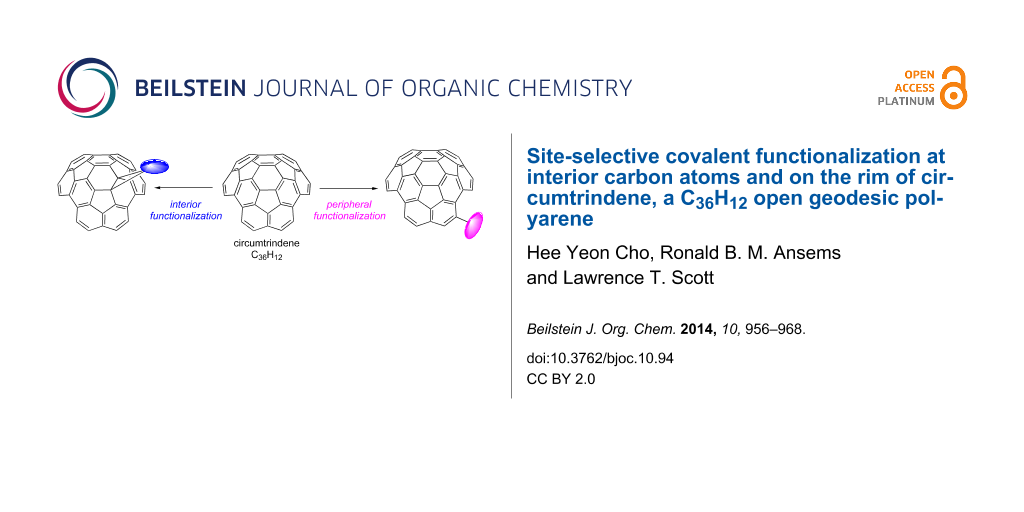
Abstract
Circumtrindene (6, C36H12), one of the largest open geodesic polyarenes ever reported, exhibits fullerene-like reactivity at its interior carbon atoms, whereas its edge carbons react like those of planar polycyclic aromatic hydrocarbons (PAHs). The Bingel–Hirsch and Prato reactions – two traditional methods for fullerene functionalization – afford derivatives of circumtrindene with one of the interior 6:6 C=C bonds modified. On the other hand, functionalization on the rim of circumtrindene can be achieved by normal electrophilic aromatic substitution, the most common reaction of planar PAHs. This peripheral functionalization has been used to extend the π-system of the polyarene by subsequent coupling reactions and to probe the magnetic environment of the concave/convex space around the hydrocarbon bowl. For both classes of functionalization, computational results are reported to complement the experimental observations.

Graphical Abstract
Introduction
Investigations into the structures and properties of geodesic polyarenes began with the synthesis of corannulene (1, C20H10) in 1966 [1,2] and were greatly stimulated by the discovery of buckminsterfullerene (2, C60) in 1985 (Figure 1) [3]. Whereas fullerenes comprising complete three-dimensional polyhedra can be classified as “closed” geodesic polyarenes, curved subunits of fullerenes are regarded as “open” geodesic polyarenes [4]. The fullerenes constitute one family of geodesic polyarenes, and bowl-shaped polycyclic aromatic hydrocarbons (PAHs) constitute another.
![[1860-5397-10-94-1]](/bjoc/content/figures/1860-5397-10-94-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Prototypical open and closed geodesic polyarenes.
Figure 1: Prototypical open and closed geodesic polyarenes.
Both open and closed geodesic polyarenes are characterized by curved π-systems composed of pyramidalized carbon atoms (Figure 2). Pyramidalization not only imposes curvature, but it also imparts significant electronic perturbations. The less parallel the alignment of p-orbitals, for example, the poorer will be their overlap, and that will lead to weaker π-bonds. Weakening a π-bond raises the energy of the bonding π molecular orbital (MO) and lowers the energy of the antibonding π*-MO. This is one important consequence of pyramidalization. A second consequence is the mixing of s-orbital character with the p-orbitals in the π-system. This hybridization lowers the energy of each atomic orbital in the π-system and thereby lowers the energy of all the derived π-MOs, i.e., both the HOMO (highest occupied molecular orbital) and the LUMO (lowest unoccupied molecular orbital) [5]. This lowering of the HOMO offsets the raising of the HOMO caused by poorer π overlap; however, lowering the LUMO as a consequence of orbital hybridization amplifies the lowering caused by the weaker π-bond. The major electronic consequence of π-system curvature, therefore, is a substantial lowering of the energy of the LUMO, while leaving the energy or the HOMO relatively unchanged.
![[1860-5397-10-94-2]](/bjoc/content/figures/1860-5397-10-94-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Planar vs pyramidalized π-system.
Figure 2: Planar vs pyramidalized π-system.
Considerable effort by a number of research groups has been devoted to the preparation of fullerene fragments and to studies of their chemical and physical properties [6-9]. Several laboratories, including the authors’, have been particularly successful in synthesizing geodesic polyarenes using flash vacuum pyrolysis (FVP) [6]. The FVP method involves slow sublimation of a starting material under vacuum, rapid passage of the gas-phase molecules through a hot zone, and subsequent capture of the products in a cold trap [10-12]. The high temperatures used in FVP (often up to 1100 °C) provide sufficient thermal energy to enable molecules to overcome high activation energy barriers.
The FVP approach takes advantage of the fact that the normal out-of-plane deformations of simple planar polyarenes become greatly amplified at high temperatures. FVP has been employed to prepare numerous highly-curved polyarenes, including corannulene (1) [[13,14], dibenzo[a,g]corannulene (3, C28H14) [15], a C3-symmetric hemifullerene (4, C30H12) [16,17], the geodesic heterocycle triphenyleno[1,12-bcd:4,5-b’c’d’:8,9-b’’c’’d’’]trithiophene (5, C18H6S3) [18], the deep buckybowl circumtrindene (6, C36H12) [19,20], and even fullerene C60 (2) (Figure 1 and Figure 3) [21,22]. Syntheses of curved PAHs are not limited, however, to FVP; non-pyrolytic, “wet chemical” methods to access fullerene fragments have also been developed in our laboratory and elsewhere [1,2,15,23-27].
![[1860-5397-10-94-3]](/bjoc/content/figures/1860-5397-10-94-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected examples of geodesic polyarenes synthesized by FVP.
Figure 3: Selected examples of geodesic polyarenes synthesized by FVP.
In addition to synthesis, the functionalization of geodesic polyarenes – by both covalent and noncovalent bonding – has attracted significant attention in recent years. The functionalization of fullerenes was intensively explored immediately following the discovery of methods for bulk preparation of fullerenes in 1990 [28]. Within the first few years, several covalent functionalization sequences were introduced that became widely used for the construction of multifunctional architectures with C60 as an integral building unit. Representative synthetic reactions of fullerene C60 (2) include cyclopropanation [29,30], [3 + 2] cycloaddition [31,32], [4 + 2] cycloaddition [33], nucleophilic addition [34], and radical addition reactions [35]. Two of the earliest and most widely used reactions, the Bingel–Hirsch reaction [29,30] and the Prato reaction [31,32] are illustrated in Scheme 1. The Bingel–Hirsch reaction affords a cyclopropanated fullerene 7, and the Prato reaction gives a [3 + 2] cycloaddition adduct 8. Concurrent with the development of covalent functionalization, numerous noncovalent functionalization methods were also investigated. In particular, the preparation of endohedral fullerenes, having metal atoms or other “guests” trapped inside the fullerene cage, opened yet another new branch of fullerene chemistry [36,37].
