Abstract
An analogue of thiamine having a furan ring in place of the thiazolium ring has been synthesised by a short and efficient route, involving gold(I)-catalysed cyclisation of an alkynyl alcohol to form the furan ring. The furan analogue of thiamine diphosphate (ThDP) was also made and tested for binding to and inhibition of pyruvate decarboxylase (PDC) from Zymomonas mobilis (overexpressed in E. coli with a N-terminal His-tag). It is a very strong inhibitor, with a Ki value of 32.5 pM. It was also shown that the furan analogue of thiamine can be functionalised at the C-2 position, which will allow access to mimics of reaction intermediates of various ThDP-dependent enzymes.

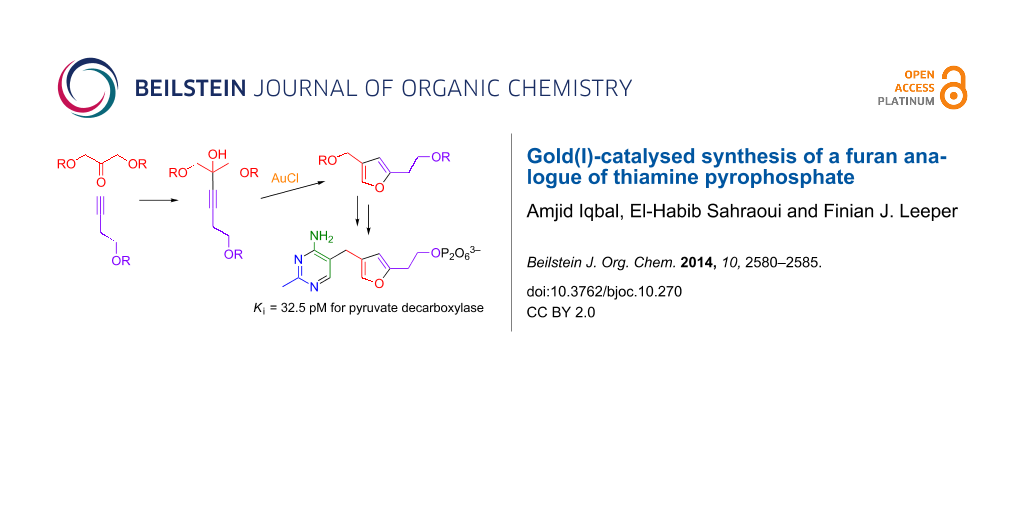
Graphical Abstract
Introduction
The biologically active form of vitamin B1 is thiamine diphosphate (ThDP, 1, Figure 1), which is an essential cofactor and involved in a number of metabolic pathways, including oxidative and non-oxidative decarboxylation of α-keto acids (e.g., pyruvate dehydrogenase, pyruvate decarboxylase), the formation of amino acid precursors (acetohydroxy acid synthase), and ketol transfer between sugars (transketolase) [1]. One common feature of ThDP-dependent enzymes is to catalyse the cleavage and formation of bonds adjacent to the carbon of a carbonyl group with the thiazolium ring of ThDP acting as an electron sink during catalysis in order to stabilise what would otherwise be an acyl carbanion in the form of an enamine intermediate [2].
![[1860-5397-10-270-1]](/bjoc/content/figures/1860-5397-10-270-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of thiamine diphosphate (ThDP, 1).
Figure 1: Structure of thiamine diphosphate (ThDP, 1).
The catalytic cycle for pyruvate decarboxylase (PDC, a representative ThDP-dependent enzyme), first proposed by Breslow [3,4], is given in Scheme 1. After formation of the ThDP ylid, the keto group of the substrate is attacked by the thiazolium C-2 carbanion to form the tetrahedral pre-decarboxylation intermediate 2-(2-lactyl)-ThDP, LThDP. It has been proposed that the covalent attachment of pyruvate to form LThDP introduces significant strain, the release of which is a driving force for the decarboxylation [5]. The effects of this type of strain on bond angles and lengths have recently been observed in a high resolution crystal structure of another ThDP-dependent enzyme, transketolase [6]. Another important factor of this decarboxylation is the presence of an electron sink in the form of the positively charged nitrogen atom. Decarboxylation gives the C-2α-carbanion/enamine resonance forms of the post decarboxylation intermediate. Protonation of this enamine intermediate at C-2α gives HEThDP and final elimination of acetaldehyde completes the catalytic cycle.
