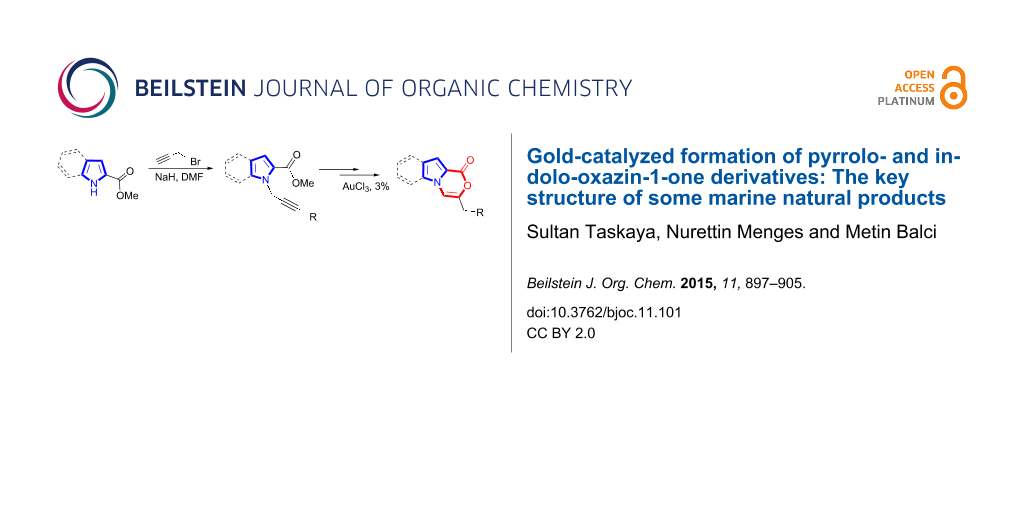
Abstract
Various N-propargylpyrrole and indolecarboxylic acids were efficiently converted into 3,4-dihydropyrrolo- and indolo-oxazin-1-one derivatives by a gold(III)-catalyzed cyclization reaction. Some of the products underwent TFA-catalyzed double bond isomerization and some did not. Cyclization reactions in the presence of alcohol catalyzed by Au(I) resulted in the formation of hemiacetals after cascade reactions.

Graphical Abstract
Introduction
Pyrrole-containing heterocycles are widely distributed within a large number of natural products and biologically active molecules [1]. These compounds possess a wide range of biological and pharmacological activities [2-5]. The oxazinone moiety is also frequently found in compounds displaying biological activities [6,7]. Pyrrolo-oxazinone structures with an oxazinone ring fused to a pyrrole are found in marine natural products, such as lukianol A (1) and lukianol B (2) (Figure 1) [8-10]. Lukianol B (2) was found to be the most potent human aldose reductase (h-ALR2) inhibitor in thousands of marine natural products screened [11]. The ningalin B alkaloid 3, having an isomeric pyrrolo-oxazinone skeleton, possesses antitumor and multidrug resistance reversal activity [12-14]. Lamellarins 4 with a pyrrolo-oxazinone substructure are also marine natural products and they inhibit the proliferation of cancer cells and therefore are promising candidates for anticancer drugs [15-20].
![[1860-5397-11-101-1]](/bjoc/content/figures/1860-5397-11-101-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of some marine natural products 1–4.
Figure 1: Structures of some marine natural products 1–4.
The design and synthesis of pharmaceutically relevant building blocks has always been a significant goal in organic synthesis. Moreover, modification of natural products is also an important approach to identify promising anticancer agents. Especially due to the bioactivity of lukianols, many groups accomplished the synthesis of these marine products and their analogues [21-26]. Despite the general and widespread interest in these structures, the core structure, 1H-pyrrolo[2,1-c][1,4]oxazin-1-one (5), is not described in the literature. However, there are relatively few synthetic routes leading to the substituted core structure 6 (Figure 2), which was generated by treatment of methyl 2-pyrrole-carboxylate with chloroacetone [2,27-30]. This core structure is also a very important intermediate for the synthesis of pyrrolo-pyrazinones as well as for pyrrolo-pyrazines [31]. Recently, Wang et al. reported the base-catalyzed intramolecular cyclization of alkynyl alcohols at high temperatures to give 1,4-oxaza heterocycles [32].