![[1860-5397-10-94-i1]](/bjoc/content/inline/1860-5397-10-94-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Covalent functionalization of fullerene C60 by the Bingel–Hirsch reaction and the Prato reaction.
Scheme 1: Covalent functionalization of fullerene C60 by the Bingel–Hirsch reaction and the Prato reaction.
Our laboratory has been interested in the covalent functionalization of open geodesic polyarenes at their interior carbon atoms (i.e., carbon atoms that are shared by three rings) [4,38]. The curved PAHs corannulene (1) and diindenochrysene (10), for example, show fullerene-type reactivity towards carbenes and nucleophiles, respectively (Scheme 2). Dichlorocarbene adds to corannulene (1) to afford predominantly the cyclopropanated product 9 [39], whereas methyllithium adds to 10, giving the 1,2-addition product 11, after alkylation of the resulting anion by methyl iodide [40]. In contrast to these examples, no planar PAH has ever been observed to suffer direct covalent bond formation at an interior carbon atom, despite the long history of aromatic hydrocarbon chemistry [41,42].
![[1860-5397-10-94-i2]](/bjoc/content/inline/1860-5397-10-94-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Fullerene-type chemistry at interior carbon atoms of corannulene (1) and diindenochrysene (10).
Scheme 2: Fullerene-type chemistry at interior carbon atoms of corannulene (1) and diindenochrysene (10).
These PAHs (1 and 10) are obviously nonplanar and have the same patterns of pentagons and hexagons as that found on the surface of fullerene C60. The distortions of the π-systems in these molecules, however, are less severe than that in C60, as indicated by their π-orbital axis vector (POAV) angles [43,44]. The POAV angles of C60 (2), corannulene (1), and diindenochrysene (10) are 11.6° (101.6 –90° = 11.6° pyramidalization), 8.2°, and 9.0°, respectively (Figure 4) [23,24,45]. Furthermore, the barrier for bowl-to-bowl inversion in corannulene (10.2 kcal/mol at −64 °C) [46] is so low that corannulene inverts rapidly even at room temperature. The inversion barrier of diindenochrysene (10) was calculated to be even lower (6.7 kcal/mol at ambient temperature by B3LYP/6-311G**) [47].
![[1860-5397-10-94-4]](/bjoc/content/figures/1860-5397-10-94-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: POAV angles of fullerene C60 (2), corannulene (1), and diindenochrysene (10).
Figure 4: POAV angles of fullerene C60 (2), corannulene (1), and diindenochrysene (10).
While there are many examples of covalent chemistry for fullerenes (i.e., closed geodesic polyarenes), covalent functionalizations of open geodesic polyarenes other than corannulene have been reported only rarely. As in fullerene chemistry, chemical derivatization of buckybowls can be expected to change the electronic, optical, magnetic, mechanical, and chemical properties of the material. For such functionalization reactions, site selectivity often becomes an issue when there is more than one site where the reaction could occur. Herein, we report methods for the site-selective covalent functionalization of circumtrindene (6), one of the most highly-curved fullerene fragments known, comprising 60% of the C60 ball.
Results and Discussion
Synthesis and properties of circumtrindene (6)
In 1996, our laboratory reported that the C36H12 bowl circumtrindene (6) could be obtained by flash vacuum pyrolysis of decacyclene (11, Scheme 3) [19]. Extreme temperatures, in the range of 1200–1300 °C, were required, and the yield was less than 1%, but the transformation gave 60% of the C60 ball in multimilligram quantities in a single step from a cheap, commercially available starting material with no reagents other than heat. Obviously, this brute-force triple-cyclodehydrogenation is not an efficient process. The yield could be improved dramatically, however, by the incorporation of substituents capable of generating aryl radicals by C–X bond homolysis in the fjord regions; the C36H12 bowl was produced in 26% yield from 3,9,15-trichlorodecacyclene (12) by FVP at 1100 °C (Scheme 3) [20].
![[1860-5397-10-94-i3]](/bjoc/content/inline/1860-5397-10-94-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of circumtrindene (6) by FVP.
Scheme 3: Synthesis of circumtrindene (6) by FVP.
The importance of strategically functionalizing hydrocarbon precursors in a way that will generate radical centers where new bonds are to be formed by FVP is dramatically illustrated by the vastly improved efficiency of this triple cyclization reaction when starting from 12 instead of 11. Unfortunately, the extra mass and polarity introduced by the chlorine atoms impedes sublimation of the substrate. A thin stream of nitrogen carrier gas leaked into the sample chamber largely overcomes this complication; however, even with this modification of the set-up, some 30% of starting material 12 decomposes in the sample chamber and fails to sublime.
The halogenated FVP precursor, 3,9,15-trichlorodecacyclene (12), was prepared in five steps from commercially available starting materials, following the same route reported in our earlier publication (Scheme 4) [20]. Cyclotrimerization of 17 to the C3-symmetric trichlorodecacyclene 12 was accomplished by a triple aldol condensation of the chloro ketone 17 [48,49]. Improvements in the procedures reported previously are described in the experimental section in the Supporting Information File 1.
![[1860-5397-10-94-i4]](/bjoc/content/inline/1860-5397-10-94-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthetic route to 3,9,15-trichlorodecacyclene (12).
Scheme 4: Synthetic route to 3,9,15-trichlorodecacyclene (12).
In agreement with DFT calculations [50] the X-ray crystal structure of circumtrindene (6) confirms that the carbon atoms comprising the top of the dome are distorted from planarity to an even greater degree than the carbon atoms in C60 [51]; the pyramidalization in circumtrindene (POAV angle = 12.1° [52], Figure 5) significantly surpasses that in fullerene C60 (POAV angle = 11.6°, Figure 4). Moreover, the X-ray structure of circumtrindene (6) reveals that the carbon–carbon bonds around the 6-membered ring at the top of the dome are not equivalent. The bond lengths vary depending on their location (Figure 5), as is also observed in fullerenes [53]. The carbon–carbon bond shared by two six-membered rings (so-called 6:6-bonds: 1.379 Å) are shorter than those shared by a 5-membered ring and a 6-membered ring (5:6-bonds: 1.430 Å). That is, the carbon–carbon bonds of the hexagonal ring in the center of circumtrindene are not the same as those in benzene. They look more like those of a “cyclohexatriene” than as those of a benzene ring.
![[1860-5397-10-94-5]](/bjoc/content/figures/1860-5397-10-94-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: POAV angle and bond lengths of circumtrindene.
Figure 5: POAV angle and bond lengths of circumtrindene.
The 1H NMR spectrum of circumtrindene (6) consists of two doublets at δ 7.58 and 7.20 ppm. Induced diamagnetic ring currents in this conjugated π-system deshield the hydrogen atoms on the rim; however, the magnitude of the effect is significantly less pronounced than that seen even in smaller PAHs that are less curved or planar [54]. Clearly, the high degree of curvature in this geodesic polyarene has a strong impact on the NMR resonances. The unequal magnitudes of the ring currents on the convex and concave faces of circumtrindene are probed and discussed in a later section.
Interior functionalization of circumtrindene
Unlike planar PAHs, circumtrindene (6) exhibits fullerene-like covalent chemistry at its “interior” carbon atoms. Two examples are reported below [55].
Bingel–Hirsch reaction
The Bingel–Hirsch reaction produces 3-membered rings by a reaction that begins with a nucleophilic addition to a fullerene (Scheme 1). It is considered to be one of the most valuable preparative methods for functionalizing fullerenes, since it changes both the solubility and the electrochemical behavior.