![[1860-5397-10-270-i1]](/bjoc/content/inline/1860-5397-10-270-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanism of pyruvate decarboxylase and structures of some previously synthesised ThDP analogues.
Scheme 1: Mechanism of pyruvate decarboxylase and structures of some previously synthesised ThDP analogues.
The 4’-aminopyrimidine ring of ThDP can interconvert among three ionization/tautomeric states, the protonated form and two neutral forms, the amino tautomer and the imino tautomer (Scheme 1) and it is believed to be the imine nitrogen atom of the latter that is the base that deprotonates C-2 to form the ylid [7,8].
Although many crystal structures of ThDP dependent enzymes exist, there still are some unanswered questions regarding mechanistic processes of these enzymes (e.g., how the substrate binds, what is the role of important catalytic groups, how are individual steps accelerated). In order to answer these questions, it would help to have snapshots of the enzyme bound to various reaction intermediates in the cycle. However, this is usually not possible when using the natural cofactor because the enzyme turns over rapidly, which in most cases prevents the trapping of reaction intermediates bound to the enzyme. One solution to this problem is to use analogues of ThDP that closely mimic the reaction intermediates of the catalytic cycle, but crucially are unable to continue the reaction mechanism. To this end we have previously synthesised inactive analogues of the ThDP cofactor, including 3-deazaThDP 2, in which the nitrogen atom of the thiazolium ring is replaced by a carbon [9,10]. 3-DeazaThDP is isoelectronic with ThDP, but the lack of positive charge means that deprotonation at C-2 is not possible.
3-DeazaThDP 2 is an almost irreversible inhibitor, which binds 25,000 times more strongly (Ki = 14 pM) than ThDP to PDC from Zymomonas mobilis (ZmPDC) and 500 times more strongly (Ki = 5 nM) to the E1 component of α-ketoglutarate dehydrogenase [10]. This extremely tight binding can be rationalised by the structural similarity between 2 and the ylid of ThDP, with increased hydrophobic interactions between the enzyme and the electrically neutral analogue compared to the positively charged ThDP [11].
The synthesis of 2 was achieved in 12 steps in reasonable yield but was lengthy and time-consuming. For this reason other analogues have also been studied. Two triazole analogues 3 (Ki = 20 pM) and 4 (Ki = 30 pM against ZmPDC) were synthesised in just four synthetic steps [12]. However, there is no possibility of attaching a substituent at the 2-position of the triazole to mimic enzymic reaction intermediates. Another analogue of ThDP 5 with a benzene ring in place of the thiazolium ring was synthesised and also found to be a very strong inhibitor of ZmPDC, binding at almost the same rate as 2 [10]. With this benzene analogue also, it would be difficult to functionalise the 2-position selectively.
In this paper, we report a short and efficient synthesis of a furan-based analogue of thiamine. This analogue was converted to its diphosphate, which is another extremely potent inhibitor of ZmPDC, and could also be functionalised at the 2-position using a Friedel–Crafts acylation.
Results and Discussion
Synthesis of ThDP analogue 17
The key step in our planned synthesis was the formation of the furan ring. Homogeneous gold-catalysed reactions have been used recently in the synthesis of furans from alkynes [13-20]. The ease with which alkynes, allenes and alkenes can be activated by Au(I) catalysts to form carbon–carbon and carbon–heteroatom bonds makes the method an appealing strategy towards diverse chemical targets [21-30]. Furthermore, the catalysts are stable to air and moisture and the reactions generally proceed under mild conditions. Aponick and co-workers reported a catalytic dehydrative cyclisation reaction of alkynyl alcohols catalysed by simple gold(I) salts [31]. The reaction proceeds rapidly under mild, open flask conditions to provide aromatic heterocycles such as furans, pyrroles and thiophenes in high yield with low catalyst loadings (Scheme 2).
![[1860-5397-10-270-i2]](/bjoc/content/inline/1860-5397-10-270-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Dehydrative cyclization catalysed by gold(I) and its presumed mechanism [31].
Scheme 2: Dehydrative cyclization catalysed by gold(I) and its presumed mechanism [31].
