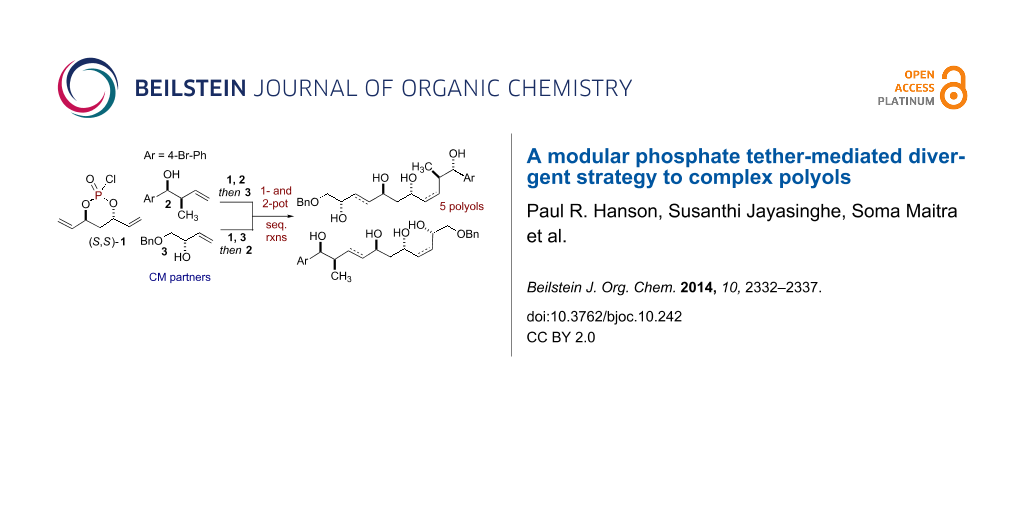
Abstract
An efficient and divergent synthesis of polyol subunits utilizing a phosphate tether-mediated, one-pot, sequential RCM/CM/reduction process is reported. A modular, 3-component coupling strategy has been developed, in which, simple “order of addition” of a pair of olefinic-alcohol components to a pseudo-C2-symmetric phosphoryl chloride, coupled with the RCM/CM/reduction protocol, yields five polyol fragments. Each of the product polyols bears a central 1,3-anti-diol subunit with differential olefinic geometries at the periphery.

Graphical Abstract
Introduction
1,3-anti-Diol subunits are a central component in several potent biologically active polyketides [1-4]. This prevalence has led to the development of various synthetic methods for their construction [5]. In particular, divergent strategies are ideal for analog generation [6-11], which in turn, can enhance the quality of screening decks in early phase drug discovery. One aim of divergent synthetic strategies is to produce multiple scaffolds from a single set of starting materials [12]. In this regard, one-pot, sequential processes [13-16] are well suited to address this challenge by forming multiple bonds and stereocenters, while invoking step- [17], atom- [18-21], green- [22,23], and pot economy [24-26]. We have previously reported phosphate tether-mediated strategies to streamline the synthesis of 1,3-anti-diol containing natural products, including recent reports employing one-pot, sequential protocols [27-32]. Herein, we report a modular, divergent approach to construct advanced polyol intermediates 10–14 and 17–21 in one- or two-pot sequences utilizing the innate properties of a phosphate tether. Taken collectively, this modular, divergent 3-component coupling strategy generates five polyol fragments, bearing differential Z- and E-olefins, from a pair of olefinic-alcohol components A and B and a pseudo-C2-symmetric phosphoryl chloride (S,S)-1. Moreover, the method relies on simple "order of addition" of components for the phosphoryl coupling, ring-closing metathesis (RCM) and cross metathesis (CM) steps of the process as outlined in Scheme 1. This protocol further highlights the utility of phosphate tether mediated desymmetrization of C2-symmetric 1,3-anti-diene-diol subunit to generate polyol scaffolds which would otherwise be difficult to produce via (Z)-and (E)-selective CM of 1,3-anti-diene-diol subunits with olefinic-alcohol components.
![[1860-5397-10-242-i1]](/bjoc/content/inline/1860-5397-10-242-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The 3-component coupling strategy.
Scheme 1: The 3-component coupling strategy.
