5)Cl]2 as Pd precursor or DCM as solvent resulted in slightly lower selectivities (entries 5–6, Table 1). Further screening of the additives revealed that NaOAc was the optimal one (b/l: 97/3, 85% ee, entry 7, Table 1). Running the reaction at 0 °C resulted in an increased enantioselectivity (b/l: 96/4, 88% ee, entry 9, Table 1). When the reaction was run at −30 °C, only a trace amount of product was formed. As for the leaving groups of allyl substrates, the cinnamyl acetate could also be tolerated to give a similar level of regio- and enantioselectivity (entry 11, Table 1). The absolute configuration of the product was assigned as (S) by comparing the sign of the optical rotation with that reported in literature [28].
Table 1: Evaluation of the ligands and optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-126-i4.svg?max-width=637&scale=1.0)
|
|||||||||
| entry | 4 or 4a’ | [Pd] | L1 | Additive | Solvent | T (°C) | Yield (%)b | 6a/7ac | ee (%)d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4a | Pd2(dba)3 | L1a | LiOAc | DCE | rt | 95 | 84/16 | 68 |
| 2 | 4a | Pd2(dba)3 | L1b | LiOAc | DCE | rt | 96 | 93/7 | 68 |
| 3 | 4a | Pd2(dba)3 | L1c | LiOAc | DCE | rt | 91 | 85/15 | 80 |
| 4 | 4a | Pd2(dba)3 | L1d | LiOAc | DCE | rt | 95 | 95/5 | 82 |
| 5 | 4a | [Pd(C3H5)Cl]2 | L1d | LiOAc | DCE | rt | 93 | 90/10 | 76 |
| 6 | 4a | Pd2(dba)3 | L1d | LiOAc | DCM | rt | 91 | 83/17 | 76 |
| 7 | 4a | Pd2(dba)3 | L1d | NaOAc | DCE | rt | 90 | 97/3 | 85 |
| 8 | 4a | Pd2(dba)3 | L1d | KOAc | DCE | rt | 91 | 91/9 | 82 |
| 9 | 4a | Pd2(dba)3 | L1d | NaOAc | DCE | 0 | 95 | 96/4 | 88 |
| 10e | 4a | Pd2(dba)3 | L1d | NaOAc | DCE | −30 | trace | nd | nd |
| 11e | 4a’ | Pd2(dba)3 | L1d | NaOAc | DCE | rt | 80 | 95/5 | 87 |
aReagents and conditions: 2.0 mol % Pd2(dba)3, 4.0 mol % ligand, 0.2 mmol allyl substrate, 0.6 mmol dimethyl malonate, 0.6 mmol BSA, 3.0 mol % additive, solvent (2 mL). bIsolated yield after 12 h. cDetermined by 1H NMR of the crude reaction mixture. dDetermined by HPLC. eReaction for 24 h.
Under the optimized reaction conditions (2 mol % of Pd2(dba)3, 4 mol % of L1d, 300 mol % of CH2(COOMe)2, 300 mol % of BSA and 3 mol % of NaOAc in DCE at 0 °C; entry 9, Table 1), the substrate scope was examined to test the generality of the reaction (Table 2). We first compared the reaction of branched substrate 5 with the linear substrate 4a. Nearly identical results were obtained indicating that the reaction proceeds via the formation of the same Pd-π-allyl intermediate. Substrates bearing either an electron-donating group or electron-withdrawing group on the aromatic ring of the aryl allyl carbonates all proceeded smoothly in full conversion within 12 h. In all cases, the reactions gave excellent regioselectivity favoring the formation of the branched products in good to excellent enantioselectivity (b/l: 93/7–99/1, 81–94% ee). It is known that the regioselectivity could be strongly influenced by electronic properties of the allyl substrates and the formation of branched products was dramatically reduced for substrates bearing electron-withdrawing groups [21]. Fortunately, with our catalytic system, substrates bearing electron-withdrawing groups were well tolerated with excellent regioselectivity and preferred formation of the branched products (b/l: 93/7–99/1, entries 8–10, and 13, Table 2). Reactions of sterically hindered 1-naphthyl allyl carbonate, 2-MeO and 2-Me-substituted cinnamyl carbonates occurred smoothly to give excellent regio- and enantioselectivity (b/l: up to 99/1, up to 94% ee, entries 3, 11, 12, Table 2). In addition, heteroaryl allyl carbonates 4e and 4f also gave good regioselectivity with slightly lower enantioselectivity (entries 6 and 7, Table 2). Good regioselectivity (b/l: 81/19) was obtained with 2-buten-3-yl carbonate as a substrate (entry 14, Table 2).