Considering the similarity between the curved π-systems of circumtrindene and fullerene C60, we speculated that the Bingel–Hirsch reaction might occur with circumtrindene (6) in the same manner as it does with fullerene C60. Gratifyingly, the cyclopropanation of circumtrindene by the Bingel–Hirsch reaction proceeds smoothly to afford 20 in good yield and with complete site selectivity (Scheme 5). The reaction was performed with five equivalents of bromomalonate and one equivalent of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) as a base in toluene at room temperature.
![[1860-5397-10-94-i5]](/bjoc/content/inline/1860-5397-10-94-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Bingel–Hirsch reaction of circumtrindene (6).
Scheme 5: Bingel–Hirsch reaction of circumtrindene (6).
Based on mechanism studies on the corresponding reaction of fullerenes, the mechanism in Scheme 6 is proposed for the Bingel–Hirsch reaction of circumtrindene. The enolate generated by deprotonation of bromomalonate 18 with DBU functions as a nucleophile; it attacks one of the strongly pyramidalized carbon atoms of circumtrindene, and subsequent intramolecular displacement of the bromide generates the cyclopropanated circumtrindene 20. As with fullerenes, this cyclopropanation reaction occurs exclusively at a 6:6-bond of circumtrindene.
![[1860-5397-10-94-i6]](/bjoc/content/inline/1860-5397-10-94-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed mechanism for the Bingel–Hirsch reaction of circumtrindene (6).
Scheme 6: Proposed mechanism for the Bingel–Hirsch reaction of circumtrindene (6).
In contrast to the highly symmetrical fullerene C60, circumtrindene has three distinct sets of 6:6-bonds. Only structure 20, however, is consistent with the 1H and 13C NMR spectra of the cyclopropanated circumtrindene obtained (Cs-symmetric with non-equivalent esters, see Supporting Information File 1). Thus, the reaction is site selective for the 6:6-bond at the point of greatest curvature on the convex surface (i.e., the carbon atoms with the greatest POAV angle, see Figure 5). Similar site selectivity for the 6:6-bond that incorporates the most pyramidalized carbon atoms has previously been observed in the functionalization of fullerene C70 [56].
As explained in the introductory section, curving a π-system lowers the energy of the LUMO as a consequence of both poorer π overlap and increased s-character in the atomic orbitals used for π-bonding. The unusually high reactivity of both fullerenes and circumtrindene towards nucleophiles is one manifestation of these lowered LUMO energies. In the initial nucleophilic attack (Scheme 6), the nucleophile adds to the site with the largest LUMO coefficient (see below for DFT calculations on the LUMO of circumtrindene).
This reaction also proceeds with excellent facial selectivity; the cyclopropane ring is formed exclusively on the convex face of circumtrindene. The different electron densities of the two faces reinforce the steric bias for addition to the convex face. As a consequence of the curved π-system, the convex face has less electron density than the concave face (see DFT calculations below), and that renders the convex side electronically more favorable for a nucleophilic attack.
Prato reaction
Cycloadditions to the π-system constitute another major family of fullerene reactions. In particular, 1,3-dipolar reagents readily add to the 6:6-bond of fullerene C60, which acts as a good dipolarophile. Among such [3 + 2] cycloadditions, the reaction with azomethine ylides has found great popularity, because it leads to versatile pyrrolidine derivatives of C60. The sources of the ylide can be iminium salts, aziridines, oxazolidines or silylated iminium compounds. In 1993, Prato and coworkers developed the protocol that is now used most commonly [31,32]. In the first step, an N-substituted amino acid (e.g., N-methylglycine) reacts with an aldehyde or ketone to generate an azomethine ylides in situ. Trapping of the ylide by a fullerene provides a fullerene-pyrrolidine derivative (8), as illustrated in Scheme 1.
In light of the great electrophilicity of circumtrindene in the Bingel–Hirsch reaction, it came as no surprise that circumtrindene can act as a good dipolarophile in a [3 + 2] cycloaddition reaction as well. Accordingly, the azomethine ylide 23 generated in situ from N-methylglycine and formaldehyde adds to circumtrindene in refluxing benzene to give the pyrrolidine derivative 24 (Scheme 7). As in the Bingel–Hirsch reaction, the 1,3-dipole reacts exclusively with the 6:6-bond in circumtrindene that contains the most pyramidalized carbon atoms in the molecule.
![[1860-5397-10-94-i7]](/bjoc/content/inline/1860-5397-10-94-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Prato reaction of circumtrindene (6).
Scheme 7: Prato reaction of circumtrindene (6).
Considering the [3 + 2] cycloaddition processes in terms of frontier molecular orbitals, the dominant interaction should be that between the HOMO of the 1,3-dipole and the LUMO of the dipolarophile. In the Prato reaction in Scheme 7, the electron-rich ylide seeks out the carbon atoms of circumtrindene with the highest LUMO coefficient. As in the Bingel–Hirsch reaction above, the Prato reaction of circumtrindene likewise shows complete facial selectivity for the convex face.
Molecular orbitals and electrostatic potentials
Both of these fullerene-type functionalization reactions at the interior carbon atoms of circumtrindene show complete site and facial selectivity. The molecule behaves as an electrophile toward the bromoenolate (deprotonated bromomalonate) in the Bingel–Hirsch reaction and as an electrophile toward the 1,3-dipole (azomethine ylide) in the Prato reaction. Consequently, the LUMO of circumtrindene is the frontier molecular orbital that should control these interior functionalizations. As noted above, the distortion of the circumtrindene π-system imposed by its geodesic curvature is expected to lower the LUMO energy of the molecule substantially, thereby enhancing its reactivity (see Figure 2).
We have calculated the LUMO of circumtrindene at the B3LPY/6-31G* level of theory, using the curved geometry obtained by optimization at the same level of theory, and find that it is doubly degenerated. Figure 6 depicts one of these antibonding orbitals in two different formats. As these images show, the LUMO of circumtrindene has its largest coefficients at the interior 6:6-bond composed of carbon atoms with the greatest degree of pyramidalization. This is precisely where the Bingel–Hirsch reaction and the Prato reaction of circumtrindene occur. The DFT calculations and the experimental results are therefore in complete agreement.
![[1860-5397-10-94-6]](/bjoc/content/figures/1860-5397-10-94-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: LUMO orbital map of circumtrindene (B3LYP/6-31G*). The darkest blue areas correspond to the regions with the largest LUMO coefficients.
Figure 6: LUMO orbital map of circumtrindene (B3LYP/6-31G*). The darkest blue areas correspond to the regions...
As another consequence of π-system curvature, the electron densities for the two different faces of circumtrindene are expected to be unequal [57]. Density functional calculations (B3LPY/6-31G*) reveal that the outer surface of circumtrindene should be relatively electron deficient, resembling that of C60, whereas the inside surface should be relatively electron rich (Figure 7). The high facial selectivity of these interior functionalization reactions, giving exclusive covalent bond formation on the convex face with electron-rich reaction partners, is also consistent with the DFT calculations.
![[1860-5397-10-94-7]](/bjoc/content/figures/1860-5397-10-94-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Electrostatic potentials on the surfaces of circumtrindene (B3LPY/6-31G*).
Figure 7: Electrostatic potentials on the surfaces of circumtrindene (B3LPY/6-31G*).