![[1860-5397-11-101-2]](/bjoc/content/figures/1860-5397-11-101-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
In this manuscript we envisaged a synthetic strategy leading to the core skeleton of marine products 6 and 7 (Figure 2) or their analogues with short reaction times and an atom-economic process using a gold-catalyzed alkyne cyclization reaction as the key step.
Pyrrole and indole, bearing one (substituted) propargyl and carboxy group was initially designed as model substrate for the construction of 6 and 7. Gold-catalyzed intramolecular cyclization of enyene carboxylic acids has been reported in the literature to give the corresponding lactones [33-40]. For example, gold(I)-catalyzed reaction of 2-(phenylethynyl)benzoic acid (8) yielded the exo- and endo-dig cyclization products, lactones 9 and 10 in 62% yield in a ratio of 6:1 (Scheme 1) [41].
![[1860-5397-11-101-i1]](/bjoc/content/inline/1860-5397-11-101-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Intramolecular gold(I)-catalyzed cyclization reaction of 8 to give 9 and 10.
Scheme 1: Intramolecular gold(I)-catalyzed cyclization reaction of 8 to give 9 and 10.
Results and Discussion
The starting compound 11 was synthesized via a slightly modified route by acetylation of pyrrole with trichloroacetyl chloride in 99% yield [42,43]. The propargyl ester 13 [44-46] was obtained in high yield from the reaction of 12 with propargyl bromide in the presence of NaH as a base (Scheme 2). Gold catalysis is an excellent method for constructing complex chemical architectures in a mild manner that would be difficult to achieve using other reactions [41,47-53]. Reaction of propargyl ester 13 with AuCl3 resulted in the formation of 14, which is an H2O (present in the reaction media) addition product to alkyne units. It has been proposed that a gold-activated water intermediate can easily be added to alkynes. The addition of water to alkyne has been reported by several research groups [54,55]. The expected cyclization product having the structure 6 or 7 was not formed.
![[1860-5397-11-101-i2]](/bjoc/content/inline/1860-5397-11-101-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 13 and its reaction with AuCl3.
Scheme 2: Synthesis of 13 and its reaction with AuCl3.
After failure of the cyclization reaction of 13 with AuCl3, even in water-free solvents, we turned our attention to the construction of the pyrrolo-oxazin-1-one skeleton with the corresponding acid 15. For this purpose, the propargyl ester 13 was first hydrolyzed with K2CO3 to acid 15 (Scheme 3) [56]. The acid 15 was reacted with various metal catalysts in chloroform at different temperatures and for different reaction times. Five different catalysts were tried (Table 1). Surprisingly, no reaction was observed when the reaction was conducted with the N-heterocyclic carbene (NHC) complex of Au(I) in chloroform (Table 1, entry 5). Reactions with InCl3 and PtCl2(PPh3)3 as catalysts gave very poor yields of exo-dig cyclization product 7 after 24 h (entries 3 and 4). AgOTf and AuCl3 were also screened and AuCl3 was identified as the optimal choice due to the shorter reaction time, high yield, and easy isolation of the product 7 (entries 1 and 2, Table 1). We next investigated the isomerization of the double bond in 7, which was treated with trifluoroacetic acid (TFA) in chloroform at room temperature to give 3-methyl-1H-pyrrolo[2,1-c][1,4]oxazin-1-one (6) [27].
![[1860-5397-11-101-i3]](/bjoc/content/inline/1860-5397-11-101-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 1: Optimization of cyclization reaction of 15.
