Abstract
With the aim of developing biocompatible and water-soluble C60 derivatives, three types of C60–peptide conjugates consisting of hydrophilic oligopeptide anchors (oligo-Lys, oligo-Glu, and oligo-Arg) were synthesized. A previously reported Prato reaction adduct of a biscarboxylic acid-substituted C60 derivative was subjected to a solid phase synthesis for amide formation with N-terminal amines of peptides on resin to successfully provide C60–peptide conjugates with one C60 and two peptide anchors as water-soluble moieties. Among three C60–peptide conjugates prepared, C60–oligo-Lys was soluble in water at neutral pH, and C60–oligo-Glu was soluble in buffer with a higher pH value, but C60–oligo-Arg was insoluble in water and most other solvents. C60–oligo-Lys and C60–oligo-Glu were characterized by 1H and 13C NMR. Photoinduced 1O2 generation was observed in the most soluble C60–oligo-Lys conjugate under visible light irradiation (527 nm) to show the potential of this highly water-soluble molecule in biological systems, for example, as a photosensitizer in photodynamic therapy.
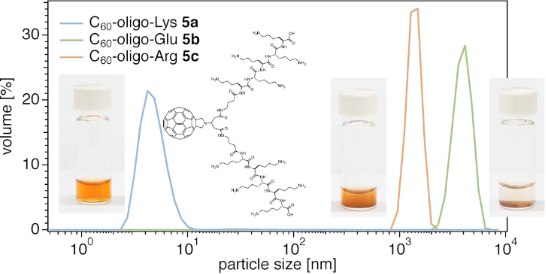
Graphical Abstract
Introduction
Since the seminal discovery in 1985 by Kroto, Smalley, Curl, and co-workers [1], fullerenes, specifically buckminsterfullerene C60, have intrigued the scientific community. The unique structure of fullerenes, characterized by a fully conjugated closed-cage structure, containing a mixture of hexagonal and pentagonal rings, have been recognized for the unique electronic [2-4], optical [5,6], and mechanical properties [7,8]. Despite the notable achievements in fullerene research and the potential applications in diverse fields, a significant obstacle remained for the use in biological studies: fullerenes are poorly soluble in polar solvents, including water and other water-miscible solvents [9]. This challenge consequently restricted the studies of fullerenes as biomaterials since related in vitro bioassay systems require water solubility of the chemicals for testing. To overcome this important obstacle, over the past decades, a variety of water-soluble fullerenes have been reported [10].
General approaches towards enhancing the water solubility of fullerenes involve either 1) covalent functionalization of the fullerene surface with polar moieties or 2) complexation with water-soluble host molecules or polymers. Related to the former approach, the Nakamura group [11], Wudl group [12], and Hirsch group [13] reported initial work in the early 1990s on water-soluble C60 derivatives by covalently attaching water-soluble moieties to the fullerene core. Subsequently, Nakamura and co-workers further developed reactions between C60 and organocopper reagents, enabling the sequential addition of functional groups to obtain penta- and decaadducts, which largely enhanced the water solubility [14,15]. As early examples of the latter approach in the 1990s, Wennerström and co-workers reported the study of supramolecular BiCAP complexation of C60 with γ-cyclodextrin (γ-CD) [16]. Shinkai and co-workers synthesized water-soluble calixarene derivatives to form water-soluble complexes with C60 [17,18]. By either chemical functionalization or complexation of the fullerene core, a number of biocompatible fullerene materials with interesting biological activities were recently prepared and reported [19-24].
We have reported water-soluble complexes of C60 with a nontoxic and nonionic polymer, poly(vinylpyrrolidone) (PVP) [25] and applied these to several in vitro biological assays to report DNA photocleavage [26] and related ROS generation [27,28], antimicrobial photoactivity [29], chondrogenesis-promoting activity [30,31], photocytotoxicity [32,33], and GST enzyme inhibition [34]. For the covalent functionalization, we have previously developed a versatile and convenient biscarboxylic acid-substituted C60 derivative (3, Figure 1) [35], which was prepared via the Prato reaction [36]. We used this derivative 3 as a starting material and synthesized a series of water-soluble C60 and C70 derivatives by covalently attaching biocompatible water-soluble polymers, such as polyethylene glycol (PEG) [37,38] and PVP [39]. Although these C60– and C70–polymer conjugates revealed high water-solubility, it was found that they, especially the PEG conjugates, formed micelle-like aggregations in aqueous solution, as observed by dynamic light scattering (DLS), cryoTEM, and concentration-dependent surface tension measurements [40]. Despite the small size (≈10 nm in hydrodynamic diameter), the aggregation of these fullerene moieties was not ideal for biological applications as photodynamic therapy photosensitizers (PDT PSs) [41] and magnetic resonance imaging contrast agents (MRI CAs) [42], which are the most relevant topics in fullerene biological studies. Aggregated fullerenes in the micelle structure may cause self-quenching of the excited state of PS fullerenes or may inhibit the approach of bulk water molecules to the fullerene core and hamper water exchange – an important factor for enhancement in MRI. Well-dispersed water-soluble C60 derivatives exhibiting less aggregation are in high demand.
![[1860-5397-20-71-1]](/bjoc/content/figures/1860-5397-20-71-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Synthesis of C60–oligopeptide conjugates 5a–c and b) synthesis of compound 3. Fulleropyrrolidine-based biscarboxylic acid derivative 3 was prepared by Prato reaction and subsequent deprotection. Compound 3 was subjected to SPPS with the peptides on trityl resin (i.e., 2a–c) to provide 4a–c. By simultaneous deprotection of peptide side chains and cleavage from resin, C60–oligo-Lys (5a), C60–oligo-Glu (5b), and C60–oligo-Arg (5c) were obtained. Reagents and conditions: i) 20% piperidine, rt, 2 × 10 min, ii) HBTU, DIPEA, in DMF, rt, overnight, and iii) trifluoroacetic acid (TFA)/triisopropylsilane (TIPS)/H2O, rt, 1.5–2 h. AA and PG stand for amino acid and protecting group, respectively. All AAs in 1a–c, 2a–c, and 4a–c were protected.