Results and Discussion
The titled divergent strategy was initiated during efforts to further explore the utility of phosphate tethers. Previous reports emphasized the utilization of phosphate tethers in chemo- and diastereoselective reactions including one-pot, sequential RCM/CM/H2 protocols and their applications in total synthesis of various natural products [27-32]. In addition, the scope of phosphate-tethered methods was further expanded via extensive RCM studies of different triene subunits utilizing stereochemically divergent olefin partners [33]. Recently, the potential of phosphate tethered facilitated processes were highlighted in the pot-economical and efficient total synthesis of the antifungal agent strictifolione, whereby two consecutive phosphate tether-mediated, one-pot, sequential protocols were employed [34].
The requisite trienes 5–7 for this study were generated via our previously reported coupling of the pseudo-C2-symmetric phosphoryl chloride (S,S)-1 with the olefinic alcohol components 2–4 [27-32]. The alcohol substrates are carefully chosen to incorporate exo-allylic methyl groups since previous RCM studies [33] showed that the productive RCM for 8-membered ring formation was observed only for the S,S- configured trienes in the presence of an exo-methyl group at the allylic position (Figure 1).
![[1860-5397-10-242-1]](/bjoc/content/figures/1860-5397-10-242-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Initial attempts were focused on generating the first set of five polyols starting from trienes 5 and 6 in a two-pot operation (Scheme 2). The first operation entailed a one-pot, sequential RCM/CM/chemoselective hydrogenation protocol [32], yielding two bicyclo[n.3.1]phosphate intermediates 8 and 9, and a second pot LiAlH4 reduction to provide the Z-configured tetraol subunits 10 and 11. Trienes 5 and 6 were generated via coupling with alcohol partners 2 and 3, respectively, and the divergent aspect of the method was introduced by simple switching of the olefinic partners in the subsequent CM reaction to afford five differentiated polyols starting from three coupling partners.
![[1860-5397-10-242-i2]](/bjoc/content/inline/1860-5397-10-242-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of polyols 10–14 in one-, two-pot sequential protocols.
Scheme 2: Synthesis of polyols 10–14 in one-, two-pot sequential protocols.
In this regard, triene 5 was first subjected to RCM in the presence of the Hoveyda–Grubbs II (HG-II) catalyst [35-37] in refluxing CH2Cl2, followed by solvent concentration and CM with allylic alcohol 3 in refluxing CH2Cl2 for two hours. It was observed that the use of CH2Cl2 was critical for the successful CM reactions in order to avoid the formation of isomerized ketone byproducts. Subsequent chemoselective diimide reduction with o-nitrobenzenesulfonylhydrazine (o-NBSH) [38-40] in CH2Cl2 at room temperature afforded bicyclo[5.3.1]phosphate 8 in 33% overall yield, representing а 70% average yield/reaction in the one-pot, sequential protocol (Scheme 2). Subsequent treatment of 8 with LiAlH4 furnished the tetraol 10 in 24% overall yield over the course of four reaction steps carried out in two pots, representing a 70% average yield per reaction.
Similarly, starting with the triene 6, a one-pot RCM/CM/chemoselective H2 was performed to obtain the bicyclo[4.3.1]phosphate 9 in 40% yield over 3 reaction steps in a one-pot operation (72% avg/rxn). In this example, the RCM reaction was performed in dicholoroethane (DCE) at 70 °C for 2 h, since lower reactivity was observed in CH2Cl2. Subsequently, phosphate 9 was treated with LiAlH4 to generate tetraol 11 in 26% overall yield in the four reactions carried out in two pots, representing a 71% average yield per reaction.
Next, a one-pot RCM/CM/LAH protocol was established to obtain two additional tetraol subunits possessing both E- and Z-olefin geometries. Triene 5, was subjected to an RCM reaction, followed by a CM reaction with allylic alcohol 3. After removing the solvent, the CM product was treated with LiAlH4 to produce tetraol 12 in 38% yield over three reaction steps in the one-pot, sequential process (73% avg/rxn) (Scheme 2). Similarly, triene 6 was subjected to an RCM reaction, followed by CM with homoallylic alcohol 2, and subsequent treatment with LiAlH4 to afford tetraol 13 in 35% yield over the three reaction steps, representing a 70% avg/rxn in the one-pot, sequential method.