| Entry | Catalyst | Time (h) | Temperature (°C) | Yield (%) |
|---|---|---|---|---|
| 1 | AuCl3 | 2 | rt | 96 |
| 2 | AgOTf | 20 | rt | 95 |
| 3 | InCl3 | 24 | 50 | 13 |
| 4 | PtCl2(PPh)3 | 26 | 50 | 5 |
| 5 | Au(L)a | 24 | rt | No reaction |
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-101-i7.svg?max-width=637&scale=1.18182)
After having obtained the optimal conditions for the Au-catalyzed cyclization of carboxylic acid 15, we attempted to determine the scope and limitation of this transformation. Then we investigated the cyclization reaction of a range of substituted N-propargyl-pyrrole-2-carboxylic acids 17, 21, 25, 29 and 33 and N-propargylindole-2-carboxylic acids 37, 41 and 45, which were synthesized by hydrolysis of the corresponding esters (Table 2). The Sonogashira cross-coupling reaction [56-61] was used for the synthesis of the desired starting materials 29 and 33. For the Sonogashira coupling reaction we used a palladium catalyst and a copper(I) cocatalyst in the presence of triphenylphosphine and triethylamine as the base. The carboxylic acids were then submitted to a gold-catalyzed cyclization reaction (Table 2).
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-101-i8.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-101-i9.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-101-i10.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-101-i11.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-101-i12.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-101-i13.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-101-i14.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-101-i15.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-101-i16.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-101-i17.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-101-i18.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-101-i19.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-101-i20.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-101-i21.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-101-i22.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-101-i23.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-101-i24.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-101-i25.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-101-i26.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-101-i27.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-101-i28.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-101-i29.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-101-i30.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-101-i31.svg?max-width=637&scale=1.0)
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-101-i32.svg?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-101-i33.svg?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-101-i34.svg?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-101-i35.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-101-i36.svg?max-width=637&scale=1.0)
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-101-i37.svg?max-width=637&scale=1.0)
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-101-i38.svg?max-width=637&scale=1.0)
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-101-i39.svg?max-width=637&scale=1.0)
The optimized reaction conditions for cyclization was employed for pyrrole- and indole-carboxylic acids. We observed that all substituted pyrrole as well as indole carboxylic acids underwent a smooth gold-catalyzed cyclization reaction to afford the corresponding oxazinones 18, 22, 26, 30, 34, 38, 42 and 46 in high yields (Table 2). Substitution at the terminal carbon atom of the propargyl group with methyl and phenyl groups did not affect the mode of the cyclization reaction.
Further investigation of cyclic products involved examining the isomerization of the exocyclic double bond. For this purpose, the cyclization products were reacted with trifloroacetic acid (TFA) to form endo-cyclic double bonds. Some of the compounds underwent double bond shifts to form an oxazinone skeleton 19, 23, 27, 39 and 43, whereas the compounds 30 and 34 did not form the corresponding endo-cyclic systems 31 and 35. In order to address this question, we carried out some DFT calculations [66].
The calculations performed in the gase phase showed that 6 is about 10.6 kcal/mol (DFT, B3LYP at 6-31G** level) and 8.1 kcal/mol (HF/6-311G** level) more stable than 7 (Figure 3). However, in the case of 30 and 31, we also found that the endo-cyclic isomer 31 is thermodynamically about 4.8 kcal/mol (DFT, B3LYP at 6-31G** level) and 3.8 kcal/mol (HF/6-311G** level) more stable than the exo-cyclic isomer 30. Although 31 is thermodynamically more stable than 30, the resistance of 30 to isomerization in the presence of TFA can be explained by the regioselective protonation of the double bond. The proton is added to the carbon atom to generate the most stable carbocation. The most stable carbocation is the benzylic carbocation which would not undergo a rearrangement.
![[1860-5397-11-101-3]](/bjoc/content/figures/1860-5397-11-101-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Geometry optimized structures of 6, 7, 30 and 31.
Figure 3: Geometry optimized structures of 6, 7, 30 and 31.
As discussed above, when the cyclization reaction of 15 was carried out with [(NHC)AuCl] complexes (Table 1, entry 5), the starting material was fully recovered. Gold N-heterocyclic carbene complexes, in conjunction with a silver salt, were found to efficiently catalyze different types of reactions [67-69]. Therefore, 15 was reacted with [(NHC)AuCl] complex in the presence of AgOTf as the cocatalyst in chloroform. Beside the expected cyclization product 7 (10%), the product 47 was formed conclusively through incorporation of ethanol present in chloroform (<1%) as the major product in 51% yield (Scheme 4). NMR spectral analysis of the mixture revealed also the formation of 48 in 39% yield, which is the hydrolysis product of 47. Attempts to isolate 47 failed. Chromatography on silica gel converted 47 to 48 in almost quantitative yield. However, the products 6 and 7 only were isolated when the reaction was carried out in EtOH-free chloroform in 83% and 17% yields, respectively (Scheme 4).