Figure 1: a) Synthesis of C60–oligopeptide conjugates 5a–c and b) synthesis of compound 3. Fulleropyrrolidine...
To address the challenge mentioned above, we developed highly water-soluble C60–peptide conjugates in this study. In addition to the water solubility introduced by the peptides, these conjugates have a superior biocompatibility compared to those with synthetic polymers, such as PEG and PVP. We utilized the previously reported biscarboxylic acid derivative 3, which was suitable for the coupling to peptides on resin, prepared by solid-phase peptide synthesis (SPPS) [35]. The detailed conditions for the amide-forming reaction were optimized using biscarboxylic acid-substituted C60 derivative 3 and a similar peptide with a primary amine derived from γ-aminobutyric acid (GABA) at the N-terminus of the Lys pentamer peptide on resin. Using the optimal reaction conditions, three types of hydrophilic peptide pentamers on resin, oligo-Lys (2a), oligo-Glu (2b), and oligo-Arg (2c), were subjected to the reaction with 3 for the synthesis of C60–peptide conjugates 5a–c (Figure 1).
Results and Discussion
Syntheses of C60–oligopeptides 5a–c
The oligopeptides 2a–c were synthesized on resin using Fmoc-protected amino acids with a standard SPPS method (Figure 1a) [43]. A moderate loading (0.4 mmol⋅g−1) of the initial amino acid was used. After synthesizing the pentamer of Lys on resin, a GABA residue was attached to the N-terminus of the peptide in order to provide a less-hindered primary amine, enabling an efficient amide conjugation reaction with biscarboxylic acid-substituted C60 derivative 3.
Compound 3 was prepared by Prato reaction of C60 and an N-glycine derivative and subsequent deprotection of the t-Bu group under acidic conditions, without affecting the C60 cage (Figure 1b). Challenges in this step included finding suitable conditions to conjugate one C60 moiety and two peptide anchors on the resin. Preliminarily, the reaction conditions were optimized using a similar peptide (GABA-(Lys)5-peptide-PEG) on resin for the reaction with compound 3. Initial trials, using 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) and N-methylmorpholine (NMM), respectively, as a coupling reagent and a base, and 5 equiv of peptide on resin (rink amide MBHA) relative to 3, provided a rather low yield (13%, isolated by HPLC), which was increased to 24% by changing the solid phase to 2-chlorotrityl chloride resin. By replacing the base with N,N-diisopropylethylamine (DIPEA), the yield was slightly increased to 28%, which became higher (32%) when HBTU was used as a coupling reagent. Finally, use of a greater excess (6.0 equiv) of peptide on resin relative to 3, and a combination of HBTU and DIPEA as the coupling reagent and base, provided the C60–peptide conjugate in an isolated yield of 41% (based on the used compound 3). The yield of C60–peptide conjugate formation decreased when using fewer equivalents of peptide on resin relative to compound 3, providing as a byproduct the C60–peptide conjugate connected to only one peptide anchor.
Based on the optimized reaction conditions outlined above, the conjugation of biscarboxylic acid-substituted C60 derivative 3 and the peptides on resin 2a–c were performed by SPPS to provide the C60–oligopeptides on resin 4a–c (Figure 1a). Subsequently, the last step of the reaction – the cleavage of the C60–peptide conjugate from the resin and the simultaneous deprotection of the amino acid residues – provided C60–peptide conjugates 5a–c with fully deprotected peptides.
The syntheses of C60–oligo-Lys (5a), C60–oligo-Glu (5b), and C60–oligo-Arg (5c) were confirmed by HRMS (Figures S2, S11, and S18, Supporting Information File 1). While C60–oligo-Lys (5a) was successfully observed by HRESIMS in a charged state of 3+ (Figure S2, Supporting Information File 1), C60–oligo-Glu (5b) was confirmed by HRMS–MALDI (Figure S11, Supporting Information File 1) due to insolubility in the acidic eluent generally used for HRESIMS (a mixture of MeOH and water containing 0.1% formic acid). C60–oligo-Arg (5c), which was not sufficiently soluble in most of the solvents, was slightly soluble in the acidic eluent, so that HRESIMS analysis provide HRMS data for a charged state of 4+ (Figure S18, Supporting Information File 1).
C60–oligo-Lys (5a), with sufficient solubility in polar solvents, was isolated by reversed-phase HPLC. C60–oligo-Glu (5b), which was soluble only in basic aqueous solution, could not be isolated by HPLC, especially in the presence of an oligo-Glu impurity, and was purified only after spin filtration. C60–oligo-Arg (5c) was not soluble in any solvent and could not be further purified.
Solubility in water
The water solubility of C60–peptide conjugates 5a–c was tested after the removal of any remaining solvent traces by lyophilization. Upon addition of Milli-Q® water (pH 7.0), C60–oligo-Lys (5a) was immediately and thoroughly solubilized. In contrast, the other conjugates, C60–oligo-Glu (5b, purified) and C60–oligo-Arg (5c, crude), did not produce transparent solutions in water at neutral pH value even by sonication (Figure 2). While C60–oligo-Glu (5b) was soluble in buffer with a higher pH value (>8.3), C60–oligo-Arg (5c) was not soluble in most polar and nonpolar solvents. In addition, 5a was not highly soluble in most nonpolar solvents, including toluene and CH2Cl2, but slightly soluble in other polar solvents, including MeOH and DMSO.
![[1860-5397-20-71-2]](/bjoc/content/figures/1860-5397-20-71-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of C60–oligo-Lys (5a), C60–oligo-Glu (5b), and C60–oligo-Arg (5c) and images of dissolved or dispensed compounds in Milli-Q® water (pH 7.0). aCrude yield.