Peripheral functionalization of circumtrindene
Since the isolation and characterization of the first polycyclic aromatic hydrocarbons in the 1800s, a tremendous range of chemical reactions on such compounds has been developed [41,42]. Most of this chemistry involves chemical modifications on the edge carbons and has been used to functionalize the existing polyarenes and/or to extend them to larger PAHs. The most common class of reactions is the electrophilic aromatic substitution; however, free radical, nucleophilic addition, reduction, and oxidation reactions are also possible. To synthesize larger PAHs that are not available from natural sources or to make existing ones in more rational ways, functionalization reactions can be followed by functional group interconversions and coupling reactions.
As discussed above, the synthesis of bowl-shaped geodesic polyarenes is non-trivial, owing to the great strain energy in such molecules. In this regard, it is generally easier to construct deeper bowls from already existing curved molecules, rather than to make them from planar building blocks. Circumtrindene (6) has a curvature comparable to C60 fullerene and, therefore, has much of the strain already built in that is needed to synthesize deeper bowls. Circumtrindene represents 60% of the network of sp2 carbons that constitute C60 fullerene; to go to 70%, 80%, and 90% of C60, the number of carbon atoms added in each step would be six, as in monoindeno-, diindeno-, and triindenocircumtrindene (25–27) (Figure 8). We envisioned that circumtrindene might be used as a building block from which to prepare deeper bowls by a peripheral functionalization method with standard aromatic chemistry.
![[1860-5397-10-94-8]](/bjoc/content/figures/1860-5397-10-94-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Monoindeno- (25), diindeno- (26), and triindenocircumtrindene (27).
Figure 8: Monoindeno- (25), diindeno- (26), and triindenocircumtrindene (27).
Such functionalization reactions will also allow us to probe the magnetic environments of the two different faces of circumtrindene. As illustrated in Figure 8, the inside surface of circumtrindene is considerably more electron rich than the outside surface. If a suitable substituent could be placed on the rim of circumtrindene that hangs probe groups over both the concave and the convex faces without restraints, it would be possible to explore the different magnetic environments above the two surfaces experimentally.
Ring extension
To extend the bowl and π-system of circumtrindene, a reasonable first step would be functionalizing the rim of circumtrindene with a group that is suitable for subsequent coupling reactions, e.g., a bromine atom (Figure 9). Since the α-positions of the naphthalene subunits should be more reactive than the crowded β-positions, the site selectivity between the two types of rim carbon atoms was not expected to present a problem.
![[1860-5397-10-94-9]](/bjoc/content/figures/1860-5397-10-94-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Two different types of rim carbon atoms on circumtrindene.
Figure 9: Two different types of rim carbon atoms on circumtrindene.
We were aware, however, that the interior carbon atoms of circumtrindene might also be brominated. Fullerenes are known to undergo bromine addition with elemental bromine in chlorinated solvents at room temperature [28]. To avoid the unwanted bromination at interior carbon atoms, the reaction conditions were carefully optimized (see Supporting Information File 1 for more details). In a reasonably dilute solution and with two equivalents of bromine, selective monobromination on the α-carbon of circumtrindene has been achieved with a quantitative yield (Scheme 8).
![[1860-5397-10-94-i8]](/bjoc/content/inline/1860-5397-10-94-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Site-selective peripheral monobromination of circumtrindene.
Scheme 8: Site-selective peripheral monobromination of circumtrindene.
With bromocircumtrindene 28 in hand, Suzuki coupling reactions were carried out with commercially available boronic acids. The reaction proceeds smoothly with 2-bromophenylboronic acid in the presence of Pd(0) and a base to furnish 29 (Scheme 9). The Suzuki reaction proceeds reasonably well, despite the potential complication of the product undergoing a subsequent Suzuki coupling with the bromine of the phenyl group. The next step of the sequence, a ring closure to indenocircumtrindene (25), was accomplished by FVP. The bromine in the ortho-position of the phenyl group is presumably lost by homolytic bond cleavage, and the resulting aryl radical then cyclizes [6]. Unfortunately, the reaction also generates a significant amount of circumtrindene by loss of the phenyl group during the FVP process [58].
![[1860-5397-10-94-i9]](/bjoc/content/inline/1860-5397-10-94-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Suzuki coupling and ring-closing reactions toward indenocircumtrindene (25).
Scheme 9: Suzuki coupling and ring-closing reactions toward indenocircumtrindene (25).
In an effort to improve on the yield over the FVP process, solution-phase ring-closure methods were also examined. All attempts to close 29 to indenocircumtrindene (25) by intramolecular arylations (Heck-type cyclization), unfortunately, were unsuccessful. Heck-type cyclizations work nicely with planar PAHs and even on moderately curved PAHs, such as corannulene [15,23,24]. Nonetheless, when applied to the indenocircumtrindene synthesis, the reaction did not give any ring-closed products. Oxidative addition of Pd(0) into one of the strained C–C bonds on the rim of circumtrindene might be competing with the desired cyclization [59]; however, we have not characterized any of the decomposition products from this reaction.
Magnetic environments
Induced diamagnetic ring currents strongly shield nuclei that are positioned over the faces of aromatic rings. In planar PAHs, the π-orbitals are perpendicular to the σ-bonds, the electrons are distributed equally on the two faces of the aromatic system, and the ring currents on the two faces are equivalent. When curvature is built into molecules, however, the electron densities of the two faces become unequal, and the shielding ability of the ring currents are no longer expected to be the same [60]. To probe the ring currents of the two faces of circumtrindene, we designed a compound with groups hanging over both the concave and the convex faces. Such a compound can be synthesized by replacing the bromo group of 28 with a 2,6-dimethyl substituted phenyl group (Scheme 10). The Suzuki coupling was again employed; compound 28 underwent the coupling reaction with 2,6-dimethylphenylboronic acid in the presence of a palladium catalyst [Pd(PPh3)4] and potassium carbonate (K2CO3) to furnish 30 in 60% yield.
![[1860-5397-10-94-i10]](/bjoc/content/inline/1860-5397-10-94-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Suzuki coupling to prepare compound 30.
Scheme 10: Suzuki coupling to prepare compound 30.
In the 1H NMR spectrum of 30, the singlets for the two methyl groups appear at very different chemical shifts: 2.29 and 1.12 ppm. The difference in chemical shift between the two singlets is 1.17 ppm. On the basis of calculations described below, we conclude that the higher field resonance (1.12 ppm) corresponds to the methyl group over the concave side, whereas the lower field resonance (2.29 ppm) corresponds to the methyl group over the convex side.
Since the walls of circumtrindene consist of three naphthalene units, a 2,6-dimethylphenyl substituted naphthalene 31 was synthesized as a reference compound [61]. The naphthalene units of circumtrindene are arched, but the naphthyl group of 31 is planar; therefore, it provides a good comparison for shielding and de-shielding effects. The 1H NMR spectrum of reference compound 31 shows that the methyl groups are magnetically equivalent, as anticipated, giving rise to a single peak at 1.88 ppm (Figure 10). A comparison between 30 and 31 reveals that the signal of the methyl group hanging over the concave side of circumtrindene is shifted upfield by 0.76 ppm, whereas that for the methyl group hanging over the convex side is shifted downfield by 0.41 ppm.
![[1860-5397-10-94-10]](/bjoc/content/figures/1860-5397-10-94-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Chemical shifts of ortho-methyl groups in 30 and 31.
Figure 10: Chemical shifts of ortho-methyl groups in 30 and 31.