![[1860-5397-11-101-i4]](/bjoc/content/inline/1860-5397-11-101-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of 15 with Au(I)/AgOTf in the presence of EtOH and CD3OD.
Scheme 4: Reaction of 15 with Au(I)/AgOTf in the presence of EtOH and CD3OD.
To reveal the mechanism for the formation of the products we carried out two different reactions. First, 15 was reacted with CD3OD under the same reaction conditions. The 1H NMR spectral analysis indicated the formation of 49 and 50 in 65% and 35% yields, respectively. Furthermore, the amount of deuterium atoms in 49 and 50 attached to the methylene group as –CH2D was about 64% (Scheme 4).
To prove whether 7 was involved as the intermediate, the cyclization product 7 was reacted with Au(I)/AgOTf with EtOH under the same reaction conditions. As a result, the products 47 and 48 were formed in yields of 39% and 61%, respectively (Scheme 5).
![[1860-5397-11-101-i5]](/bjoc/content/inline/1860-5397-11-101-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of 7 with Au(I)/AgOTf in the presence of EtOH.
Scheme 5: Reaction of 7 with Au(I)/AgOTf in the presence of EtOH.
Based on all this information obtained, we propose the following gold-catalyzed cascade reaction mechanism. The proposed catalytic cycle was initiated with π-activation of the triple bond by the carbene-based cationic gold species to form the intermediate 51, which triggers a gold-promoted intramolecular addition of a carboxy group to the alkyne functionality to give methylene-oxazin-1-one derivative 7 (or 18) (Scheme 6). In the next step, the double bond is activated by π-coordination of the gold(I) catalyst. This enables a nucleophilic attack by the alcohol oxygen, which affords hemiacetal 47. In case of internal alkynes, two isomeric E- and Z-oxazinones during the cyclization reaction can be formed. However, we observed exclusive formation of a single isomer. Recently, Michelet et al. [70] and others [41,71,72] demonstrated selectively the formation of Z-lactones. The anti intramolecular addition of the carboxylic acid to the gold-alkyne intermediate 51, forms an intermediate 52 where the substituent is in the endo-position. Removal of gold results in the formation of Z-isomer. Therefore, we assign the endo-configuration to the formed oxazinones.
![[1860-5397-11-101-i6]](/bjoc/content/inline/1860-5397-11-101-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed reaction mechanism for the intramolecular gold-catalyzed cyclization followed by EtOH addition.
Scheme 6: Proposed reaction mechanism for the intramolecular gold-catalyzed cyclization followed by EtOH addi...
Conclusion
We have developed a general synthetic methodology of pyrrolo- and indolo-oxazin-1-one derivatives. The key step was a gold-catalyzed cyclization reaction of N-propargyl-substituted pyrrole and indole carboxylic acid derivatives. The hydroxy groups of carboxylic acids attacked the activated triple bond to form 6-exo-dig cyclization products, oxazin-1-one derivatives. Some of the exo-cyclic double bonds underwent isomerization to endo-cyclic compounds upon treatment with TFA, while some did not. DFT studies supported our findings. Moreover, cyclization reactions in the presence of alcohol formed hemiacetal derivatives after gold-catalyzed cascade reactions.
References
-
Gossauer, A. Pyrrole. Houben-Weyl – Methods of Organic Chemistry; Thieme: Stuttgart, 1994; Vol. E6a/1, pp 556 ff.
See for a review.