Figure 2: Structure of C60–oligo-Lys (5a), C60–oligo-Glu (5b), and C60–oligo-Arg (5c) and images of dissolved...
The solubility of C60–peptide conjugates 5a–c was in line with DLS data of the aqueous solutions or dispersions. While 5a (blue line) revealed an extremely small hydrodynamic diameter (<10 nm) by DLS, 5b (green line) and 5c (purple line) revealed the presence of larger aggregates in water (pH 7.0), with a hydrodynamic diameters larger than 1 µm (Figure 3). In buffer at a higher pH value (e.g., pH 9, TRIS buffer), C60–oligo-Glu (5b) showed much smaller aggregation (dotted green line, ≈12 nm), providing a transparent solution, while C60–oligo-Arg (5c) remained insoluble over the tested pH value range (4.0–9.2). This was presumably due to the strong cation–π interactions between the cationic Arg moieties and the aromatic C60, which is generally enhanced in polar environments [44-46]. The list of the solvents used to solubilize the molecules is summarized in Table S1, Supporting Information File 1.
![[1860-5397-20-71-3]](/bjoc/content/figures/1860-5397-20-71-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: DLS diagrams of C60–peptide conjugates 5a (1 mM, in Milli-Q® water), 5b (1 mM, in Milli-Q® water or in pH 9.0 TRIS buffer), and 5c (1 mM, in Milli-Q® water). Particle size: mean, nm (polydispersity index, PDI): 5a: 4.8 (0.757), 5b: 3974 (0.906), 5c (crude): 1382 (0.115) in Milli-Q® water and 5b: 11.8 (1.000) in pH 9.0 TRIS buffer. Milli-Q® water was pH 7.0.
Figure 3: DLS diagrams of C60–peptide conjugates 5a (1 mM, in Milli-Q® water), 5b (1 mM, in Milli-Q® water or...
Spectral characterizations of 5a and 5b
The absorption spectra of C60–oligo-Lys (5a) and C60–oligo-Glu (5b) were recorded in Milli-Q® water (pH 7.0) and in TRIS buffer (pH 9.0), respectively (Figure 4). The spectrum of C60–oligo-Lys (5a) was in good agreement with that typically observed for C60 monoadduct derivatives [47]. The electronic spectra of the fulleropyrrolidines were characterized by notable π–π* absorption in the UV region. Additionally, 5a exhibited broad absorption in the visible region with relatively low intensity as well as a distinctive sharp peak at around 430 nm [48]. However, those features were not observed in the spectrum of C60–oligo-Glu (5b), presumably due to the aggregation [32].
![[1860-5397-20-71-4]](/bjoc/content/figures/1860-5397-20-71-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis spectra of C60–peptide conjugates 5a and 5b (20 μM in Milli-Q® water for 5a and in pH 9.0 TRIS buffer for 5b).
Figure 4: UV–vis spectra of C60–peptide conjugates 5a and 5b (20 μM in Milli-Q® water for 5a and in pH 9.0 TR...
The 1H NMR spectrum of C60–oligo-Lys (5a) was recorded in D2O. As shown in Figure 5, the spectrum of 5a (upper spectrum) clearly shows peaks for protons corresponding to the fulleropyrrolidine part, which are in line with the peaks in the spectrum of the C60 Prato monoadduct (lower spectrum), linker part, and oligo-Lys side chain part (α, β, γ, δ, and ε). The observed splitting of the protons a and b was presumably due to a diastereotopic effect of the methine proton c, similar to the spectrum of the monoadduct.
![[1860-5397-20-71-5]](/bjoc/content/figures/1860-5397-20-71-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 1H NMR spectrum of C60–peptide conjugate 5a in D2O (above) and of the precursor monoadduct in CDCl3 (bottom) at 600 MHz.
Figure 5: 1H NMR spectrum of C60–peptide conjugate 5a in D2O (above) and of the precursor monoadduct in CDCl3...
Figure 6a shows the 13C NMR spectra of 5a in D2O and of the monoadduct in CDCl3. Together with the 1H NMR, COSY, HSQC, and HMBC spectra (Figures S3–S9, Supporting Information File 1), all peaks corresponding to the pyrrolidine part, linker part, and oligo-Lys part were assigned as shown in the chemical structure. In the sp2 region of 5a (Figure 6a, top), 17 signals (1C × 3 + 2C × 13) were observed similarly to the monoadduct (Figure 6a, bottom), which corresponds to the fullerene core in a characteristic manner for a C2v-symmetric structure with a [6,6]-addition pattern. In the expanded spectrum of 5a in the aromatic region (Figure 6b, top), several intense signals (corresponding to 2C) were observed as split peaks (highlighted by purple arrows). A similar situation was observed in the expanded spectrum of 5b (Figure 6b, middle, measured in pyridine). This phenomenon suggests a symmetry break in the carbon cage moiety of the C60–peptide conjugate upon the addition of chiral peptide anchors to the C60 core. Together with the HRESIMS results (Figure S2, Supporting Information File 1), it was confirmed that the highly water-soluble compound 5a was successfully synthesized.
![[1860-5397-20-71-6]](/bjoc/content/figures/1860-5397-20-71-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: 13C NMR spectrum of C60–peptide conjugate 5a in D2O and of the precursor monoadduct in CDCl3 at 150 MHz (a) and expansion of the sp2 carbon region (b). The asterisks in (a) correspond to a TFA impurity. Purple arrows in (b) indicate the split peaks. The 1H NMR spectrum of purified 5b was measured in D2O with 2% NaOD (Figure S12, Supporting Information File 1). 13C NMR analysis of the same sample was not possible due to the high ionic strength of the solution. NMR characterization was performed using a crude sample of 5b, with the penta-Glu impurity being soluble in pyridine-d5 (Figures S13–S17, Supporting Information File 1).