This RCM/CM/LAH procedure was further merged with global hydrogenation, whereby the resulting tetraols 12 and 13, after one-pot, sequential RCM/CM/LAH protocol, were separately treated with o-NBSH to obtain tetraol 14 in 26% yield over the four reaction steps in a two-pot operation starting from triene 5 (72% avg/rxn). Utilizing this two-pot sequential protocol, the same tetraol 14 was obtained starting from two different trienes (5 and 6) and reacting with two different CM partners. It should be noted, that after phosphate tether removal, treatment of tetraol with o-NBSH (20 equiv), resulted in the global reduction of both E- and Z-olefins in very good yields, while in contrast, diimide reduction in the presence of phosphate intermediates did not hydrogenate the endocyclic olefin even when large excesses of diimide reagent were employed (30 equiv). This empirical result further substantiates the protecting group ability of the phosphate tether for the endocyclic Z-olefin.
Next, our attempts were focused on generating the second set of five polyols starting from trienes 6 and 7. Utilizing the aforementioned strategy detailed in Scheme 2, triene 7 was subjected to the one-pot, sequential RCM/CM/chemoselective H2 procedure and subsequent LiAlH4 reduction to generate tetraol 17 in 24% yield over 4 reaction steps in a two-pot operation (70% avg/rxn) (Scheme 3). Further, starting with the same triene 6, which was previously used in Scheme 2, but utilizing a different cross-metathesis partner (homoallylic alcohol 4), a different tetraol 18 was generated in 23% yield over the four reaction steps in a two-pot operation (69% avg/rxn) (Scheme 3).
![[1860-5397-10-242-i3]](/bjoc/content/inline/1860-5397-10-242-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of polyols 17–21 in one-, two-pot sequential protocols.
Scheme 3: Synthesis of polyols 17–21 in one-, two-pot sequential protocols.
In a similar manner, starting with triene 7, RCM and subsequent CM with allylic alcohol 3, followed by tether removal with LiAlH4, were performed to obtain tetraol 19 in 42% yield over three reaction steps in the one-pot, sequential operation (75% avg/rxn). Triene 6 was next subjected to RCM, followed by CM with homoallylic alcohol 4 and LiAlH4 reduction to furnish tetraol 20 in an overall 40% yield over three reaction steps in a one-pot operation (72% avg/rxn). Tetraols 19 and 20 were separately subjected to a global hydrogenation using o-NBSH to afford tetraol 21 in 34% yield over four reaction steps in a two-pot operation starting from triene 6 (77% avg/rxn).
Conclusion
In conclusion, we have reported one- or two-pot sequential methods mediated by a phosphate tether to generate a diverse array of polyol molecules utilizing readily prepared trienes 5, 6 and 7 and CM partners 2, 3, and 4. This divergent method was established by taking advantage of the innate properties of phosphate tethers to provide efficient syntheses of the stereochemically-rich polyol subunits 10–14 and 17–21.
Supporting Information
| Supporting Information File 1: Experimental section. | ||
| Format: PDF | Size: 1.6 MB | Download |
Acknowledgements
This investigation was generously supported by funds provided by the National Institute of General Medical Sciences (NIH R01 GM077309). The authors thank Dr. Justin Douglas and Sarah Neuenswander (KU) for assistance with NMR measurements and Dr. Todd Williams (KU) for HRMS analysis. The authors also thank the University of Kansas and the State of Kansas for support of our program. The authors also thank Materia, Inc. for supplying the metathesis catalyst.
References
-
Staunton, J.; Weissman, K. J. Nat. Prod. Rep. 2001, 18, 380–416. doi:10.1039/a909079g
Return to citation in text: [1] -
Gupta, P.; Mahajan, N.; Taneja, S. C. Catal. Sci. Technol. 2013, 3, 2462–2480. doi:10.1039/c3cy00125c
Return to citation in text: [1] -
Rychnovsky, S. D. Chem. Rev. 1995, 95, 2021–2040. doi:10.1021/cr00038a011
Return to citation in text: [1] -
Friestad, G. K.; Sreenilayam, G. Org. Lett. 2010, 12, 5016–5019. doi:10.1021/ol1021417
Return to citation in text: [1] -
Bode, E. S.; Wolberg, M.; Müller, M. Synthesis 2006, 557–588. doi:10.1055/s-2006-926315
See for a review polyol synthesis.