Return to citation in text: [1] -
Meng, Y.; Wang, G.; Li, Y.; Hou, K.; Yuan, Y.; Zhang, L.-J.; Song, H.-R.; Shi, W. Chin. Chem. Lett. 2013, 24, 619–621. doi:10.1016/j.cclet.2013.04.020
Return to citation in text: [1] [2] -
Han, S.; Siegel, D. S.; Morrison, K. C.; Hergenrother, P. J.; Movassaghi, M. J. Org. Chem. 2013, 78, 11970–11984. doi:10.1021/jo4020112
Return to citation in text: [1] -
Krishnaiah, P.; Reddy, V. L. N.; Venkataramana, G.; Ravinder, K.; Srinivasulu, M.; Raju, T. V.; Ravikumar, K.; Chandrasekar, D.; Ramakrishna, S.; Venkateswarlu, Y. J. Nat. Prod. 2004, 67, 1168–1171. doi:10.1021/np030503t
Return to citation in text: [1] -
Rochais, C.; Lisowski, V.; Dallemagne, P.; Rault, S. Bioorg. Med. Chem. 2006, 14, 8162–8175. doi:10.1016/j.bmc.2006.09.022
Return to citation in text: [1] -
Benaamane, N.; Nedjar-Kolli, B.; Bentarzi, Y.; Hammal, L.; Geronikaki, A.; Eleftheriou, P.; Lagunin, A. Bioorg. Med. Chem. 2008, 16, 3059–3066. doi:10.1016/j.bmc.2007.12.033
Return to citation in text: [1] -
Kern, J. C.; Terefenko, E. A.; Fensome, A.; Unwallla, R.; Wrobel, J.; Zhu, Y.; Cohen, J.; Winneker, R.; Zhang, Z.; Zhang, P. Bioorg. Med. Chem. Lett. 2007, 17, 189–192. doi:10.1016/j.bmcl.2006.09.060
Return to citation in text: [1] -
Yoshida, W. Y.; Lee, K. K.; Carrol, A. R.; Scheuer, P. J. Helv. Chim. Acta 1992, 75, 1721–1725. doi:10.1002/hlca.19920750525
Return to citation in text: [1] -
Lu, Y.; Arndtsen, B. A. Org. Lett. 2009, 11, 1369–1372. doi:10.1021/ol900185n
Return to citation in text: [1] -
Takamura, K.; Matsuo, H.; Tanaka, A.; Tanaka, J.; Fukuda, T.; Ishibashi, F.; Iwao, M. Tetrahedron 2013, 69, 2782–2788. doi:10.1016/j.tet.2013.01.077
Return to citation in text: [1] -
Manzanaro, S.; Salvá, J.; de la Fuente, J. Á. J. Nat. Prod. 2006, 69, 1485–1487. doi:10.1021/np0503698
Return to citation in text: [1] -
Kang, H.; Fenical, W. J. Org. Chem. 1997, 62, 3254–3262. doi:10.1021/jo962132+
Return to citation in text: [1] -
Hasse, K.; Willis, A. C.; Banwell, M. G. Aust. J. Chem. 2009, 62, 683–691. doi:10.1071/CH09158
Return to citation in text: [1] -
Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Org. Lett. 2011, 13, 312–315. doi:10.1021/ol1027877
Return to citation in text: [1] -
Andersen, R. J.; Faulkner, D. J.; Cun-heng, H.; Van Duyne, G. D.; Clardy, J. J. Am. Chem. Soc. 1985, 107, 5492–5495. doi:10.1021/ja00305a027
Return to citation in text: [1] -
Carroll, A. R.; Bowden, B. F.; Coll, J. C. Aust. J. Chem. 1993, 46, 489–501. doi:10.1071/CH9930489
Return to citation in text: [1] -
Urban, S.; Butler, M. S.; Capon, R. J. Aust. J. Chem. 1994, 47, 1919–1924. doi:10.1071/CH9941919
Return to citation in text: [1] -
Fan, H.; Peng, J.; Hamann, M. T.; Hu, J.-F. Chem. Rev. 2008, 108, 264–287. doi:10.1021/cr078199m
Return to citation in text: [1] -
Chittchang, M.; Paul, G. M.; Ploypradith, P.; Ruchirawat, S. Eur. J. Med. Chem. 2010, 45, 2165–2172. doi:10.1016/j.ejmech.2010.01.053
Return to citation in text: [1] -
Ueda, K.; Amaike, K.; Maceiczyk, R. M.; Itami, K.; Yamaguchi, J. J. Am. Chem. Soc. 2014, 136, 13226–13232. doi:10.1021/ja508449y