Figure 6: 13C NMR spectrum of C60–peptide conjugate 5a in D2O and of the precursor monoadduct in CDCl3 at 150...
1O2 generation under visible light irradiation
To preliminarily evaluate the synthesized C60–oligo-Lys (5a) as a PS, generation of singlet oxygen was measured by the ESR spin trapping method under irradiation of visible light (527 nm green LED). 4-Oxo-TEMP was used as a spin trapping reagent to form an adduct with 1O2, i.e., 4-oxo-TEMPO, which was observed by ESR (Figure 7b). As shown in Figure 7a, upon visible light irradiation, three peaks corresponding to 4-oxo-TEMPO were observed in the solution of C60–oligo-Lys (5a), similar to the results with rose bengal, a standard compound for 1O2 generation. By taking into account that the absorption intensity of 5a at 527 nm used for the photoirradiation was ≈10 times smaller than that of rose bengal, it was suggested that 1O2 was sufficiently present in the solution of 5a.
![[1860-5397-20-71-7]](/bjoc/content/figures/1860-5397-20-71-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: a) X-band ESR spectra of the 4-oxo-TEMP adduct with 1O2 generated by C60–oligo-Lys (5a) and rose bengal (RB), respectively, in aqueous solutions under irradiation with a green LED lamp (527 nm) for 1 min or 10 min. PS: 40 μM, 4-oxo-TEMP: 80 mM, in 60 mM phosphate buffer (pH 7.0). Measurement conditions: temperature 296 K, microwave frequency 10.03 GHz, microwave power 10 mW, receiver gain 5.0 × 104, modulation amplitude 1.00 G, modulation frequency 100 KHz, sweep time 83.89 s, number of scans: 1. b) Scheme for the photoinduced 1O2 generation by C60 and reaction with spin trapping reagent 4-oxo-TEMP to form 4-oxo-TEMPO.
Figure 7: a) X-band ESR spectra of the 4-oxo-TEMP adduct with 1O2 generated by C60–oligo-Lys (5a) and rose be...
Conclusion
Starting from biscarboxylic acid-substituted fulleropyrrolidine derivative 3, the three C60–oligopeptides 5a–c were synthesized through SPPS. Between these conjugates, C60–oligo-Lys (5a) had sufficient solubility and a very small hydrodynamic diameter in a neutral aqueous medium, as shown by DLS analysis. Visible-light-induced 1O2 generation by C60–oligo-Lys (5a) was confirmed by ESR spin trapping. This suggests the high potential of 5a as a basis for a fullerene-derived PS in biological applications.
Experimental
Synthesis of oligopeptides on resin 1a–c
The oligopeptides on resin 1a–c were synthesized via a general SPPS protocol. The first addition of Fmoc-AA(PG)-OH to the 2-chlorotrityl resin was performed in the presence of DIPEA (2 equiv) in CH2Cl2, followed by capping with a CH2Cl2/MeOH/DIPEA (18:2:1, v/v) mixture to quench any remaining unreacted chlorotrityl moieties on the resin surface. Subsequently, four additional Fmoc-AA(PG)-OH residues and one Fmoc-GABA-OH residue were added to the resin to provide the peptides on resin 1a–c. Each coupling step was carried out in the presence of HCTU (4 equiv) and NMM (8 equiv) in DMF. Each Fmoc deprotection step of the peptide N-terminus was conducted by the repeated treatment of the peptide on resin with 20% piperidine in DMF (2 × 10 min). After each coupling reaction, the resin was washed with DMF.
Synthesis of C60–peptide conjugates 5a–c
The synthetic details and corresponding spectra for the C60–peptide conjugates 5a–c are shown in Supporting Information File 1. The optimization of the conditions for the reaction between the peptides on resin and fulleropyrrolidine 3 are described in the Results and Discussion section above. These were used to prepare the C60–peptide conjugates on resin 4a–c from 2a–c. The C60–oligopeptides 5a–c were obtained by cleavage from resin and simultaneous deprotection of the PGs on the amino acid side chains through the addition of by the addition of a mixture of TFA and TIPS in water. The most soluble conjugate, C60–oligo-Lys (5a), was purified by reversed-phase HPLC, while C60–oligo-Glu (5b) was purified by spin filtration.
C60–oligo-Lys (5a) was obtained in a yield of 32% for the total peptide synthesis and characterized by HRESIMS. HRESIMS (m/z): [M + 3H]3+ calcd for C135H148N23O16, 782.3819; found, 782.3821.
C60–oligo-Glu (5b) was obtained in a yield of 36% for the total peptide synthesis and characterized by HRMS–MALDI. HRMS–MALDI (m/z): [M + H]+ calcd for C125H96N13O36, 2354.6075; found, 2354.6008.
C60–oligo-Arg (5c) was obtained in a crude yield of 66% for the total peptide synthesis and characterized by HRESIMS. HRESIMS (m/z): [M + 4H]4+ calcd for C135H149N43O16, 657.0536; found, 657.0540.