Return to citation in text: [1] -
Mori, Y.; Suzuki, M. Tetrahedron Lett. 1989, 30, 4387–4388. doi:10.1016/S0040-4039(00)99367-5
Return to citation in text: [1] -
Liu, D.; Kozmin, S. A. Org. Lett. 2002, 4, 3005–3007. doi:10.1021/ol026433j
Return to citation in text: [1] -
Zacuto, M. J.; Leighton, J. L. Org. Lett. 2005, 7, 5525–5527. doi:10.1021/ol052373g
Return to citation in text: [1] -
Aurrecoechea, J. M.; Suerro, R.; de Torres, E. J. Org. Chem. 2006, 71, 8767–8778. doi:10.1021/jo0614487
Return to citation in text: [1] -
Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem. – Eur. J. 2013, 19, 6398–6408. doi:10.1002/chem.201300198
Return to citation in text: [1] -
Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020–3023. doi:10.1021/ja5003247
Return to citation in text: [1] -
Burke, M. D.; Schreiber, S. L. Angew. Chem., Int. Ed. 2004, 43, 46–58. doi:10.1002/anie.200300626
See for diversity oriented synthesis.
Return to citation in text: [1] -
Louie, J.; Bielawski, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 11312–11313. doi:10.1021/ja016431e
See for tandem, metathesis/hydrogenation.
Return to citation in text: [1] -
Scholte, A. A.; An, M.-H.; Snapper, M. L. Org. Lett. 2006, 8, 4759–4762. doi:10.1021/ol061837n
See for RCM/oxidation.
Return to citation in text: [1] -
Seigal, B. A.; Fajardo, C.; Snapper, M. L. J. Am. Chem. Soc. 2005, 127, 16329–16332. doi:10.1021/ja055806j
See for RCM/Kharasch addition.
Return to citation in text: [1] -
Ferrié, L.; Bouzbouz, S.; Cossy, J. Org. Lett. 2009, 11, 5446–5448. doi:10.1021/ol9021386
See for other tandem metathesis processes.
Return to citation in text: [1] -
Wender, P. A.; Verma, V. A.; Paxton, T. J.; Pillow, T. H. Acc. Chem. Res. 2008, 41, 40–49. doi:10.1021/ar700155p
Return to citation in text: [1] -
Trost, B. M. Science 1991, 254, 1471–1477. doi:10.1126/science.1962206
Return to citation in text: [1] -
Trost, B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. doi:10.1002/anie.199502591
Return to citation in text: [1] -
Young, S. I.; Baran, P. S. Nat. Chem. 2009, 1, 193–205. doi:10.1038/nchem.216
See for reviews on protecting group-free synthesis.