Table 2: Regio- and enantioselective allylic alkylation of monosubstituted allyl substrates.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-126-i5.svg?max-width=637&scale=1.0)
|
|||||
| entry | R | T (°C) | Yield (%)b | 6/7c | ee (%)d |
|---|---|---|---|---|---|
| 1 | 4a, Ph | 0 | 95 | 96/4 | 88 |
| 2 | 5 | 0 | 93 | 95/5 | 87 |
| 3 | 4b, 1-naphthyl | 0 | 95 | 99/1 | 92 |
| 4 | 4c, 4-MeC6H4 | 0 | 93 | 95/5 | 85 |
| 5 | 4d, 4-MeOC6H4 | 0 | 96 | 95/5 | 82 |
| 6 | 4e, 2-thienyl | 0 | 94 | 98/2 | 70 |
| 7 | 4f, 2-furyl | rt | 90 | 83/17 | 65 |
| 8 | 4g, 4-ClC6H4 | 0 | 91 | 96/4 | 83 |
| 9 | 4h, 4-BrC6H4 | 0 | 90 | 99/1 | 83 |
| 10 | 4i, 2-FC6H4 | rt | 90 | 93/7 | 81 |
| 11 | 4j, 2-MeOC6H4 | 0 | 95 | 99/1 | 92 |
| 12 | 4k, 2-MeC6H4 | 0 | 91 | 97/3 | 94 |
| 13 | 4l, 3-ClC6H4 | 0 | 90 | 93/7 | 88 |
| 14e | 4m, methyl | 0 | 96 | 81/19 | ND |
aReagents and conditions: 2.0 mol % Pd2(dba)3, 4.0 mol % L1d, 0.5 mmol allyl substrate, 1.5 mmol dimethyl malonate, 1.5 mmol BSA, 3.0 mol % NaOAc, DCE (5 mL). bIsolated yield after 12 h. cDetermined by 1H NMR of the crude. dDetermined by HPLC. e[Pd(C3H5)Cl]2 as the Pd precursor.
We conducted some control experiments to probe the effect of the bis(trifluoromethyl) group in the ligands (Scheme 3). With ferrocence-based biphenyl phosphine-oxazoline L2 as the ligand, the Pd-catalyzed allylic alkylation of cinnamyl carbonate with dimethyl malonate afforded the linear product as the major product (b/l: 40/60). Whereas the corresponding ligand L1d with two CF3 groups (instead of two phenyl groups) at the P atom improved the regioselectivity significantly (b/l: 96/4). A preliminary explanation was described in Figure 2. In addition to the effect of different metals, there are at least two additional factors controlling the regioselectivity of the allylic alkylation reaction. The steric factor favors path a since the terminal allylic carbon is less hindered. In contrast, when the R group has the ability to stabilize the carbocation, the electronic factor would favor the formation of the branched product (path b). The phosphorus atom has a stronger trans effect comparing with the oxazoline nitrogen, indicating that the carbon trans to phosphorus atom bears more electropositivity [46]. This fact may be responsible for the preferred placement of the substituted allylic carbon in the trans position to the phosphorus atom to better stabilize the electropositivity of the carbon. When the nucleophile attacks the more electropositive substituted allylic carbon terminus, a branched product will be formed. The introduction of the CF3 group on the phosphorus atom further increases the trans influence of the P(CF3)2 moiety and enhances the electronic factor, providing a better branched-product selectivity. Further experimental studies and computational investigation are still needed to confirm this hypothesis.
![[1860-5397-10-126-i3]](/bjoc/content/inline/1860-5397-10-126-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Comparison of the effect of ligands in the reaction.
Scheme 3: Comparison of the effect of ligands in the reaction.
![[1860-5397-10-126-2]](/bjoc/content/figures/1860-5397-10-126-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Preliminary explanation of the regioselectivity.
Figure 2: Preliminary explanation of the regioselectivity.