To assign the two NMR signals in the circumtrindene derivative, computational studies of the 1H NMR chemical shifts were conducted (GIAO method at the B3LYP/6-31G** level of theory). The results of the calculation show good agreement between the experimentally obtained chemical shifts and those calculated, both for 30 and for reference compound 31 (Table 1). The data clearly indicate that the resonance of the methyl group over the concave face is the one shifted upfield by 0.76 ppm and that the signal for the methyl group over the convex face is the one shifted downfield by 0.41 ppm. Thus, the concave face is substantially more shielding than the convex face. These data reveal a striking difference in the magnetic environments over the two different faces – convex and concave – of circumtrindene, which is a dramatic consequence of the curved π-system.
Table 1: Calculated (B3LYP/6-31G**) and experimentally obtained NMR chemical shifts of methyl groups in compounds 30 and 31.
| Compound | δexp (ppm) | δcalc (ppm) |
|---|---|---|
| 1-(2,6-dimethylphenyl)naphthalene (31) | 1.88 | 1.94 |
| 1-(2,6-dimethylphenyl)circumtrindene (30-endo CH3) | 1.12 | 1.13 |
| 1-(2,6-dimethylphenyl)circumtrindene (30-exo CH3) | 2.29 | 2.32 |
Conclusion
Bowl-shaped geodesic polyarenes can be considered the missing links between the “classic” flat PAHs and the spheroidal fullerenes. The present study has shown that open geodesic polyarenes can exhibit chemistry inherent to both classes of aromatics. The curved π-system induces unequal electron densities and unique magnetic environments on the two faces of circumtrindene, significant strain energy in the molecule, and non-identical bond lengths. The lowered LUMO energy and the interior localization of the LUMO coefficients enabled site-selective interior functionalization of circumtrindene by fullerene-type chemistry. On the other hand, the edge carbons, which are not present in fullerenes, still possess reactivity of common planar PAHs. Peripheral functionalization was conducted on those edge carbons, which allowed the π-system to be extended to make a larger bowl indenocircumtrindene (25), and the magnetic environments of the two different faces were probed. Computational studies not only supported the experimental data but also provided deeper insights into this curved polycyclic aromatic fullerene fragment.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data for all new compounds. | ||
| Format: PDF | Size: 2.0 MB | Download |
References
-
Barth, W. E.; Lawton, R. G. J. Am. Chem. Soc. 1966, 88, 380–381. doi:10.1021/ja00954a049
Return to citation in text: [1] [2] -
Lawton, R. G.; Barth, W. E. J. Am. Chem. Soc. 1971, 93, 1730–1745. doi:10.1021/ja00736a028
Return to citation in text: [1] [2] -
Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162–163. doi:10.1038/318162a0
Return to citation in text: [1] -
Scott, L. T.; Bronstein, H. E.; Preda, D. V.; Ansems, R. B. M.; Bratcher, M. S.; Hagen, S. Pure Appl. Chem. 1999, 71, 209–219. doi:10.1351/pac199971020209
Return to citation in text: [1] [2] -
Fleming, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Chichester, 2010. doi:10.1002/9780470689493
Return to citation in text: [1] -
Tsefrikas, V. M.; Scott, L. T. Chem. Rev. 2006, 106, 4868–4884. doi:10.1021/cr050553y
Return to citation in text: [1] [2] [3] -
Wu, Y.-T.; Siegel, J. S. Chem. Rev. 2006, 106, 4843–4867. doi:10.1021/cr050554q
Return to citation in text: [1] -
Sygula, A.; Rabideau, P. W. In Carbon-Rich Compounds; Haley, M. M.; Tykwinski, R. R., Eds.; Wiley-VCH : Weinheim, 2006; pp 529–565.
Return to citation in text: [1] -
Petrukhina, M. A.; Scott, L. T., Eds. Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry; John Wiley & Sons: Hoboken, 2011. doi:10.1002/9781118011263
Return to citation in text: [1] -
Brown, R. F. C. Pyrolytic Methods in Organic Chemistry: Application of Flow and Flash Vacuum Pyrolytic Techniques; Academic Press: New York, 1980.
Return to citation in text: [1] -
Brown, R. F. C. Pure Appl. Chem. 1990, 62, 1981–1986. doi:10.1351/pac199062101981
Return to citation in text: [1] -
Necula, A.; Scott, L. T. J. Anal. Appl. Pyrolysis 2000, 54, 65–87. doi:10.1016/S0165-2370(99)00085-6
Return to citation in text: [1] -
Scott, L. T.; Hashemi, M. M.; Meyer, D. T.; Warren, H. B. J. Am. Chem. Soc. 1991, 113, 7082–7084. doi:10.1021/ja00018a082
Return to citation in text: [1] -
Scott, L. T.; Cheng, P.-C.; Hashemi, M. M.; Bratcher, M. S.; Meyer, D. T.; Warren, H. B. J. Am. Chem. Soc. 1997, 119, 10963–10968. doi:10.1021/ja972019g
Return to citation in text: [1] -
Reisch, H. A.; Bratcher, M. S.; Scott, L. T. Org. Lett. 2000, 2, 1427–1430. doi:10.1021/ol005755p
Return to citation in text: [1] [2] [3] -
Abdourazak, A. H.; Marcinow, Z.; Sygula, A.; Sygula, R.; Rabideau, P. W. J. Am. Chem. Soc. 1995, 117, 6410–6411. doi:10.1021/ja00128a052
Return to citation in text: [1] -
Hagen, S.; Bratcher, M. S.; Erickson, M. S.; Zimmermann, G.; Scott, L. T. Angew. Chem., Int. Ed. Engl. 1997, 36, 406–408. doi:10.1002/anie.199704061
Return to citation in text: [1] -
Imamura, K.; Takimiya, K.; Otsubo, T.; Aso, Y. Chem. Commun. 1999, 1859–1860. doi:10.1039/a905860e
Return to citation in text: [1] -
Scott, L. T.; Bratcher, M. S.; Hagen, S. J. Am. Chem. Soc. 1996, 118, 8743–8744. doi:10.1021/ja9621511
Return to citation in text: [1] [2] -
Ansems, R. B. M.; Scott, L. T. J. Am. Chem. Soc. 2000, 122, 2719–2724. doi:10.1021/ja993028n
Return to citation in text: [1] [2] [3] -
Scott, L. T.; Boorum, M. M.; McMahon, B. J.; Hagen, S.; Mack, J.; Blank, J.; Wegner, H.; de Meijere, A. Science 2002, 295, 1500–1503. doi:10.1126/science.1068427
Return to citation in text: [1] -
Scott, L. T. Angew. Chem., Int. Ed. 2004, 43, 4994–5007. doi:10.1002/anie.200400661
Return to citation in text: [1] -
Jackson, E. A.; Steinberg, B. D.; Bancu, M.; Wakamiya, A.; Scott, L. T. J. Am. Chem. Soc. 2007, 129, 484–485. doi:10.1021/ja067487h
Return to citation in text: [1] [2] [3] -
Steinberg, B. D.; Jackson, E. A.; Filatov, A. S.; Wakamiya, A.; Petrukhina, M. A.; Scott, L. T. J. Am. Chem. Soc. 2009, 131, 10537–10545. doi:10.1021/ja9031852
Return to citation in text: [1] [2] [3] -
Sygula, A.; Xu, G.; Marcinow, Z.; Rabideau, P. W. Tetrahedron 2001, 57, 3637–3644. doi:10.1016/S0040-4020(01)00251-4
Return to citation in text: [1] -
Sakurai, H.; Daiko, T.; Hirao, T. Science 2003, 301, 1878–1882. doi:10.1126/science.1088290
Return to citation in text: [1] -
Butterfield, A. M.; Gilomen, B.; Siegel, J. S. Org. Process Res. Dev. 2012, 16, 664–676. doi:10.1021/op200387s
Return to citation in text: [1] -
Hirsch, A.; Brettreich, M. Fullerenes; Wiley-VCH: Weinheim, 2004.