Supporting Information
| Supporting Information File 1: Details for the synthesis of 5a–c and intermediates as well as spectral data. | ||
| Format: PDF | Size: 2.3 MB | Download |
Acknowledgements
The authors thank Prof. Jean-Christophe Leroux at ETH Zürich for his help with DLS measurements. The authors thank Dr. Marc-Olivier Ebert at ETH Zürich for his help with NMR measurements and discussions. The authors thank the Molecular and Biomolecular Analytical Service (MoBiAS) at ETH Zürich for their support with HRMS measurements.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Kroto, H. W.; Heath, J. R.; O’Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162–163. doi:10.1038/318162a0
Return to citation in text: [1] -
Xu, J.; Buin, A.; Ip, A. H.; Li, W.; Voznyy, O.; Comin, R.; Yuan, M.; Jeon, S.; Ning, Z.; McDowell, J. J.; Kanjanaboos, P.; Sun, J.-P.; Lan, X.; Quan, L. N.; Kim, D. H.; Hill, I. G.; Maksymovych, P.; Sargent, E. H. Nat. Commun. 2015, 6, 7081. doi:10.1038/ncomms8081
Return to citation in text: [1] -
Chiang, C.-H.; Wu, C.-G. Nat. Photonics 2016, 10, 196–200. doi:10.1038/nphoton.2016.3
Return to citation in text: [1] -
Chai, Y.; Liu, L.; Xu, Y.; Liu, X.; Wang, C.; Bo, Y.; Zhang, Y.; Wang, Z.; Weng, Y.; Guldi, D. M.; Wu, B.; Wang, C. J. Am. Chem. Soc. 2023, 145, 14190–14195. doi:10.1021/jacs.3c03486
Return to citation in text: [1] -
Yu, L.; Xu, J.; Peng, B.; Qin, G.; Su, G. J. Phys. Chem. Lett. 2022, 13, 11622–11629. doi:10.1021/acs.jpclett.2c02702
Return to citation in text: [1] -
Eklund, P. C.; Rao, A. M.; Wang, Y.; Zhou, P.; Wang, K.-A.; Holden, J. M.; Dresselhaus, M. S.; Dresselhaus, G. Thin Solid Films 1995, 257, 211–232. doi:10.1016/0040-6090(94)05706-0
Return to citation in text: [1] -
Pawlak, R.; Kawai, S.; Fremy, S.; Glatzel, T.; Meyer, E. ACS Nano 2011, 5, 6349–6354. doi:10.1021/nn201462g
Return to citation in text: [1] -
David, W. I. F.; Ibberson, R. M.; Matthewman, J. C.; Prassides, K.; Dennis, T. J. S.; Hare, J. P.; Kroto, H. W.; Taylor, R.; Walton, D. R. M. Nature 1991, 353, 147–149. doi:10.1038/353147a0
Return to citation in text: [1] -
Ruoff, R. S.; Tse, D. S.; Malhotra, R.; Lorents, D. C. J. Phys. Chem. 1993, 97, 3379–3383. doi:10.1021/j100115a049
Return to citation in text: [1] -
Nakamura, E.; Isobe, H. Acc. Chem. Res. 2003, 36, 807–815. doi:10.1021/ar030027y
Return to citation in text: [1] -
Tokuyama, H.; Yamago, S.; Nakamura, E.; Shiraki, T.; Sugiura, Y. J. Am. Chem. Soc. 1993, 115, 7918–7919. doi:10.1021/ja00070a064
Return to citation in text: [1] -
Sijbesma, R.; Srdanov, G.; Wudl, F.; Castoro, J. A.; Wilkins, C.; Friedman, S. H.; DeCamp, D. L.; Kenyon, G. L. J. Am. Chem. Soc. 1993, 115, 6510–6512. doi:10.1021/ja00068a006
Return to citation in text: [1] -
Lamparth, I.; Hirsch, A. J. Chem. Soc., Chem. Commun. 1994, 1727–1728. doi:10.1039/c39940001727
Return to citation in text: [1] -
Sawamura, M.; Iikura, H.; Nakamura, E. J. Am. Chem. Soc. 1996, 118, 12850–12851. doi:10.1021/ja962681x
Return to citation in text: [1] -
Matsuo, Y.; Nakamura, E. Chem. Rev. 2008, 108, 3016–3028. doi:10.1021/cr0684218
Return to citation in text: [1] -
Andersson, T.; Nilsson, K.; Sundahl, M.; Westman, G.; Wennerström, O. J. Chem. Soc., Chem. Commun. 1992, 604–606. doi:10.1039/c39920000604
Return to citation in text: [1] -
Ikeda, A.; Nobukuni, S.; Udzu, H.; Zhong, Z.; Shinkai, S. Eur. J. Org. Chem. 2000, 3287–3293. doi:10.1002/1099-0690(200010)2000:19<3287::aid-ejoc3287>3.0.co;2-r
Return to citation in text: [1] -
Ikeda, A.; Shinkai, S. Chem. Rev. 1997, 97, 1713–1734. doi:10.1021/cr960385x
Return to citation in text: [1] -
Garbuio, L.; Antonello, S.; Guryanov, I.; Li, Y.; Ruzzi, M.; Turro, N. J.; Maran, F. J. Am. Chem. Soc. 2012, 134, 10628–10637. doi:10.1021/ja303696s
Return to citation in text: [1] -
Jennepalli, S.; Hammer, K. A.; Riley, T. V.; Pyne, S. G.; Keller, P. A. Eur. J. Org. Chem. 2015, 195–201. doi:10.1002/ejoc.201403046
Return to citation in text: [1] -