Conclusion
In summary, we have synthesized a class of novel and efficient bis(trifluoromethyl)phosphine-oxazolines as π-acceptor ligands which have shown good to excellent regio- and enantioselectivity for the Pd-catalyzed asymmetric allylic alkylation reaction of monosubstituted allyl carbonates. Further studies on the synthesis of 1-[bis(perfluoroalkyl)phosphine]-1’-oxazolinylferrocene ligands and their applications in asymmetric catalysis are ongoing in our lab.
Supporting Information
| Supporting Information File 1: Experimental, characterization data and spectra. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395. doi:10.1021/cr9409804
Return to citation in text: [1] -
Pfaltz, A.; Lautens, M. In Comprehensive Asymmetric Catalysis; Pfaltz, A.; Yamamoto, H., Eds.; Springer: New York, 1999; Vol. 2, pp 833 ff.
Return to citation in text: [1] -
Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. doi:10.1021/cr020027w
Return to citation in text: [1] -
Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258. doi:10.1002/anie.200605113
Return to citation in text: [1] -
Förster, S.; Helmchen, G.; Kazmaier, U. In Catalytic Asymmetric Synthesis, 3rd ed.; Ojima, I., Ed.; Wiley: Hoboken, 2010; pp 497 ff.
Return to citation in text: [1] -
Hayashi, T.; Kishi, K.; Yamamoto, A.; Ito, Y. Tetrahedron Lett. 1990, 31, 1743. doi:10.1016/S0040-4039(00)88870-X
Return to citation in text: [1] -
Trost, B. M.; Krische, M. J.; Radinov, R.; Zanoni, G. J. Am. Chem. Soc. 1996, 118, 6297. doi:10.1021/ja960649x
Return to citation in text: [1] -
Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 1998, 120, 9074. doi:10.1021/ja981142k
Return to citation in text: [1] -
Trost, B. M.; Tsui, H.-C.; Toste, F. D. J. Am. Chem. Soc. 2000, 122, 3534. doi:10.1021/ja994326n
Return to citation in text: [1] -
Vyskočil, Š.; Smrčina, M.; Hanuš, V.; Polášek, M.; Kočovský, P. J. Org. Chem. 1998, 63, 7738. doi:10.1021/jo980757x
Return to citation in text: [1] -
Tenaglia, A.; Heumann, A. Angew. Chem., Int. Ed. 1999, 38, 2180. doi:10.1002/(SICI)1521-3773(19990802)38:15<2180::AID-ANIE2180>3.0.CO;2-A
Return to citation in text: [1] -
Nakano, H.; Okuyama, Y.; Hongo, H. Tetrahedron Lett. 2000, 41, 4615. doi:10.1016/S0040-4039(00)00674-2
Return to citation in text: [1] -
Boele, M. D. K.; Kamer, P. C. J.; Lutz, M.; Spek, A. L.; de Vries, J. G.; van Leeuwen, P. W. N. M.; van Strijdonck, G. P. F. Chem.–Eur. J. 2004, 10, 6232. doi:10.1002/chem.200400154
Return to citation in text: [1] -
Pàmies, O.; Diéguez, M.; Claver, C. J. Am. Chem. Soc. 2005, 127, 3646. doi:10.1021/ja0425738
Return to citation in text: [1] -
Diéguez, M.; Pàmies, O.; Claver, C. Adv. Synth. Catal. 2005, 347, 1257. doi:10.1002/adsc.200505013
Return to citation in text: [1] -
Diéguez, M.; Pàmies, O.; Claver, C. J. Organomet. Chem. 2006, 691, 2257. doi:10.1016/j.jorganchem.2005.11.024
Return to citation in text: [1] -