Return to citation in text: [1] [2] -
Bingel, C. Chem. Ber. 1993, 126, 1957–1959. doi:10.1002/cber.19931260829
Return to citation in text: [1] [2] -
Camps, X.; Hirsch, A. J. Chem. Soc., Perkin Trans. 1 1997, 1595–1596. doi:10.1039/a702055d
Return to citation in text: [1] [2] -
Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1993, 115, 9798–9799. doi:10.1021/ja00074a056
Return to citation in text: [1] [2] [3] -
Prato, M.; Maggini, M. Acc. Chem. Res. 1998, 31, 519–526. doi:10.1021/ar970210p
Return to citation in text: [1] [2] [3] -
Rubin, Y.; Khan, S.; Freedberg, D. I.; Yeretzian, C. J. Am. Chem. Soc. 1993, 115, 344–345. doi:10.1021/ja00054a049
Return to citation in text: [1] -
Hirsch, A.; Soi, A.; Karfunhel, H. R. Angew. Chem., Int. Ed. Engl. 1992, 31, 766–768. doi:10.1002/anie.199207661
Return to citation in text: [1] -
McEwen, C. N.; McKay, R. G.; Larsen, B. S. J. Am. Chem. Soc. 1992, 114, 4412–4414. doi:10.1021/ja00037a064
Return to citation in text: [1] -
Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2008, 6083–6094. doi:10.1039/b811738a
Return to citation in text: [1] -
Murata, M.; Murata, Y.; Komatsu, K. Molecular Surgery toward Organic Synthesis of Endohedral Fullerenes. In Chemistry of Nanocarbons; Akasaka, T.; Wudl, F.; Nagase, S., Eds.; John Wiley & Sons: Chichester, 2010; pp 215–237. doi:10.1002/9780470660188.ch8
Return to citation in text: [1] -
Carbon atoms on the rims of PAHs can be either methine carbons, with a hydrogen atom or a substituent attached, (belonging to only one ring) or quaternary (belonging to two rings); “interior” carbon atoms belong to three rings.
Return to citation in text: [1] -
Preda, D. V.; Scott, L. T. Tetrahedron Lett. 2000, 41, 9633–9637. doi:10.1016/S0040-4039(00)01734-2
Return to citation in text: [1] -
Bronstein, H. E.; Scott, L. T. J. Org. Chem. 2008, 73, 88–93. doi:10.1021/jo7018224
Return to citation in text: [1] -
Harvey, R. G. Polycyclic Aromatic Hydrocarbons; Wiley-VCH: New York, N. Y., 1997.
Return to citation in text: [1] [2] -
Clar, E. Polycyclic Hydrocarbons; Academic Press: New York, 1964.
Return to citation in text: [1] [2] -
Haddon, R. C.; Scott, L. T. Pure Appl. Chem. 1986, 58, 137–142.
Return to citation in text: [1] -
Haddon, R. C. J. Am. Chem. Soc. 1987, 109, 1676–1685. doi:10.1021/ja00240a013
Return to citation in text: [1] -
Bronstein, H. E.; Choi, N.; Scott, L. T. J. Am. Chem. Soc. 2002, 124, 8870–8875. doi:10.1021/ja0123148
Return to citation in text: [1] -
Scott, L. T.; Hashemi, M. M.; Bratcher, M. S. J. Am. Chem. Soc. 1992, 114, 1920–1921. doi:10.1021/ja00031a079
Return to citation in text: [1] -
Eisenberg, D.; Shenhar, R.; Rabinovitz, M. Anions of Buckybowls. In Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry; Petrukhina, M. A.; Scott, L. T., Eds.; John Wiley & Sons: Hoboken, 2011; pp 63–93. doi:10.1002/9781118011263.ch3
Return to citation in text: [1] -
Boorum, M. M.; Scott, L. T. The synthesis of tris-annulated benzenes by aldol trimerization of cyclic ketones. In Modern Arene Chemistry; Astruc, D., Ed.; Wiley-VCH: Weinheim, 2002; pp 20–31. doi:10.1002/3527601767.ch1
Return to citation in text: [1] -
Amick, A. W.; Scott, L. T. J. Org. Chem. 2007, 72, 3412–3418. doi:10.1021/jo070080q
Return to citation in text: [1] -
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2010.
All the calculations were performed at the B3LYP/6-31G** level of theory.
Return to citation in text: [1] -
Forkey, D. M.; Attar, S.; Noll, B. C.; Koerner, R.; Olmstead, M. M.; Balch, A. L. J. Am. Chem. Soc. 1997, 119, 5766–5767. doi:10.1021/ja970845j
Return to citation in text: [1] -
The POAV angle at the largest curved point of the molecule was considered.
Return to citation in text: [1] -
Bühl, M.; Hirsch, A. Chem. Rev. 2001, 101, 1153–1184. doi:10.1021/cr990332q
See for more details on the X-ray structure of fullerene-C60, which shows that the 6:6-bond of C60 is 1.355 Å; the 6:5-bond is 1.467 Å.
Return to citation in text: [1] -
The 1H NMR spectrum of corannulene (C20H10) shows a single resonance at 7.8 ppm, and the 1H NMR spectrum of coronene (C24H12) shows a single resonance at 8.9 ppm.
Return to citation in text: [1] -
Ansems, R. B. M. Ph.D. Thesis, Boston College, Chestnut Hill, Massachusetts, 2004.
See for additional examples.
Return to citation in text: [1] -
Hawkins, J. M.; Meyer, A.; Solow, M. A. J. Am. Chem. Soc. 1993, 115, 7499–7500. doi:10.1021/ja00069a058
Return to citation in text: [1] -
Klärner, F.-G.; Panitzky, J.; Preda, D.; Scott, L. T. J. Mol. Model. 2000, 6, 318–327. doi:10.1007/PL00010733
Return to citation in text: [1] -
Xue, X. Ph.D. Thesis, Boston College, Chestnut Hill, Massachusetts, 2008.
See for an example of an investigation of the phenyl-loss mechanism is currently on-going in the authors’ laboratory. We have observed similar phenyl loss processes in other FVP reactions with phenyl substituted substrates.