Following this synthetic strategy, it was decided to explore the cyclisation of alkyne tetraol 11, which should give furan 12 with hydroxyethyl and hydroxymethyl substituents at the 2- and 4-position respectively (Scheme 3). This could then be converted into a furan analogue of ThDP.
![[1860-5397-10-270-i3]](/bjoc/content/inline/1860-5397-10-270-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of furan 12. Reagents and conditions: (i) TBDMS-Cl, N-methylimidazole. (ii) n-BuLi, −78 °C, then 9 and to rt, 55% (iii) TBAF, THF, 79%. (iv) AuCl, THF, 85%.
Scheme 3: Synthesis of furan 12. Reagents and conditions: (i) TBDMS-Cl, N-methylimidazole. (ii) n-BuLi, −78 °...
The first step in the synthesis of tetraol 11 was the protection of 3-butynol (6) and 1,3-dihydroxyacetone (8) as their TBDMS ethers, 7 and 9. The alkyne 7 was then coupled with ketone 9 using n-BuLi. TBAF-mediated deprotection gave unsaturated alcohol 11, the substrate for dehydrative cyclization with AuCl. The order of addition, reaction time and temperature were all found to be important for this reaction: the best yield was obtained when a solution of 11 in THF was added at 0 °C to the dry AuCl. The mixture was stirred for 10 min and then filtered through Celite to remove the catalyst. Using this method an 85% yield of furan 12 could be obtained. Delay in filtration or increase in temperature caused the catalyst to decompose and made the product difficult to purify. Subsequent to our synthesis of 12 a very similar synthesis of the same compound was published by Deslongchamps and coworkers [32], which only differed in that the cyclisation of 11 to 12 in excellent yield was catalysed by Hg(II), instead of Au(I).
With furan 12 in hand, it was then transformed into the furan analogue 17 of ThDP by the same route as previously used for the synthesis of deazaThDP 2 [9,10], Scheme 4. Thus selective oxidation of the benzylic alcohol with MnO2, condensation with 3-anilinopropionitrile, and then reaction with acetamidine gives the thiamine analogue 15. Tosylation of the alcohol and displacement of tosylate by the diphosphate trianion worked well to give ThDP analogue 17.
![[1860-5397-10-270-i4]](/bjoc/content/inline/1860-5397-10-270-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the furan analogue 17 of ThDP. Reagents and conditions: (i) MnO2, CHCl3, 72%; (ii) PhNHCH2CH2CN, NaOMe, DMSO, MeOH, 76%; (iii) CH3C(NH)NH2·HCl, NaOEt, EtOH, 65%; (iv) TsCl, pyridine, −5 °C, 72%; (v) TBA3HPP, MeCN, 4 °C, 30%; (vi) AcCl, AlCl3, DCM, 67%.
Scheme 4: Synthesis of the furan analogue 17 of ThDP. Reagents and conditions: (i) MnO2, CHCl3, 72%; (ii) PhN...
A further objective was to show that the furan ring could be substituted at C-2. To this end furan 15 was reacted with acetyl chloride and AlCl3 and the 2-acetylfuran 18 was obtained in 67% yield. 1H,13C correlations in the HMBC spectrum of 18 proved that C-2 and not C-4 had been acetylated, with a key correlation being from the first CH2 of the CH2CH2OAc side-chain (C-5a) to the furan carbon that has a hydrogen attached (C-4). This selective acylation of C-2 will allow the addition of substituents at this position that match the substituents in the enzymic reactions, such as the 2-hydroxyethyl group found in HEThDP (Scheme 1).
Inhibition of pyruvate decarboxylase by furan 17
The furan analogue of ThDP, 17, was tested as an inhibitor of pyruvate decarboxylase from Zymomonas mobilis (ZmPDC). ZmPDC is a tetramer, made up of a dimer of dimers, and there are four active sites per tetramer located at the interface between the subunits in each dimer [33]. One of the main reasons for using this enzyme for our study is that, unlike most other PDCs (e.g., from yeast), it does not show allosteric activation by its own substrate and thus gives normal Michaelis–Menten kinetics [34-38], and also it shows good stability. Previously the native enzyme expressed in Escherichia coli has been used in such studies but, in order to simplify the purification, in this study the gene was cloned into a pET28a vector, to give the enzyme an N-terminal His6-tag. This form of the protein was overexpressed in high yield in E. coli, and was easily purified on a Ni2+-NTA column. The His6-tag does not appear to affect the enzymic activity.