Raluy, E.; Diéguez, M.; Pàmies, O. J. Org. Chem. 2007, 72, 2842. doi:10.1021/jo062311j
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7
Return to citation in text: [1] -
Popa, D.; Puigjaner, C.; Gómez, M.; Benet-Buchholz, J.; Vidal-Ferran, A.; Pericàs, M. A. Adv. Synth. Catal. 2007, 349, 2265. doi:10.1002/adsc.200600599
Return to citation in text: [1] -
Cheung, H. Y.; Yu, W.-Y.; Au-Yeung, T. T. L.; Zhou, Z.; Chan, A. S. C. Adv. Synth. Catal. 2009, 351, 1412. doi:10.1002/adsc.200900086
Return to citation in text: [1] -
Rosas-Hernández, A.; Vargas-Malvaez, E.; Martin, E.; Crespi, L.; Bayón, J. C. J. Mol. Catal. A 2010, 328, 68. doi:10.1016/j.molcata.2010.06.001
Return to citation in text: [1] [2] -
Gual, A.; Castillón, S.; Pàmies, O.; Diéguez, M.; Claver, C. Dalton Trans. 2011, 40, 2852. doi:10.1039/c0dt01067g
Return to citation in text: [1] -
Hayashi, T.; Kawatsura, M.; Uozumi, Y. Chem. Commun. 1997, 561. doi:10.1039/a700045f
Return to citation in text: [1] [2] -
Hayashi, T.; Kawatsura, M.; Uozumi, Y. J. Am. Chem. Soc. 1998, 120, 1681. doi:10.1021/ja973150r
Return to citation in text: [1] [2] -
Prétôt, R.; Pfaltz, A. Angew. Chem., Int. Ed. 1998, 37, 323. doi:10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T
Return to citation in text: [1] [2] -
Hilgraf, R.; Pfaltz, A. Synlett 1999, 1814. doi:10.1055/s-1999-2939
Return to citation in text: [1] [2] -
Hilgraf, R.; Pfaltz, A. Adv. Synth. Catal. 2005, 347, 61. doi:10.1002/adsc.200404168
Return to citation in text: [1] [2] -
You, S.-L.; Zhu, X.-Z.; Luo, Y.-M.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2001, 123, 7471. doi:10.1021/ja016121w
Return to citation in text: [1] [2] [3] -
Zheng, W.-H.; Sun, N.; Hou, X.-L. Org. Lett. 2005, 7, 5151. doi:10.1021/ol051882f
Return to citation in text: [1] [2] -
Zheng, W.-H.; Zheng, B.-H.; Zhang, Y.; Hou, X.-L. J. Am. Chem. Soc. 2007, 129, 7718. doi:10.1021/ja071098l
Return to citation in text: [1] [2] -
Fang, P.; Ding, C.-H.; Hou, X.-L.; Dai, L.-X. Tetrahedron: Asymmetry 2010, 21, 1176. doi:10.1016/j.tetasy.2010.03.045
Return to citation in text: [1] [2] -
Chen, J.-P.; Ding, C.-H.; Liu, W.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2010, 132, 15493. doi:10.1021/ja106703y
Return to citation in text: [1] [2] -
Yang, X.-F.; Yu, W.-H.; Ding, C.-H.; Ding, Q.-P.; Wan, S.-L.; Hou, X.-L.; Dai, L.-X.; Wang, P.-J. J. Org. Chem. 2013, 78, 6503. doi:10.1021/jo400663d
Return to citation in text: [1] [2] -
Hu, Z.; Li, Y.; Liu, K.; Shen, Q. J. Org. Chem. 2012, 77, 7957. doi:10.1021/jo3011717
Return to citation in text: [1] [2] -
Liu, W.-B.; Zheng, C.; Zhuo, C.-X.; Dai, L.-X.; You, S.-L. J. Am. Chem. Soc. 2012, 134, 4812. doi:10.1021/ja210923k
Return to citation in text: [1] -
Liu, W.-B.; Zhang, X.; Dai, L.-X.; You, S.-L. Angew. Chem., Int. Ed. 2012, 51, 5183. doi:10.1002/anie.201200649
Return to citation in text: [1] -