Return to citation in text: [1] -
Hoffmann, R. W. Synthesis 2006, 3531–3541. doi:10.1055/s-2006-950311
Return to citation in text: [1] -
Lipshutz, B. H.; Huang, S.; Leong, W. W. Y.; Zhong, G.; Isley, N. A. J. Am. Chem. Soc. 2012, 134, 19985–19988. doi:10.1021/ja309409e
Return to citation in text: [1] -
Sheldon, R. A.; Arends, I. W. C. E.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, 2007; pp 1–47. doi:10.1002/9783527611003.ch1
Return to citation in text: [1] -
Ishikawa, H.; Suzuki, T.; Hayashi, Y. Angew. Chem., Int. Ed. 2009, 48, 1304–1307. doi:10.1002/anie.200804883
Return to citation in text: [1] -
Ishikawa, H.; Honma, M.; Hayashi, Y. Angew. Chem., Int. Ed. 2011, 50, 2824–2827. doi:10.1002/anie.201006204
Return to citation in text: [1] -
Hayashi, Y.; Umemiya, S. Angew. Chem., Int. Ed. 2013, 52, 3450–3452. doi:10.1002/anie.201209380
Return to citation in text: [1] -
Whitehead, A.; McReynolds, M. D.; Moore, J. D.; Hanson, P. R. Org. Lett. 2005, 7, 3375–3378. doi:10.1021/ol0512886
Return to citation in text: [1] [2] [3] -
Thomas, C. D.; McParland, J. P.; Hanson, P. R. Eur. J. Org. Chem. 2009, 5487–5500. doi:10.1002/ejoc.200900560
Return to citation in text: [1] [2] [3] -
Venukadasula, P. K. M.; Chegondi, R.; Maitra, S.; Hanson, P. R. Org. Lett. 2010, 12, 1556–1559. doi:10.1021/ol1002913
Return to citation in text: [1] [2] [3] -
Hanson, P. R.; Chegondi, R.; Nguyen, J.; Thomas, C. D.; Waetzig, J.; Whitehead, A. J. Org. Chem. 2011, 76, 4358–4370. doi:10.1021/jo2003506
Return to citation in text: [1] [2] [3] -
Chegondi, R.; Tan, M. M. L.; Hanson, P. R. J. Org. Chem. 2011, 76, 3909–3916. doi:10.1021/jo200337v
Return to citation in text: [1] [2] [3] -
Venukadasula, P. K. M.; Chegondi, R.; Suryn, G. M.; Hanson, P. R. Org. Lett. 2012, 14, 2634–2637. doi:10.1021/ol301007h
Return to citation in text: [1] [2] [3] [4] -
Chegondi, R.; Maitra, S.; Markley, J. L.; Hanson, P. R. Chem. – Eur. J. 2013, 19, 8088–8093. doi:10.1002/chem.201300913
Return to citation in text: [1] [2] -
Jayasinghe, S.; Venukadasula, P. K. M.; Hanson, P. R. Org. Lett. 2014, 16, 122–125. doi:10.1021/ol403110p
Return to citation in text: [1] -
Kingsbury, J. S.; Harrity, J. P. A.; Bonitatebus, P. J., Jr.; Hoveyda, A. H. J. Am. Chem. Soc. 1999, 121, 791–799. doi:10.1021/ja983222u
Return to citation in text: [1] -
Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g
Return to citation in text: [1] -
Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett. 2000, 41, 9973–9976. doi:10.1016/S0040-4039(00)01808-6
Return to citation in text: [1] -
Myers, A. G.; Zheng, B.; Movassaghi, M. J. Org. Chem. 1997, 62, 7507. doi:10.1021/jo9710137
Return to citation in text: [1] -
Haukaas, M. H.; O’Doherty, G. A. Org. Lett. 2002, 4, 1771–1774. doi:10.1021/ol025844x
Return to citation in text: [1] -
Buszek, K. R.; Brown, N. J. Org. Chem. 2007, 72, 3125–3128. doi:10.1021/jo0622173
Return to citation in text: [1]
| 35. | Kingsbury, J. S.; Harrity, J. P. A.; Bonitatebus, P. J., Jr.; Hoveyda, A. H. J. Am. Chem. Soc. 1999, 121, 791–799. doi:10.1021/ja983222u |
| 36. | Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g |