Ousaka, N.; Mamiya, F.; Iwata, Y.; Nishimura, K.; Yashima, E. Angew. Chem., Int. Ed. 2017, 56, 791–795. doi:10.1002/anie.201611349
Return to citation in text: [1] -
Dostalova, S.; Moulick, A.; Milosavljevic, V.; Guran, R.; Kominkova, M.; Cihalova, K.; Heger, Z.; Blazkova, L.; Kopel, P.; Hynek, D.; Vaculovicova, M.; Adam, V.; Kizek, R. Monatsh. Chem. 2016, 147, 905–918. doi:10.1007/s00706-016-1675-0
Return to citation in text: [1] -
Liu, J.-H.; Cao, L.; Luo, P. G.; Yang, S.-T.; Lu, F.; Wang, H.; Meziani, M. J.; Haque, S. A.; Liu, Y.; Lacher, S.; Sun, Y.-P. ACS Appl. Mater. Interfaces 2010, 2, 1384–1389. doi:10.1021/am100037y
Return to citation in text: [1] -
Li, Y.; Biswas, R.; Kopcha, W. P.; Dubroca, T.; Abella, L.; Sun, Y.; Crichton, R. A.; Rathnam, C.; Yang, L.; Yeh, Y.-W.; Kundu, K.; Rodríguez‐Fortea, A.; Poblet, J. M.; Lee, K.-B.; Hill, S.; Zhang, J. Angew. Chem., Int. Ed. 2023, 62, e202380362. doi:10.1002/anie.202380362
Return to citation in text: [1] -
Yamakoshi, Y. N.; Yagami, T.; Fukuhara, K.; Sueyoshi, S.; Miyata, N. J. Chem. Soc., Chem. Commun. 1994, 517–518. doi:10.1039/c39940000517
Return to citation in text: [1] -
Yamakoshi, Y. N.; Yagami, T.; Sueyoshi, S.; Miyata, N. J. Org. Chem. 1996, 61, 7236–7237. doi:10.1021/jo961210q
Return to citation in text: [1] -
Yamakoshi, Y.; Sueyoshi, S.; Fukuhara, K.; Miyata, N.; Masumizu, T.; Kohno, M. J. Am. Chem. Soc. 1998, 120, 12363–12364. doi:10.1021/ja9823969
Return to citation in text: [1] -
Yamakoshi, Y.; Umezawa, N.; Ryu, A.; Arakane, K.; Miyata, N.; Goda, Y.; Masumizu, T.; Nagano, T. J. Am. Chem. Soc. 2003, 125, 12803–12809. doi:10.1021/ja0355574
Return to citation in text: [1] -
Kai, Y.; Komazawa, Y.; Miyajima, A.; Miyata, N.; Yamakoshi, Y. Fullerenes, Nanotubes, Carbon Nanostruct. 2003, 11, 79–87. doi:10.1081/fst-120018664
Return to citation in text: [1] -
Tsuchiya, T.; Yamakoshi, Y. N.; Miyata, N. Biochem. Biophys. Res. Commun. 1995, 206, 885–894. doi:10.1006/bbrc.1995.1126
Return to citation in text: [1] -
Tsuchiya, T.; Oguri, I.; Nakajima Yamakoshi, Y.; Miyata, N. Fullerene Sci. Technol. 1996, 4, 989–999. doi:10.1080/10641229608001157
Return to citation in text: [1] -
Sakai, A.; Yamakoshi, Y. N.; Miyata, N. Fullerene Sci. Technol. 1995, 3, 377–388. doi:10.1080/153638x9508543792
Return to citation in text: [1] [2] -
Sakai, A.; Yamakoshi, Y.; Miyata, N. Fullerene Sci. Technol. 1999, 7, 743–756. doi:10.1080/10641229909351375
Return to citation in text: [1] -
Iwata, N.; Mukai, T.; Yamakoshi, Y. N.; Haraa, S.; Yanase, T.; Shoji, M.; Endo, T.; Miyata, N. Fullerene Sci. Technol. 1998, 6, 213–226. doi:10.1080/10641229809350196
Return to citation in text: [1] -
Aroua, S.; Schweizer, W. B.; Yamakoshi, Y. Org. Lett. 2014, 16, 1688–1691. doi:10.1021/ol500363r
Return to citation in text: [1] [2] -
Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1993, 115, 9798–9799. doi:10.1021/ja00074a056
Return to citation in text: [1] -
Aroua, S.; Tiu, E. G. V.; Ishikawa, T.; Yamakoshi, Y. Helv. Chim. Acta 2016, 99, 805–813. doi:10.1002/hlca.201600171
Return to citation in text: [1] -
Liosi, K.; Stasyuk, A. J.; Masero, F.; Voityuk, A. A.; Nauser, T.; Mougel, V.; Solà, M.; Yamakoshi, Y. JACS Au 2021, 1, 1601–1611. doi:10.1021/jacsau.1c00239
Return to citation in text: [1] -
Aroua, S.; Tiu, E. G. V.; Ayer, M.; Ishikawa, T.; Yamakoshi, Y. Polym. Chem. 2015, 6, 2616–2619. doi:10.1039/c4py01333f
Return to citation in text: [1] -
Tiu, E. G. V.; Liosi, K.; Aroua, S.; Yamakoshi, Y. J. Mater. Chem. B 2017, 5, 6676–6680. doi:10.1039/c7tb00829e
Return to citation in text: [1] -
Hamblin, M. R. Photochem. Photobiol. Sci. 2018, 17, 1515–1533. doi:10.1039/c8pp00195b
Return to citation in text: [1] -
Shu, C.; Corwin, F. D.; Zhang, J.; Chen, Z.; Reid, J. E.; Sun, M.; Xu, W.; Sim, J. H.; Wang, C.; Fatouros, P. P.; Esker, A. R.; Gibson, H. W.; Dorn, H. C. Bioconjugate Chem. 2009, 20, 1186–1193. doi:10.1021/bc900051d
Return to citation in text: [1] -
Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Mol. Biotechnol. 2006, 33, 239–254. doi:10.1385/mb:33:3:239