Return to citation in text: [1] -
Shaltout, R. M.; Sygula, R.; Sygula, A.; Fronczek, F. R.; Stanley, G. G.; Rabideau, P. W. J. Am. Chem. Soc. 1998, 120, 835–836. doi:10.1021/ja973454d
Return to citation in text: [1] -
Monaco, G.; Scott, L. T.; Zanasi, R. J. Phys. Chem. A 2008, 112, 8136–8147. doi:10.1021/jp8038779
Return to citation in text: [1] -
Lipshutz, B. H.; Siegmann, K.; Garcia, E.; Kayser, F. J. Am. Chem. Soc. 1993, 115, 9276–9282. doi:10.1021/ja00073a051
Return to citation in text: [1]
| 43. | Haddon, R. C.; Scott, L. T. Pure Appl. Chem. 1986, 58, 137–142. |
| 44. | Haddon, R. C. J. Am. Chem. Soc. 1987, 109, 1676–1685. doi:10.1021/ja00240a013 |
| 23. | Jackson, E. A.; Steinberg, B. D.; Bancu, M.; Wakamiya, A.; Scott, L. T. J. Am. Chem. Soc. 2007, 129, 484–485. doi:10.1021/ja067487h |
| 24. | Steinberg, B. D.; Jackson, E. A.; Filatov, A. S.; Wakamiya, A.; Petrukhina, M. A.; Scott, L. T. J. Am. Chem. Soc. 2009, 131, 10537–10545. doi:10.1021/ja9031852 |
| 45. | Bronstein, H. E.; Choi, N.; Scott, L. T. J. Am. Chem. Soc. 2002, 124, 8870–8875. doi:10.1021/ja0123148 |
| 46. | Scott, L. T.; Hashemi, M. M.; Bratcher, M. S. J. Am. Chem. Soc. 1992, 114, 1920–1921. doi:10.1021/ja00031a079 |
| 51. | Forkey, D. M.; Attar, S.; Noll, B. C.; Koerner, R.; Olmstead, M. M.; Balch, A. L. J. Am. Chem. Soc. 1997, 119, 5766–5767. doi:10.1021/ja970845j |
| 48. | Boorum, M. M.; Scott, L. T. The synthesis of tris-annulated benzenes by aldol trimerization of cyclic ketones. In Modern Arene Chemistry; Astruc, D., Ed.; Wiley-VCH: Weinheim, 2002; pp 20–31. doi:10.1002/3527601767.ch1 |
| 49. | Amick, A. W.; Scott, L. T. J. Org. Chem. 2007, 72, 3412–3418. doi:10.1021/jo070080q |
| 50. |
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2010.
All the calculations were performed at the B3LYP/6-31G** level of theory. |
| 20. | Ansems, R. B. M.; Scott, L. T. J. Am. Chem. Soc. 2000, 122, 2719–2724. doi:10.1021/ja993028n |
| 20. | Ansems, R. B. M.; Scott, L. T. J. Am. Chem. Soc. 2000, 122, 2719–2724. doi:10.1021/ja993028n |
| 47. | Eisenberg, D.; Shenhar, R.; Rabinovitz, M. Anions of Buckybowls. In Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry; Petrukhina, M. A.; Scott, L. T., Eds.; John Wiley & Sons: Hoboken, 2011; pp 63–93. doi:10.1002/9781118011263.ch3 |
| 19. | Scott, L. T.; Bratcher, M. S.; Hagen, S. J. Am. Chem. Soc. 1996, 118, 8743–8744. doi:10.1021/ja9621511 |
| 53. |
Bühl, M.; Hirsch, A. Chem. Rev. 2001, 101, 1153–1184. doi:10.1021/cr990332q
See for more details on the X-ray structure of fullerene-C60, which shows that the 6:6-bond of C60 is 1.355 Å; the 6:5-bond is 1.467 Å. |
| 54. | The 1H NMR spectrum of corannulene (C20H10) shows a single resonance at 7.8 ppm, and the 1H NMR spectrum of coronene (C24H12) shows a single resonance at 8.9 ppm. |
| 55. |
Ansems, R. B. M. Ph.D. Thesis, Boston College, Chestnut Hill, Massachusetts, 2004.
See for additional examples. |
| 58. |
Xue, X. Ph.D. Thesis, Boston College, Chestnut Hill, Massachusetts, 2008.
See for an example of an investigation of the phenyl-loss mechanism is currently on-going in the authors’ laboratory. We have observed similar phenyl loss processes in other FVP reactions with phenyl substituted substrates. |
| 15. | Reisch, H. A.; Bratcher, M. S.; Scott, L. T. Org. Lett. 2000, 2, 1427–1430. doi:10.1021/ol005755p |
| 23. | Jackson, E. A.; Steinberg, B. D.; Bancu, M.; Wakamiya, A.; Scott, L. T. J. Am. Chem. Soc. 2007, 129, 484–485. doi:10.1021/ja067487h |
| 24. | Steinberg, B. D.; Jackson, E. A.; Filatov, A. S.; Wakamiya, A.; Petrukhina, M. A.; Scott, L. T. J. Am. Chem. Soc. 2009, 131, 10537–10545. doi:10.1021/ja9031852 |
| 6. | Tsefrikas, V. M.; Scott, L. T. Chem. Rev. 2006, 106, 4868–4884. doi:10.1021/cr050553y |
| 57. | Klärner, F.-G.; Panitzky, J.; Preda, D.; Scott, L. T. J. Mol. Model. 2000, 6, 318–327. doi:10.1007/PL00010733 |
| 41. | Harvey, R. G. Polycyclic Aromatic Hydrocarbons; Wiley-VCH: New York, N. Y., 1997. |
| 42. | Clar, E. Polycyclic Hydrocarbons; Academic Press: New York, 1964. |
| 56. | Hawkins, J. M.; Meyer, A.; Solow, M. A. J. Am. Chem. Soc. 1993, 115, 7499–7500. doi:10.1021/ja00069a058 |
| 31. | Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1993, 115, 9798–9799. doi:10.1021/ja00074a056 |
| 32. | Prato, M.; Maggini, M. Acc. Chem. Res. 1998, 31, 519–526. doi:10.1021/ar970210p |
| 60. | Monaco, G.; Scott, L. T.; Zanasi, R. J. Phys. Chem. A 2008, 112, 8136–8147. doi:10.1021/jp8038779 |
| 61. | Lipshutz, B. H.; Siegmann, K.; Garcia, E.; Kayser, F. J. Am. Chem. Soc. 1993, 115, 9276–9282. doi:10.1021/ja00073a051 |
| 59. | Shaltout, R. M.; Sygula, R.; Sygula, A.; Fronczek, F. R.; Stanley, G. G.; Rabideau, P. W. J. Am. Chem. Soc. 1998, 120, 835–836. doi:10.1021/ja973454d |
| 1. | Barth, W. E.; Lawton, R. G. J. Am. Chem. Soc. 1966, 88, 380–381. doi:10.1021/ja00954a049 |
| 2. | Lawton, R. G.; Barth, W. E. J. Am. Chem. Soc. 1971, 93, 1730–1745. doi:10.1021/ja00736a028 |
| 6. | Tsefrikas, V. M.; Scott, L. T. Chem. Rev. 2006, 106, 4868–4884. doi:10.1021/cr050553y |
| 7. | Wu, Y.-T.; Siegel, J. S. Chem. Rev. 2006, 106, 4843–4867. doi:10.1021/cr050554q |
| 8. | Sygula, A.; Rabideau, P. W. In Carbon-Rich Compounds; Haley, M. M.; Tykwinski, R. R., Eds.; Wiley-VCH : Weinheim, 2006; pp 529–565. |
| 9. | Petrukhina, M. A.; Scott, L. T., Eds. Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry; John Wiley & Sons: Hoboken, 2011. doi:10.1002/9781118011263 |