| 37. | Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett. 2000, 41, 9973–9976. doi:10.1016/S0040-4039(00)01808-6 |
| 38. | Myers, A. G.; Zheng, B.; Movassaghi, M. J. Org. Chem. 1997, 62, 7507. doi:10.1021/jo9710137 |
| 39. | Haukaas, M. H.; O’Doherty, G. A. Org. Lett. 2002, 4, 1771–1774. doi:10.1021/ol025844x |
| 40. | Buszek, K. R.; Brown, N. J. Org. Chem. 2007, 72, 3125–3128. doi:10.1021/jo0622173 |
| 1. | Staunton, J.; Weissman, K. J. Nat. Prod. Rep. 2001, 18, 380–416. doi:10.1039/a909079g |
| 2. | Gupta, P.; Mahajan, N.; Taneja, S. C. Catal. Sci. Technol. 2013, 3, 2462–2480. doi:10.1039/c3cy00125c |
| 3. | Rychnovsky, S. D. Chem. Rev. 1995, 95, 2021–2040. doi:10.1021/cr00038a011 |
| 4. | Friestad, G. K.; Sreenilayam, G. Org. Lett. 2010, 12, 5016–5019. doi:10.1021/ol1021417 |
| 13. |
Louie, J.; Bielawski, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 11312–11313. doi:10.1021/ja016431e
See for tandem, metathesis/hydrogenation. |
| 14. |
Scholte, A. A.; An, M.-H.; Snapper, M. L. Org. Lett. 2006, 8, 4759–4762. doi:10.1021/ol061837n
See for RCM/oxidation. |
| 15. |
Seigal, B. A.; Fajardo, C.; Snapper, M. L. J. Am. Chem. Soc. 2005, 127, 16329–16332. doi:10.1021/ja055806j
See for RCM/Kharasch addition. |
| 16. |
Ferrié, L.; Bouzbouz, S.; Cossy, J. Org. Lett. 2009, 11, 5446–5448. doi:10.1021/ol9021386
See for other tandem metathesis processes. |
| 33. | Chegondi, R.; Maitra, S.; Markley, J. L.; Hanson, P. R. Chem. – Eur. J. 2013, 19, 8088–8093. doi:10.1002/chem.201300913 |
| 12. |
Burke, M. D.; Schreiber, S. L. Angew. Chem., Int. Ed. 2004, 43, 46–58. doi:10.1002/anie.200300626
See for diversity oriented synthesis. |
| 32. | Venukadasula, P. K. M.; Chegondi, R.; Suryn, G. M.; Hanson, P. R. Org. Lett. 2012, 14, 2634–2637. doi:10.1021/ol301007h |
| 6. | Mori, Y.; Suzuki, M. Tetrahedron Lett. 1989, 30, 4387–4388. doi:10.1016/S0040-4039(00)99367-5 |
| 7. | Liu, D.; Kozmin, S. A. Org. Lett. 2002, 4, 3005–3007. doi:10.1021/ol026433j |
| 8. | Zacuto, M. J.; Leighton, J. L. Org. Lett. 2005, 7, 5525–5527. doi:10.1021/ol052373g |
| 9. | Aurrecoechea, J. M.; Suerro, R.; de Torres, E. J. Org. Chem. 2006, 71, 8767–8778. doi:10.1021/jo0614487 |
| 10. | Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem. – Eur. J. 2013, 19, 6398–6408. doi:10.1002/chem.201300198 |
| 11. | Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020–3023. doi:10.1021/ja5003247 |
| 34. | Jayasinghe, S.; Venukadasula, P. K. M.; Hanson, P. R. Org. Lett. 2014, 16, 122–125. doi:10.1021/ol403110p |
| 5. |
Bode, E. S.; Wolberg, M.; Müller, M. Synthesis 2006, 557–588. doi:10.1055/s-2006-926315
See for a review polyol synthesis. |
| 27. | Whitehead, A.; McReynolds, M. D.; Moore, J. D.; Hanson, P. R. Org. Lett. 2005, 7, 3375–3378. doi:10.1021/ol0512886 |
| 28. | Thomas, C. D.; McParland, J. P.; Hanson, P. R. Eur. J. Org. Chem. 2009, 5487–5500. doi:10.1002/ejoc.200900560 |
| 29. | Venukadasula, P. K. M.; Chegondi, R.; Maitra, S.; Hanson, P. R. Org. Lett. 2010, 12, 1556–1559. doi:10.1021/ol1002913 |
| 30. | Hanson, P. R.; Chegondi, R.; Nguyen, J.; Thomas, C. D.; Waetzig, J.; Whitehead, A. J. Org. Chem. 2011, 76, 4358–4370. doi:10.1021/jo2003506 |