Return to citation in text: [1] -
Gromiha, M. M.; Santhosh, C.; Ahmad, S. Int. J. Biol. Macromol. 2004, 34, 203–211. doi:10.1016/j.ijbiomac.2004.04.003
Return to citation in text: [1] -
Pletneva, E. V.; Laederach, A. T.; Fulton, D. B.; Kostić, N. M. J. Am. Chem. Soc. 2001, 123, 6232–6245. doi:10.1021/ja010401u
Return to citation in text: [1] -
Marforio, T. D.; Calza, A.; Mattioli, E. J.; Zerbetto, F.; Calvaresi, M. Int. J. Mol. Sci. 2021, 22, 11567. doi:10.3390/ijms222111567
Return to citation in text: [1] -
Guldi, D. M. J. Phys. Chem. A 1997, 101, 3895–3900. doi:10.1021/jp9702863
Return to citation in text: [1] -
Guldi, D. M.; Prato, M. Acc. Chem. Res. 2000, 33, 695–703. doi:10.1021/ar990144m
Return to citation in text: [1]
| 35. | Aroua, S.; Schweizer, W. B.; Yamakoshi, Y. Org. Lett. 2014, 16, 1688–1691. doi:10.1021/ol500363r |
| 43. | Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Mol. Biotechnol. 2006, 33, 239–254. doi:10.1385/mb:33:3:239 |
| 44. | Gromiha, M. M.; Santhosh, C.; Ahmad, S. Int. J. Biol. Macromol. 2004, 34, 203–211. doi:10.1016/j.ijbiomac.2004.04.003 |
| 45. | Pletneva, E. V.; Laederach, A. T.; Fulton, D. B.; Kostić, N. M. J. Am. Chem. Soc. 2001, 123, 6232–6245. doi:10.1021/ja010401u |
| 46. | Marforio, T. D.; Calza, A.; Mattioli, E. J.; Zerbetto, F.; Calvaresi, M. Int. J. Mol. Sci. 2021, 22, 11567. doi:10.3390/ijms222111567 |
| 1. | Kroto, H. W.; Heath, J. R.; O’Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162–163. doi:10.1038/318162a0 |
| 9. | Ruoff, R. S.; Tse, D. S.; Malhotra, R.; Lorents, D. C. J. Phys. Chem. 1993, 97, 3379–3383. doi:10.1021/j100115a049 |
| 26. | Yamakoshi, Y. N.; Yagami, T.; Sueyoshi, S.; Miyata, N. J. Org. Chem. 1996, 61, 7236–7237. doi:10.1021/jo961210q |
| 7. | Pawlak, R.; Kawai, S.; Fremy, S.; Glatzel, T.; Meyer, E. ACS Nano 2011, 5, 6349–6354. doi:10.1021/nn201462g |
| 8. | David, W. I. F.; Ibberson, R. M.; Matthewman, J. C.; Prassides, K.; Dennis, T. J. S.; Hare, J. P.; Kroto, H. W.; Taylor, R.; Walton, D. R. M. Nature 1991, 353, 147–149. doi:10.1038/353147a0 |
| 27. | Yamakoshi, Y.; Sueyoshi, S.; Fukuhara, K.; Miyata, N.; Masumizu, T.; Kohno, M. J. Am. Chem. Soc. 1998, 120, 12363–12364. doi:10.1021/ja9823969 |
| 28. | Yamakoshi, Y.; Umezawa, N.; Ryu, A.; Arakane, K.; Miyata, N.; Goda, Y.; Masumizu, T.; Nagano, T. J. Am. Chem. Soc. 2003, 125, 12803–12809. doi:10.1021/ja0355574 |
| 5. | Yu, L.; Xu, J.; Peng, B.; Qin, G.; Su, G. J. Phys. Chem. Lett. 2022, 13, 11622–11629. doi:10.1021/acs.jpclett.2c02702 |
| 6. | Eklund, P. C.; Rao, A. M.; Wang, Y.; Zhou, P.; Wang, K.-A.; Holden, J. M.; Dresselhaus, M. S.; Dresselhaus, G. Thin Solid Films 1995, 257, 211–232. doi:10.1016/0040-6090(94)05706-0 |
| 19. | Garbuio, L.; Antonello, S.; Guryanov, I.; Li, Y.; Ruzzi, M.; Turro, N. J.; Maran, F. J. Am. Chem. Soc. 2012, 134, 10628–10637. doi:10.1021/ja303696s |
| 20. | Jennepalli, S.; Hammer, K. A.; Riley, T. V.; Pyne, S. G.; Keller, P. A. Eur. J. Org. Chem. 2015, 195–201. doi:10.1002/ejoc.201403046 |
| 21. | Ousaka, N.; Mamiya, F.; Iwata, Y.; Nishimura, K.; Yashima, E. Angew. Chem., Int. Ed. 2017, 56, 791–795. doi:10.1002/anie.201611349 |
| 22. | Dostalova, S.; Moulick, A.; Milosavljevic, V.; Guran, R.; Kominkova, M.; Cihalova, K.; Heger, Z.; Blazkova, L.; Kopel, P.; Hynek, D.; Vaculovicova, M.; Adam, V.; Kizek, R. Monatsh. Chem. 2016, 147, 905–918. doi:10.1007/s00706-016-1675-0 |
| 23. | Liu, J.-H.; Cao, L.; Luo, P. G.; Yang, S.-T.; Lu, F.; Wang, H.; Meziani, M. J.; Haque, S. A.; Liu, Y.; Lacher, S.; Sun, Y.-P. ACS Appl. Mater. Interfaces 2010, 2, 1384–1389. doi:10.1021/am100037y |
| 24. | Li, Y.; Biswas, R.; Kopcha, W. P.; Dubroca, T.; Abella, L.; Sun, Y.; Crichton, R. A.; Rathnam, C.; Yang, L.; Yeh, Y.-W.; Kundu, K.; Rodríguez‐Fortea, A.; Poblet, J. M.; Lee, K.-B.; Hill, S.; Zhang, J. Angew. Chem., Int. Ed. 2023, 62, e202380362. doi:10.1002/anie.202380362 |