| 5. | Fleming, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Chichester, 2010. doi:10.1002/9780470689493 |
| 29. | Bingel, C. Chem. Ber. 1993, 126, 1957–1959. doi:10.1002/cber.19931260829 |
| 30. | Camps, X.; Hirsch, A. J. Chem. Soc., Perkin Trans. 1 1997, 1595–1596. doi:10.1039/a702055d |
| 4. | Scott, L. T.; Bronstein, H. E.; Preda, D. V.; Ansems, R. B. M.; Bratcher, M. S.; Hagen, S. Pure Appl. Chem. 1999, 71, 209–219. doi:10.1351/pac199971020209 |
| 21. | Scott, L. T.; Boorum, M. M.; McMahon, B. J.; Hagen, S.; Mack, J.; Blank, J.; Wegner, H.; de Meijere, A. Science 2002, 295, 1500–1503. doi:10.1126/science.1068427 |
| 22. | Scott, L. T. Angew. Chem., Int. Ed. 2004, 43, 4994–5007. doi:10.1002/anie.200400661 |
| 3. | Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162–163. doi:10.1038/318162a0 |
| 1. | Barth, W. E.; Lawton, R. G. J. Am. Chem. Soc. 1966, 88, 380–381. doi:10.1021/ja00954a049 |
| 2. | Lawton, R. G.; Barth, W. E. J. Am. Chem. Soc. 1971, 93, 1730–1745. doi:10.1021/ja00736a028 |
| 15. | Reisch, H. A.; Bratcher, M. S.; Scott, L. T. Org. Lett. 2000, 2, 1427–1430. doi:10.1021/ol005755p |
| 23. | Jackson, E. A.; Steinberg, B. D.; Bancu, M.; Wakamiya, A.; Scott, L. T. J. Am. Chem. Soc. 2007, 129, 484–485. doi:10.1021/ja067487h |
| 24. | Steinberg, B. D.; Jackson, E. A.; Filatov, A. S.; Wakamiya, A.; Petrukhina, M. A.; Scott, L. T. J. Am. Chem. Soc. 2009, 131, 10537–10545. doi:10.1021/ja9031852 |
| 25. | Sygula, A.; Xu, G.; Marcinow, Z.; Rabideau, P. W. Tetrahedron 2001, 57, 3637–3644. doi:10.1016/S0040-4020(01)00251-4 |
| 26. | Sakurai, H.; Daiko, T.; Hirao, T. Science 2003, 301, 1878–1882. doi:10.1126/science.1088290 |
| 27. | Butterfield, A. M.; Gilomen, B.; Siegel, J. S. Org. Process Res. Dev. 2012, 16, 664–676. doi:10.1021/op200387s |
| 15. | Reisch, H. A.; Bratcher, M. S.; Scott, L. T. Org. Lett. 2000, 2, 1427–1430. doi:10.1021/ol005755p |
| 18. | Imamura, K.; Takimiya, K.; Otsubo, T.; Aso, Y. Chem. Commun. 1999, 1859–1860. doi:10.1039/a905860e |
| 13. | Scott, L. T.; Hashemi, M. M.; Meyer, D. T.; Warren, H. B. J. Am. Chem. Soc. 1991, 113, 7082–7084. doi:10.1021/ja00018a082 |
| 14. | Scott, L. T.; Cheng, P.-C.; Hashemi, M. M.; Bratcher, M. S.; Meyer, D. T.; Warren, H. B. J. Am. Chem. Soc. 1997, 119, 10963–10968. doi:10.1021/ja972019g |
| 19. | Scott, L. T.; Bratcher, M. S.; Hagen, S. J. Am. Chem. Soc. 1996, 118, 8743–8744. doi:10.1021/ja9621511 |
| 20. | Ansems, R. B. M.; Scott, L. T. J. Am. Chem. Soc. 2000, 122, 2719–2724. doi:10.1021/ja993028n |
| 10. | Brown, R. F. C. Pyrolytic Methods in Organic Chemistry: Application of Flow and Flash Vacuum Pyrolytic Techniques; Academic Press: New York, 1980. |
| 11. | Brown, R. F. C. Pure Appl. Chem. 1990, 62, 1981–1986. doi:10.1351/pac199062101981 |
| 12. | Necula, A.; Scott, L. T. J. Anal. Appl. Pyrolysis 2000, 54, 65–87. doi:10.1016/S0165-2370(99)00085-6 |
| 6. | Tsefrikas, V. M.; Scott, L. T. Chem. Rev. 2006, 106, 4868–4884. doi:10.1021/cr050553y |
| 16. | Abdourazak, A. H.; Marcinow, Z.; Sygula, A.; Sygula, R.; Rabideau, P. W. J. Am. Chem. Soc. 1995, 117, 6410–6411. doi:10.1021/ja00128a052 |
| 17. | Hagen, S.; Bratcher, M. S.; Erickson, M. S.; Zimmermann, G.; Scott, L. T. Angew. Chem., Int. Ed. Engl. 1997, 36, 406–408. doi:10.1002/anie.199704061 |
| 34. | Hirsch, A.; Soi, A.; Karfunhel, H. R. Angew. Chem., Int. Ed. Engl. 1992, 31, 766–768. doi:10.1002/anie.199207661 |
| 31. | Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1993, 115, 9798–9799. doi:10.1021/ja00074a056 |
| 32. | Prato, M.; Maggini, M. Acc. Chem. Res. 1998, 31, 519–526. doi:10.1021/ar970210p |
| 33. | Rubin, Y.; Khan, S.; Freedberg, D. I.; Yeretzian, C. J. Am. Chem. Soc. 1993, 115, 344–345. doi:10.1021/ja00054a049 |
| 40. | Bronstein, H. E.; Scott, L. T. J. Org. Chem. 2008, 73, 88–93. doi:10.1021/jo7018224 |
| 41. | Harvey, R. G. Polycyclic Aromatic Hydrocarbons; Wiley-VCH: New York, N. Y., 1997. |
| 42. | Clar, E. Polycyclic Hydrocarbons; Academic Press: New York, 1964. |
| 4. | Scott, L. T.; Bronstein, H. E.; Preda, D. V.; Ansems, R. B. M.; Bratcher, M. S.; Hagen, S. Pure Appl. Chem. 1999, 71, 209–219. doi:10.1351/pac199971020209 |
| 38. | Carbon atoms on the rims of PAHs can be either methine carbons, with a hydrogen atom or a substituent attached, (belonging to only one ring) or quaternary (belonging to two rings); “interior” carbon atoms belong to three rings. |
| 39. | Preda, D. V.; Scott, L. T. Tetrahedron Lett. 2000, 41, 9633–9637. doi:10.1016/S0040-4039(00)01734-2 |
| 31. | Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1993, 115, 9798–9799. doi:10.1021/ja00074a056 |
| 32. | Prato, M.; Maggini, M. Acc. Chem. Res. 1998, 31, 519–526. doi:10.1021/ar970210p |
| 36. | Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2008, 6083–6094. doi:10.1039/b811738a |
| 37. | Murata, M.; Murata, Y.; Komatsu, K. Molecular Surgery toward Organic Synthesis of Endohedral Fullerenes. In Chemistry of Nanocarbons; Akasaka, T.; Wudl, F.; Nagase, S., Eds.; John Wiley & Sons: Chichester, 2010; pp 215–237. doi:10.1002/9780470660188.ch8 |
| 35. | McEwen, C. N.; McKay, R. G.; Larsen, B. S. J. Am. Chem. Soc. 1992, 114, 4412–4414. doi:10.1021/ja00037a064 |
| 29. | Bingel, C. Chem. Ber. 1993, 126, 1957–1959. doi:10.1002/cber.19931260829 |
| 30. | Camps, X.; Hirsch, A. J. Chem. Soc., Perkin Trans. 1 1997, 1595–1596. doi:10.1039/a702055d |
© 2014 Cho et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)