Ye, K.-Y.; Dai, L.-X.; You, S.-L. Org. Biomol. Chem. 2012, 10, 5932. doi:10.1039/c2ob00036a
Return to citation in text: [1] -
Ye, K.-Y.; Zhao, Z.-A.; Lai, Z.-W.; Dai, L.-X.; You, S.-L. Synthesis 2013, 45, 2109. doi:10.1055/s-0033-1339187
Return to citation in text: [1] -
Dong, T.-Y.; Lai, L.-L. J. Organomet. Chem. 1996, 509, 131. doi:10.1016/0022-328X(95)05813-5
Return to citation in text: [1] -
Chesney, A.; Bryce, M. R.; Chubb, R. W. J.; Batsanov, A. S.; Howard, J. A. K. Synthesis 1998, 413. doi:10.1055/s-1998-2051
Return to citation in text: [1] -
Park, J.; Quan, Z.; Lee, S.; Ahn, K. H.; Cho, C.-W. J. Organomet. Chem. 1999, 584, 140. doi:10.1016/S0022-328X(99)00125-4
Return to citation in text: [1] -
Tworowska, I.; Dabkowski, W.; Michalski, J. Angew. Chem., Int. Ed. 2001, 40, 2898. doi:10.1002/1521-3773(20010803)40:15<2898::AID-ANIE2898>3.0.CO;2-I
Return to citation in text: [1] -
Murphy-Jolly, M. B.; Lewis, L. C.; Caffyn, A. J. M. Chem. Commun. 2005, 4479. doi:10.1039/B507752D
Return to citation in text: [1] -
Eisenberger, P.; Kieltsch, I.; Armanino, N.; Togni, A. Chem. Commun. 2008, 1575. doi:10.1039/B801424H
Return to citation in text: [1] -
Armanino, N.; Koller, R.; Togni, A. Organometallics 2010, 29, 1771. doi:10.1021/om100015s
Return to citation in text: [1] -
You, S.-L.; Hou, X.-L.; Dai, L.-X.; Yu, Y.-H.; Xia, W. J. Org. Chem. 2002, 67, 4684. doi:10.1021/jo016330z
Return to citation in text: [1]
| 1. | Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395. doi:10.1021/cr9409804 |
| 2. | Pfaltz, A.; Lautens, M. In Comprehensive Asymmetric Catalysis; Pfaltz, A.; Yamamoto, H., Eds.; Springer: New York, 1999; Vol. 2, pp 833 ff. |
| 3. | Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. doi:10.1021/cr020027w |
| 4. | Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258. doi:10.1002/anie.200605113 |
| 5. | Förster, S.; Helmchen, G.; Kazmaier, U. In Catalytic Asymmetric Synthesis, 3rd ed.; Ojima, I., Ed.; Wiley: Hoboken, 2010; pp 497 ff. |
| 25. | Prétôt, R.; Pfaltz, A. Angew. Chem., Int. Ed. 1998, 37, 323. doi:10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T |
| 26. | Hilgraf, R.; Pfaltz, A. Synlett 1999, 1814. doi:10.1055/s-1999-2939 |
| 27. | Hilgraf, R.; Pfaltz, A. Adv. Synth. Catal. 2005, 347, 61. doi:10.1002/adsc.200404168 |
| 23. | Hayashi, T.; Kawatsura, M.; Uozumi, Y. Chem. Commun. 1997, 561. doi:10.1039/a700045f |
| 24. | Hayashi, T.; Kawatsura, M.; Uozumi, Y. J. Am. Chem. Soc. 1998, 120, 1681. doi:10.1021/ja973150r |
| 10. | Vyskočil, Š.; Smrčina, M.; Hanuš, V.; Polášek, M.; Kočovský, P. J. Org. Chem. 1998, 63, 7738. doi:10.1021/jo980757x |
| 11. | Tenaglia, A.; Heumann, A. Angew. Chem., Int. Ed. 1999, 38, 2180. doi:10.1002/(SICI)1521-3773(19990802)38:15<2180::AID-ANIE2180>3.0.CO;2-A |
| 12. | Nakano, H.; Okuyama, Y.; Hongo, H. Tetrahedron Lett. 2000, 41, 4615. doi:10.1016/S0040-4039(00)00674-2 |