The normal coupled enzyme assay for PDC was used, in which PDC catalyses the decarboxylation of pyruvate to acetaldehyde and alcohol dehydrogenase (ADH) converts the acetaldehyde to ethanol using NADH (Scheme 5). The oxidation of NADH to NAD+ is monitored by UV spectroscopy at 340 nm.
![[1860-5397-10-270-i5]](/bjoc/content/inline/1860-5397-10-270-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to follow the time-course of binding of furan 17, various concentrations (1–8 µM) were incubated with apo-PDC in a Mg2+-containing buffer and aliquots were taken out at timed intervals (1–30 min) and added to the assay solution containing excess ThDP (100 µM). Under these conditions any apo-PDC that does not already have the furan analogue bound will bind ThDP and thus show activity. The plot of the percentage residual activity against time is shown in Figure 2. For all concentrations of inhibitor the enzyme lost all activity within 30 min, though the inhibition is faster at higher concentrations.
![[1860-5397-10-270-2]](/bjoc/content/figures/1860-5397-10-270-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Time course inactivation of ZmPDC by various concentrations of furan 17.
Figure 2: Time course inactivation of ZmPDC by various concentrations of furan 17.
The data-points in Figure 2 fit poorly onto a simple exponential curve, however, a double exponential with 60% of the activity being lost at a fast rate and 40% being lost more slowly fits reasonably well; the equation used is
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-270-i6.svg?max-width=637&scale=1.18182)
The faster of these two phases (k1) cannot be measured accurately from this data as it is almost all over within the first minute at all but the lowest concentrations of inhibitor. However, an estimated value of the second order rate constant kon for the initial binding of 17 is ca. 1 µM−1 min−1.
The apparent two-stage inhibition for PDC was reported by Mann et al. for the benzene-based ThDP analogue 5 [10] and the same effect was also observed by Erixon et al. using triazole analogues 3 and 4 [37] and also with a thiazolone analogue by Kluger et al. [38]. It may be due to a slow conformational change after the initial binding or due to communication between the two active sites in each dimer.
In order to measure a Ki value for furan 17 the enzyme has to be allowed to reach its equilibrium in a competition between binding ThDP and binding the analogue, and then the level of residual activity will indicate the proportion of active sites that have ThDP bound. Therefore, ZmPDC was inactivated by furan 17 (1 µM) and then a large excess of ThDP (1 mM) was added and the recovery in activity was followed over 3 days. Some activity was slowly recovered (Figure 3) but this reached a plateau within 24 hours at 8.5% of the untreated control (which remained fully active over this period). The steady activity regained in the presence of inhibitor is therefore the equilibrium position between ThDP binding and inhibitor binding.
![[1860-5397-10-270-3]](/bjoc/content/figures/1860-5397-10-270-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Recovery of activity for ZmPDC inhibited by furan 17 (1.0 µM) and then incubated with ThDP (1.0 mM). The activity is given as a percentage of the initial activity of uninhibited enzyme.
Figure 3: Recovery of activity for ZmPDC inhibited by furan 17 (1.0 µM) and then incubated with ThDP (1.0 mM)...
The time-course for recovery of activity fits nicely to single exponential equation with an apparent first order rate constant of 0.13 h−1. Using the ratio of recovered activity over unrecovered activity at equilibrium (8.5:91.5), the relative concentrations of ThDP and inhibitor (1000:1) and the previously reported KD value for ThDP (0.35 µM) [39], one can calculate that the Ki value of furan analogue 17 is 0.35 × (8.5/91.5) × (1/1000) µM = 32.5 pM. This Ki value is more than 10,000-fold lower than the KD value for ThDP and is in the same range as previously reported for triazole analogues of ThDP (30 and 20 pM) [12,37]. However, the furan analogue of ThDP has the advantage over the triazole analogues that it can be functionalised at the C-2 position.
Conclusion
In conclusion, we report here the short synthesis of a furan analogue of thiamine, which could be diphosphorylated to give an analogue of ThDP. Biological evaluation of this diphosphate showed that it is a very strong inhibitor of ZmPDC with picomolar affinity. Friedel–Crafts acetylation of the furan at C-2 was successful which opens the possibility of synthesising furan analogues of the enzymic reaction intermediates.