| 2. | Xu, J.; Buin, A.; Ip, A. H.; Li, W.; Voznyy, O.; Comin, R.; Yuan, M.; Jeon, S.; Ning, Z.; McDowell, J. J.; Kanjanaboos, P.; Sun, J.-P.; Lan, X.; Quan, L. N.; Kim, D. H.; Hill, I. G.; Maksymovych, P.; Sargent, E. H. Nat. Commun. 2015, 6, 7081. doi:10.1038/ncomms8081 |
| 3. | Chiang, C.-H.; Wu, C.-G. Nat. Photonics 2016, 10, 196–200. doi:10.1038/nphoton.2016.3 |
| 4. | Chai, Y.; Liu, L.; Xu, Y.; Liu, X.; Wang, C.; Bo, Y.; Zhang, Y.; Wang, Z.; Weng, Y.; Guldi, D. M.; Wu, B.; Wang, C. J. Am. Chem. Soc. 2023, 145, 14190–14195. doi:10.1021/jacs.3c03486 |
| 25. | Yamakoshi, Y. N.; Yagami, T.; Fukuhara, K.; Sueyoshi, S.; Miyata, N. J. Chem. Soc., Chem. Commun. 1994, 517–518. doi:10.1039/c39940000517 |
| 13. | Lamparth, I.; Hirsch, A. J. Chem. Soc., Chem. Commun. 1994, 1727–1728. doi:10.1039/c39940001727 |
| 16. | Andersson, T.; Nilsson, K.; Sundahl, M.; Westman, G.; Wennerström, O. J. Chem. Soc., Chem. Commun. 1992, 604–606. doi:10.1039/c39920000604 |
| 32. | Sakai, A.; Yamakoshi, Y. N.; Miyata, N. Fullerene Sci. Technol. 1995, 3, 377–388. doi:10.1080/153638x9508543792 |
| 12. | Sijbesma, R.; Srdanov, G.; Wudl, F.; Castoro, J. A.; Wilkins, C.; Friedman, S. H.; DeCamp, D. L.; Kenyon, G. L. J. Am. Chem. Soc. 1993, 115, 6510–6512. doi:10.1021/ja00068a006 |
| 17. | Ikeda, A.; Nobukuni, S.; Udzu, H.; Zhong, Z.; Shinkai, S. Eur. J. Org. Chem. 2000, 3287–3293. doi:10.1002/1099-0690(200010)2000:19<3287::aid-ejoc3287>3.0.co;2-r |
| 18. | Ikeda, A.; Shinkai, S. Chem. Rev. 1997, 97, 1713–1734. doi:10.1021/cr960385x |
| 11. | Tokuyama, H.; Yamago, S.; Nakamura, E.; Shiraki, T.; Sugiura, Y. J. Am. Chem. Soc. 1993, 115, 7918–7919. doi:10.1021/ja00070a064 |
| 10. | Nakamura, E.; Isobe, H. Acc. Chem. Res. 2003, 36, 807–815. doi:10.1021/ar030027y |
| 14. | Sawamura, M.; Iikura, H.; Nakamura, E. J. Am. Chem. Soc. 1996, 118, 12850–12851. doi:10.1021/ja962681x |
| 15. | Matsuo, Y.; Nakamura, E. Chem. Rev. 2008, 108, 3016–3028. doi:10.1021/cr0684218 |
| 48. | Guldi, D. M.; Prato, M. Acc. Chem. Res. 2000, 33, 695–703. doi:10.1021/ar990144m |
| 32. | Sakai, A.; Yamakoshi, Y. N.; Miyata, N. Fullerene Sci. Technol. 1995, 3, 377–388. doi:10.1080/153638x9508543792 |
| 33. | Sakai, A.; Yamakoshi, Y.; Miyata, N. Fullerene Sci. Technol. 1999, 7, 743–756. doi:10.1080/10641229909351375 |
| 29. | Kai, Y.; Komazawa, Y.; Miyajima, A.; Miyata, N.; Yamakoshi, Y. Fullerenes, Nanotubes, Carbon Nanostruct. 2003, 11, 79–87. doi:10.1081/fst-120018664 |
| 30. | Tsuchiya, T.; Yamakoshi, Y. N.; Miyata, N. Biochem. Biophys. Res. Commun. 1995, 206, 885–894. doi:10.1006/bbrc.1995.1126 |
| 31. | Tsuchiya, T.; Oguri, I.; Nakajima Yamakoshi, Y.; Miyata, N. Fullerene Sci. Technol. 1996, 4, 989–999. doi:10.1080/10641229608001157 |
| 41. | Hamblin, M. R. Photochem. Photobiol. Sci. 2018, 17, 1515–1533. doi:10.1039/c8pp00195b |
| 42. | Shu, C.; Corwin, F. D.; Zhang, J.; Chen, Z.; Reid, J. E.; Sun, M.; Xu, W.; Sim, J. H.; Wang, C.; Fatouros, P. P.; Esker, A. R.; Gibson, H. W.; Dorn, H. C. Bioconjugate Chem. 2009, 20, 1186–1193. doi:10.1021/bc900051d |
| 39. | Aroua, S.; Tiu, E. G. V.; Ayer, M.; Ishikawa, T.; Yamakoshi, Y. Polym. Chem. 2015, 6, 2616–2619. doi:10.1039/c4py01333f |
| 40. | Tiu, E. G. V.; Liosi, K.; Aroua, S.; Yamakoshi, Y. J. Mater. Chem. B 2017, 5, 6676–6680. doi:10.1039/c7tb00829e |
| 36. | Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1993, 115, 9798–9799. doi:10.1021/ja00074a056 |
| 37. | Aroua, S.; Tiu, E. G. V.; Ishikawa, T.; Yamakoshi, Y. Helv. Chim. Acta 2016, 99, 805–813. doi:10.1002/hlca.201600171 |
| 38. | Liosi, K.; Stasyuk, A. J.; Masero, F.; Voityuk, A. A.; Nauser, T.; Mougel, V.; Solà, M.; Yamakoshi, Y. JACS Au 2021, 1, 1601–1611. doi:10.1021/jacsau.1c00239 |
| 34. | Iwata, N.; Mukai, T.; Yamakoshi, Y. N.; Haraa, S.; Yanase, T.; Shoji, M.; Endo, T.; Miyata, N. Fullerene Sci. Technol. 1998, 6, 213–226. doi:10.1080/10641229809350196 |
| 35. | Aroua, S.; Schweizer, W. B.; Yamakoshi, Y. Org. Lett. 2014, 16, 1688–1691. doi:10.1021/ol500363r |
© 2024 Ma et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.