| 13. | Boele, M. D. K.; Kamer, P. C. J.; Lutz, M.; Spek, A. L.; de Vries, J. G.; van Leeuwen, P. W. N. M.; van Strijdonck, G. P. F. Chem.–Eur. J. 2004, 10, 6232. doi:10.1002/chem.200400154 |
| 14. | Pàmies, O.; Diéguez, M.; Claver, C. J. Am. Chem. Soc. 2005, 127, 3646. doi:10.1021/ja0425738 |
| 15. | Diéguez, M.; Pàmies, O.; Claver, C. Adv. Synth. Catal. 2005, 347, 1257. doi:10.1002/adsc.200505013 |
| 16. | Diéguez, M.; Pàmies, O.; Claver, C. J. Organomet. Chem. 2006, 691, 2257. doi:10.1016/j.jorganchem.2005.11.024 |
| 17. | Raluy, E.; Diéguez, M.; Pàmies, O. J. Org. Chem. 2007, 72, 2842. doi:10.1021/jo062311j |
| 18. | Goldfuss, B.; Löschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 19. | Popa, D.; Puigjaner, C.; Gómez, M.; Benet-Buchholz, J.; Vidal-Ferran, A.; Pericàs, M. A. Adv. Synth. Catal. 2007, 349, 2265. doi:10.1002/adsc.200600599 |
| 20. | Cheung, H. Y.; Yu, W.-Y.; Au-Yeung, T. T. L.; Zhou, Z.; Chan, A. S. C. Adv. Synth. Catal. 2009, 351, 1412. doi:10.1002/adsc.200900086 |
| 21. | Rosas-Hernández, A.; Vargas-Malvaez, E.; Martin, E.; Crespi, L.; Bayón, J. C. J. Mol. Catal. A 2010, 328, 68. doi:10.1016/j.molcata.2010.06.001 |
| 22. | Gual, A.; Castillón, S.; Pàmies, O.; Diéguez, M.; Claver, C. Dalton Trans. 2011, 40, 2852. doi:10.1039/c0dt01067g |
| 23. | Hayashi, T.; Kawatsura, M.; Uozumi, Y. Chem. Commun. 1997, 561. doi:10.1039/a700045f |
| 24. | Hayashi, T.; Kawatsura, M.; Uozumi, Y. J. Am. Chem. Soc. 1998, 120, 1681. doi:10.1021/ja973150r |
| 25. | Prétôt, R.; Pfaltz, A. Angew. Chem., Int. Ed. 1998, 37, 323. doi:10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T |
| 26. | Hilgraf, R.; Pfaltz, A. Synlett 1999, 1814. doi:10.1055/s-1999-2939 |
| 27. | Hilgraf, R.; Pfaltz, A. Adv. Synth. Catal. 2005, 347, 61. doi:10.1002/adsc.200404168 |
| 28. | You, S.-L.; Zhu, X.-Z.; Luo, Y.-M.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2001, 123, 7471. doi:10.1021/ja016121w |
| 29. | Zheng, W.-H.; Sun, N.; Hou, X.-L. Org. Lett. 2005, 7, 5151. doi:10.1021/ol051882f |
| 30. | Zheng, W.-H.; Zheng, B.-H.; Zhang, Y.; Hou, X.-L. J. Am. Chem. Soc. 2007, 129, 7718. doi:10.1021/ja071098l |
| 31. | Fang, P.; Ding, C.-H.; Hou, X.-L.; Dai, L.-X. Tetrahedron: Asymmetry 2010, 21, 1176. doi:10.1016/j.tetasy.2010.03.045 |
| 32. | Chen, J.-P.; Ding, C.-H.; Liu, W.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2010, 132, 15493. doi:10.1021/ja106703y |
| 33. | Yang, X.-F.; Yu, W.-H.; Ding, C.-H.; Ding, Q.-P.; Wan, S.-L.; Hou, X.-L.; Dai, L.-X.; Wang, P.-J. J. Org. Chem. 2013, 78, 6503. doi:10.1021/jo400663d |
| 34. | Hu, Z.; Li, Y.; Liu, K.; Shen, Q. J. Org. Chem. 2012, 77, 7957. doi:10.1021/jo3011717 |
| 46. | You, S.-L.; Hou, X.-L.; Dai, L.-X.; Yu, Y.-H.; Xia, W. J. Org. Chem. 2002, 67, 4684. doi:10.1021/jo016330z |
| 6. | Hayashi, T.; Kishi, K.; Yamamoto, A.; Ito, Y. Tetrahedron Lett. 1990, 31, 1743. doi:10.1016/S0040-4039(00)88870-X |