Supporting Information
| Supporting Information File 1: Experimental section along with 1H and 13C NMR spectra for all the compounds synthesised. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Schellenberger, A. Biochim. Biophys. Acta 1998, 1385, 177–186. doi:10.1016/S0167-4838(98)00067-3
Return to citation in text: [1] -
Kluger, R. Chem. Rev. 1987, 87, 863–876. doi:10.1021/cr00081a001
Return to citation in text: [1] -
Breslow, R. J. Am. Chem. Soc. 1957, 79, 1762–1763. doi:10.1021/ja01564a064
Return to citation in text: [1] -
Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719–3726. doi:10.1021/ja01547a064
Return to citation in text: [1] -
Jordan, F. Nat. Prod. Rep. 2003, 20, 184–201. doi:10.1039/b111348h
Return to citation in text: [1] -
Lüdtke, S.; Neumann, P.; Erixon, K. M.; Leeper, F. J.; Kluger, R.; Ficner, R.; Tittmann, K. Nat. Chem. 2013, 5, 762–767. doi:10.1038/nchem.1728
Return to citation in text: [1] -
Nemeria, N. S.; Chakraborty, S.; Balakrishnan, A.; Jordan, F. FEBS J. 2009, 276, 2432–2446. doi:10.1111/j.1742-4658.2009.06964.x
Return to citation in text: [1] -
Nemeria, N. S.; Chakraborty, S.; Baykal, A.; Korotchkina, L. G.; Patel, M. S.; Jordan, F. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 78–82. doi:10.1073/pnas.0609973104
Return to citation in text: [1] -
Hawksley, D.; Griffin, D. A.; Leeper, F. J. J. Chem. Soc., Perkin Trans. 1 2001, 144–148. doi:10.1039/b006962k
Return to citation in text: [1] [2] -
Mann, S.; Perez Melero, C.; Hawksley, D.; Leeper, F. J. Org. Biomol. Chem. 2004, 2, 1732–1741. doi:10.1039/b403619k
Return to citation in text: [1] [2] [3] [4] [5] -
Jordan, F.; Li, H. J.; Brown, A. Biochemistry 1999, 38, 6369–6373. doi:10.1021/bi990373g
Return to citation in text: [1] -
Erixon, K.; Dabalos, C.; Leeper, F. J. Chem. Commun. 2007, 960–962. doi:10.1039/b615861g
Return to citation in text: [1] [2] -
Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F
Return to citation in text: [1] -
Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d
Return to citation in text: [1] -
Istrate, F. M.; Gagosz, F. Beilstein J. Org. Chem. 2011, 7, 878–885. doi:10.3762/bjoc.7.100
Return to citation in text: [1] -
Li, E.; Yao, W.; Xie, X.; Wang, C.; Shao, Y.; Li, Y. Org. Biomol. Chem. 2012, 10, 2960–2965. doi:10.1039/c2ob07173h
Return to citation in text: [1] -
Hoffmann, M.; Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. Beilstein J. Org. Chem. 2013, 9, 1774–1780. doi:10.3762/bjoc.9.206
Return to citation in text: [1] -
Wang, T.; Shi, S.; Hansmann, M. M.; Rettenmeier, E.; Rudolph, M.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2014, 53, 3715–3719. doi:10.1002/anie.201310146
Return to citation in text: [1] -
Wang, T.; Shi, S.; Rudolph, M.; Hashmi, A. S. K. Adv. Synth. Catal. 2014, 356, 2337–2342. doi:10.1002/adsc.201400356
Return to citation in text: [1] -
Wang, T.; Huang, L.; Shi, S.; Rudolph, M.; Hashmi, A. S. K. Chem. – Eur. J. 2014, in press. doi:10.1002/chem.201404229
Return to citation in text: [1] -
Muzart, J. Tetrahedron 2008, 64, 5815–5849. doi:10.1016/j.tet.2008.04.018
Return to citation in text: [1] -
Shen, H. C. Tetrahedron 2008, 64, 3885–3903. doi:10.1016/j.tet.2008.01.081
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] -
Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592
Return to citation in text: [1] -
Hashmi, A. S. K. Catal. Today 2007, 122, 211–214. doi:10.1016/j.cattod.2006.10.006
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c
Return to citation in text: [1] -
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454
Return to citation in text: [1] -
Hoffmann-Röder, A.; Krause, N. Org. Biomol. Chem. 2005, 3, 387–391. doi:10.1039/b416516k
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m
Return to citation in text: [1] [2] -
Ravindar, K.; Reddy, M. S.; Deslongchamps, P. Org. Lett. 2011, 13, 3178–3181. doi:10.1021/ol201102x
Return to citation in text: [1] -
Lindqvist, Y.; Schneider, G. Curr. Opin. Struct. Biol. 1993, 3, 896–901. doi:10.1016/0959-440X(93)90153-C
Return to citation in text: [1] -
Candy, J. M.; Duggleby, R. G. Biochim. Biophys. Acta 1998, 1385, 323–338. doi:10.1016/S0167-4838(98)00077-6
Return to citation in text: [1] -
Dobritzsch, D.; König, S.; Schneider, G.; Lu, G. J. Biol. Chem. 1998, 273, 20196–20204. doi:10.1074/jbc.273.32.20196
Return to citation in text: [1] -
Dyda, F.; Furey, W.; Swaminathan, S.; Sax, M.; Farrenkopf, B.; Jordan, F. Biochemistry 1993, 32, 6165–6170. doi:10.1021/bi00075a008
Return to citation in text: [1] -
Erixon, K. M.; Dabalos, C. L.; Leeper, F. J. Org. Biomol. Chem. 2008, 6, 3561–3572. doi:10.1039/b806580b
Return to citation in text: [1] [2] [3] -
Kluger, R.; Gish, G.; Kauffman, G. J. Biol. Chem. 1984, 259, 8960–8965.
Return to citation in text: [1] [2] -
Diefenbach, R. J.; Duggleby, R. G. Biochem. J. 1991, 276, 439–445.
Return to citation in text: [1]
| 34. | Candy, J. M.; Duggleby, R. G. Biochim. Biophys. Acta 1998, 1385, 323–338. doi:10.1016/S0167-4838(98)00077-6 |
| 35. | Dobritzsch, D.; König, S.; Schneider, G.; Lu, G. J. Biol. Chem. 1998, 273, 20196–20204. doi:10.1074/jbc.273.32.20196 |
| 36. | Dyda, F.; Furey, W.; Swaminathan, S.; Sax, M.; Farrenkopf, B.; Jordan, F. Biochemistry 1993, 32, 6165–6170. doi:10.1021/bi00075a008 |
| 37. | Erixon, K. M.; Dabalos, C. L.; Leeper, F. J. Org. Biomol. Chem. 2008, 6, 3561–3572. doi:10.1039/b806580b |
| 38. | Kluger, R.; Gish, G.; Kauffman, G. J. Biol. Chem. 1984, 259, 8960–8965. |
| 9. | Hawksley, D.; Griffin, D. A.; Leeper, F. J. J. Chem. Soc., Perkin Trans. 1 2001, 144–148. doi:10.1039/b006962k |
| 10. | Mann, S.; Perez Melero, C.; Hawksley, D.; Leeper, F. J. Org. Biomol. Chem. 2004, 2, 1732–1741. doi:10.1039/b403619k |
| 33. | Lindqvist, Y.; Schneider, G. Curr. Opin. Struct. Biol. 1993, 3, 896–901. doi:10.1016/0959-440X(93)90153-C |
| 1. | Schellenberger, A. Biochim. Biophys. Acta 1998, 1385, 177–186. doi:10.1016/S0167-4838(98)00067-3 |
| 6. | Lüdtke, S.; Neumann, P.; Erixon, K. M.; Leeper, F. J.; Kluger, R.; Ficner, R.; Tittmann, K. Nat. Chem. 2013, 5, 762–767. doi:10.1038/nchem.1728 |
| 31. | Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m |
| 32. | Ravindar, K.; Reddy, M. S.; Deslongchamps, P. Org. Lett. 2011, 13, 3178–3181. doi:10.1021/ol201102x |
| 3. | Breslow, R. J. Am. Chem. Soc. 1957, 79, 1762–1763. doi:10.1021/ja01564a064 |
| 4. | Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719–3726. doi:10.1021/ja01547a064 |