| 31. | Chegondi, R.; Tan, M. M. L.; Hanson, P. R. J. Org. Chem. 2011, 76, 3909–3916. doi:10.1021/jo200337v |
| 32. | Venukadasula, P. K. M.; Chegondi, R.; Suryn, G. M.; Hanson, P. R. Org. Lett. 2012, 14, 2634–2637. doi:10.1021/ol301007h |
| 24. | Ishikawa, H.; Suzuki, T.; Hayashi, Y. Angew. Chem., Int. Ed. 2009, 48, 1304–1307. doi:10.1002/anie.200804883 |
| 25. | Ishikawa, H.; Honma, M.; Hayashi, Y. Angew. Chem., Int. Ed. 2011, 50, 2824–2827. doi:10.1002/anie.201006204 |
| 26. | Hayashi, Y.; Umemiya, S. Angew. Chem., Int. Ed. 2013, 52, 3450–3452. doi:10.1002/anie.201209380 |
| 27. | Whitehead, A.; McReynolds, M. D.; Moore, J. D.; Hanson, P. R. Org. Lett. 2005, 7, 3375–3378. doi:10.1021/ol0512886 |
| 28. | Thomas, C. D.; McParland, J. P.; Hanson, P. R. Eur. J. Org. Chem. 2009, 5487–5500. doi:10.1002/ejoc.200900560 |
| 29. | Venukadasula, P. K. M.; Chegondi, R.; Maitra, S.; Hanson, P. R. Org. Lett. 2010, 12, 1556–1559. doi:10.1021/ol1002913 |
| 30. | Hanson, P. R.; Chegondi, R.; Nguyen, J.; Thomas, C. D.; Waetzig, J.; Whitehead, A. J. Org. Chem. 2011, 76, 4358–4370. doi:10.1021/jo2003506 |
| 31. | Chegondi, R.; Tan, M. M. L.; Hanson, P. R. J. Org. Chem. 2011, 76, 3909–3916. doi:10.1021/jo200337v |
| 32. | Venukadasula, P. K. M.; Chegondi, R.; Suryn, G. M.; Hanson, P. R. Org. Lett. 2012, 14, 2634–2637. doi:10.1021/ol301007h |
| 22. | Lipshutz, B. H.; Huang, S.; Leong, W. W. Y.; Zhong, G.; Isley, N. A. J. Am. Chem. Soc. 2012, 134, 19985–19988. doi:10.1021/ja309409e |
| 23. | Sheldon, R. A.; Arends, I. W. C. E.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, 2007; pp 1–47. doi:10.1002/9783527611003.ch1 |
| 33. | Chegondi, R.; Maitra, S.; Markley, J. L.; Hanson, P. R. Chem. – Eur. J. 2013, 19, 8088–8093. doi:10.1002/chem.201300913 |
| 18. | Trost, B. M. Science 1991, 254, 1471–1477. doi:10.1126/science.1962206 |
| 19. | Trost, B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. doi:10.1002/anie.199502591 |
| 20. |
Young, S. I.; Baran, P. S. Nat. Chem. 2009, 1, 193–205. doi:10.1038/nchem.216
See for reviews on protecting group-free synthesis. |
| 21. | Hoffmann, R. W. Synthesis 2006, 3531–3541. doi:10.1055/s-2006-950311 |
| 17. | Wender, P. A.; Verma, V. A.; Paxton, T. J.; Pillow, T. H. Acc. Chem. Res. 2008, 41, 40–49. doi:10.1021/ar700155p |
| 27. | Whitehead, A.; McReynolds, M. D.; Moore, J. D.; Hanson, P. R. Org. Lett. 2005, 7, 3375–3378. doi:10.1021/ol0512886 |
| 28. | Thomas, C. D.; McParland, J. P.; Hanson, P. R. Eur. J. Org. Chem. 2009, 5487–5500. doi:10.1002/ejoc.200900560 |
| 29. | Venukadasula, P. K. M.; Chegondi, R.; Maitra, S.; Hanson, P. R. Org. Lett. 2010, 12, 1556–1559. doi:10.1021/ol1002913 |
| 30. | Hanson, P. R.; Chegondi, R.; Nguyen, J.; Thomas, C. D.; Waetzig, J.; Whitehead, A. J. Org. Chem. 2011, 76, 4358–4370. doi:10.1021/jo2003506 |
| 31. | Chegondi, R.; Tan, M. M. L.; Hanson, P. R. J. Org. Chem. 2011, 76, 3909–3916. doi:10.1021/jo200337v |
| 32. | Venukadasula, P. K. M.; Chegondi, R.; Suryn, G. M.; Hanson, P. R. Org. Lett. 2012, 14, 2634–2637. doi:10.1021/ol301007h |
© 2014 Hanson et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)