| 7. | Trost, B. M.; Krische, M. J.; Radinov, R.; Zanoni, G. J. Am. Chem. Soc. 1996, 118, 6297. doi:10.1021/ja960649x |
| 8. | Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 1998, 120, 9074. doi:10.1021/ja981142k |
| 9. | Trost, B. M.; Tsui, H.-C.; Toste, F. D. J. Am. Chem. Soc. 2000, 122, 3534. doi:10.1021/ja994326n |
| 39. | Dong, T.-Y.; Lai, L.-L. J. Organomet. Chem. 1996, 509, 131. doi:10.1016/0022-328X(95)05813-5 |
| 40. | Chesney, A.; Bryce, M. R.; Chubb, R. W. J.; Batsanov, A. S.; Howard, J. A. K. Synthesis 1998, 413. doi:10.1055/s-1998-2051 |
| 41. | Park, J.; Quan, Z.; Lee, S.; Ahn, K. H.; Cho, C.-W. J. Organomet. Chem. 1999, 584, 140. doi:10.1016/S0022-328X(99)00125-4 |
| 28. | You, S.-L.; Zhu, X.-Z.; Luo, Y.-M.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2001, 123, 7471. doi:10.1021/ja016121w |
| 35. | Liu, W.-B.; Zheng, C.; Zhuo, C.-X.; Dai, L.-X.; You, S.-L. J. Am. Chem. Soc. 2012, 134, 4812. doi:10.1021/ja210923k |
| 36. | Liu, W.-B.; Zhang, X.; Dai, L.-X.; You, S.-L. Angew. Chem., Int. Ed. 2012, 51, 5183. doi:10.1002/anie.201200649 |
| 37. | Ye, K.-Y.; Dai, L.-X.; You, S.-L. Org. Biomol. Chem. 2012, 10, 5932. doi:10.1039/c2ob00036a |
| 38. | Ye, K.-Y.; Zhao, Z.-A.; Lai, Z.-W.; Dai, L.-X.; You, S.-L. Synthesis 2013, 45, 2109. doi:10.1055/s-0033-1339187 |
| 21. | Rosas-Hernández, A.; Vargas-Malvaez, E.; Martin, E.; Crespi, L.; Bayón, J. C. J. Mol. Catal. A 2010, 328, 68. doi:10.1016/j.molcata.2010.06.001 |
| 34. | Hu, Z.; Li, Y.; Liu, K.; Shen, Q. J. Org. Chem. 2012, 77, 7957. doi:10.1021/jo3011717 |
| 28. | You, S.-L.; Zhu, X.-Z.; Luo, Y.-M.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2001, 123, 7471. doi:10.1021/ja016121w |
| 29. | Zheng, W.-H.; Sun, N.; Hou, X.-L. Org. Lett. 2005, 7, 5151. doi:10.1021/ol051882f |
| 30. | Zheng, W.-H.; Zheng, B.-H.; Zhang, Y.; Hou, X.-L. J. Am. Chem. Soc. 2007, 129, 7718. doi:10.1021/ja071098l |
| 31. | Fang, P.; Ding, C.-H.; Hou, X.-L.; Dai, L.-X. Tetrahedron: Asymmetry 2010, 21, 1176. doi:10.1016/j.tetasy.2010.03.045 |
| 32. | Chen, J.-P.; Ding, C.-H.; Liu, W.; Hou, X.-L.; Dai, L.-X. J. Am. Chem. Soc. 2010, 132, 15493. doi:10.1021/ja106703y |
| 33. | Yang, X.-F.; Yu, W.-H.; Ding, C.-H.; Ding, Q.-P.; Wan, S.-L.; Hou, X.-L.; Dai, L.-X.; Wang, P.-J. J. Org. Chem. 2013, 78, 6503. doi:10.1021/jo400663d |
| 42. | Tworowska, I.; Dabkowski, W.; Michalski, J. Angew. Chem., Int. Ed. 2001, 40, 2898. doi:10.1002/1521-3773(20010803)40:15<2898::AID-ANIE2898>3.0.CO;2-I |
| 43. | Murphy-Jolly, M. B.; Lewis, L. C.; Caffyn, A. J. M. Chem. Commun. 2005, 4479. doi:10.1039/B507752D |
| 44. | Eisenberger, P.; Kieltsch, I.; Armanino, N.; Togni, A. Chem. Commun. 2008, 1575. doi:10.1039/B801424H |
| 45. | Armanino, N.; Koller, R.; Togni, A. Organometallics 2010, 29, 1771. doi:10.1021/om100015s |
© 2014 Lai et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)