| 21. | Muzart, J. Tetrahedron 2008, 64, 5815–5849. doi:10.1016/j.tet.2008.04.018 |
| 22. | Shen, H. C. Tetrahedron 2008, 64, 3885–3903. doi:10.1016/j.tet.2008.01.081 |
| 23. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 24. | Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592 |
| 25. | Hashmi, A. S. K. Catal. Today 2007, 122, 211–214. doi:10.1016/j.cattod.2006.10.006 |
| 26. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c |
| 27. | Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454 |
| 28. | Hoffmann-Röder, A.; Krause, N. Org. Biomol. Chem. 2005, 3, 387–391. doi:10.1039/b416516k |
| 29. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c |
| 30. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 12. | Erixon, K.; Dabalos, C.; Leeper, F. J. Chem. Commun. 2007, 960–962. doi:10.1039/b615861g |
| 37. | Erixon, K. M.; Dabalos, C. L.; Leeper, F. J. Org. Biomol. Chem. 2008, 6, 3561–3572. doi:10.1039/b806580b |
| 31. | Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m |
| 11. | Jordan, F.; Li, H. J.; Brown, A. Biochemistry 1999, 38, 6369–6373. doi:10.1021/bi990373g |
| 10. | Mann, S.; Perez Melero, C.; Hawksley, D.; Leeper, F. J. Org. Biomol. Chem. 2004, 2, 1732–1741. doi:10.1039/b403619k |
| 10. | Mann, S.; Perez Melero, C.; Hawksley, D.; Leeper, F. J. Org. Biomol. Chem. 2004, 2, 1732–1741. doi:10.1039/b403619k |
| 13. | Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F |
| 14. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d |
| 15. | Istrate, F. M.; Gagosz, F. Beilstein J. Org. Chem. 2011, 7, 878–885. doi:10.3762/bjoc.7.100 |
| 16. | Li, E.; Yao, W.; Xie, X.; Wang, C.; Shao, Y.; Li, Y. Org. Biomol. Chem. 2012, 10, 2960–2965. doi:10.1039/c2ob07173h |
| 17. | Hoffmann, M.; Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. Beilstein J. Org. Chem. 2013, 9, 1774–1780. doi:10.3762/bjoc.9.206 |
| 18. | Wang, T.; Shi, S.; Hansmann, M. M.; Rettenmeier, E.; Rudolph, M.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2014, 53, 3715–3719. doi:10.1002/anie.201310146 |
| 19. | Wang, T.; Shi, S.; Rudolph, M.; Hashmi, A. S. K. Adv. Synth. Catal. 2014, 356, 2337–2342. doi:10.1002/adsc.201400356 |
| 20. | Wang, T.; Huang, L.; Shi, S.; Rudolph, M.; Hashmi, A. S. K. Chem. – Eur. J. 2014, in press. doi:10.1002/chem.201404229 |
| 9. | Hawksley, D.; Griffin, D. A.; Leeper, F. J. J. Chem. Soc., Perkin Trans. 1 2001, 144–148. doi:10.1039/b006962k |
| 10. | Mann, S.; Perez Melero, C.; Hawksley, D.; Leeper, F. J. Org. Biomol. Chem. 2004, 2, 1732–1741. doi:10.1039/b403619k |
| 10. | Mann, S.; Perez Melero, C.; Hawksley, D.; Leeper, F. J. Org. Biomol. Chem. 2004, 2, 1732–1741. doi:10.1039/b403619k |
| 7. | Nemeria, N. S.; Chakraborty, S.; Balakrishnan, A.; Jordan, F. FEBS J. 2009, 276, 2432–2446. doi:10.1111/j.1742-4658.2009.06964.x |
| 8. | Nemeria, N. S.; Chakraborty, S.; Baykal, A.; Korotchkina, L. G.; Patel, M. S.; Jordan, F. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 78–82. doi:10.1073/pnas.0609973104 |
| 12. | Erixon, K.; Dabalos, C.; Leeper, F. J. Chem. Commun. 2007, 960–962. doi:10.1039/b615861g |
| 37. | Erixon, K. M.; Dabalos, C. L.; Leeper, F. J. Org. Biomol. Chem. 2008, 6, 3561–3572. doi:10.1039/b806580b |
© 2014 Iqbal et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)