Return to citation in text: [1] -
Mathew, P.; Asokan, C. W. Tetrahedron Lett. 2005, 46, 475–478. doi:10.1016/j.tetlet.2004.11.076
Return to citation in text: [1] -
Fürstner, A.; Weintritt, H.; Hupperts, A. J. Org. Chem. 1995, 60, 6637–6641. doi:10.1021/jo00125a068
Return to citation in text: [1] -
Boger, D. L.; Boyce, C. W.; Labroli, M. A.; Sehon, C. A.; Jin, Q. J. Am. Chem. Soc. 1999, 121, 54–62. doi:10.1021/ja982078+
Return to citation in text: [1] -
Banwell, M. G.; Flynn, B. L.; Hamel, E.; Hockles, D. C. R. Chem. Commun. 1997, 207–208. doi:10.1039/a606793j
Return to citation in text: [1] -
Hinze, C.; Kreipl, A.; Terpin, A.; Steglich, W. Synthesis 2007, 608–612. doi:10.1055/s-2007-965876
Return to citation in text: [1] -
Peschko, C.; Winklhofer, C.; Terpin, A.; Steglich, W. Synthesis 2006, 3048–3057. doi:10.1055/s-2006-950191
Return to citation in text: [1] -
Dumas, D. J. J. Org. Chem. 1988, 53, 4650–4653. doi:10.1021/jo00255a002
Return to citation in text: [1] [2] -
Khan, S. T.; Yu, P.; Hua, E.; Ali, S. N.; Nisa, M. Acta Crystallogr., Sect. E 2010, 66, o/711. doi:10.1107/S1600536810006951
Return to citation in text: [1] -
Reddy, G. S.; Rajasekhar, K.; Praveen, C.; Mukkanti, K.; Reddy, P. P. Synth. Commun. 2008, 38, 3884–3893. doi:10.1080/00397910802238742
Return to citation in text: [1] -
Vaillard, V. A.; Rossi, R. A.; Martín, S. E. Org. Biomol. Chem. 2011, 9, 4927–4935. doi:10.1039/c1ob05269a
Return to citation in text: [1] -
Çetinkaya, Y.; Balci, M. Tetrahedron Lett. 2014, 55, 6698–6702. doi:10.1016/j.tetlet.2014.10.044
Return to citation in text: [1] -
Vandavasi, J. K.; Hu, W.-P.; Senadi, G. C.; Boominathan, S. S. K.; Chen, H.-Y.; Wang, J.-J. Eur. J. Org. Chem. 2014, 6219–6226. doi:10.1002/ejoc.201402818
Return to citation in text: [1] -
Hou, Q.; Zhang, Z.; Kong, F.; Wang, S.; Wang, H.; Yao, Z.-J. Chem. Commun. 2013, 49, 695–697. doi:10.1039/C2CC36245G
Return to citation in text: [1] -
Hoffmann, M.; Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. Beilstein J. Org. Chem. 2013, 9, 1774–1780. doi:10.3762/bjoc.9.206
Return to citation in text: [1] -
Seo, H.; Jun, M. E.; Egorova, O. A.; Lee, K.-H.; Kim, K.-T.; Ahn, K. H. Org. Lett. 2012, 14, 5062–5065. doi:10.1021/ol302291c
Return to citation in text: [1] -
Yoshikawa, T.; Shindo, M. Org. Lett. 2009, 11, 5378–5381. doi:10.1021/ol902086t
Return to citation in text: [1] -
Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Org. Lett. 2006, 8, 5517–5520. doi:10.1021/ol062190+
Return to citation in text: [1] -
Harkat, H.; Weibel, J.-M.; Pale, P. Tetrahedron Lett. 2006, 47, 6273–6276. doi:10.1016/j.tetlet.2006.06.129
Return to citation in text: [1] -
Yao, T.; Larock, R. C. J. Org. Chem. 2003, 68, 5936–5942. doi:10.1021/jo034308v
Return to citation in text: [1] -
Biagetti, M.; Bellina, F.; Carpita, A.; Stabile, P.; Rossi, R. Tetrahedron 2002, 58, 5023–5038. doi:10.1016/S0040-4020(02)00469-6
Return to citation in text: [1] -
Marchal, E.; Uriac, P.; Legouin, B.; Toupet, L.; van de Weghe, P. Tetrahedron 2007, 63, 9979–9990. doi:10.1016/j.tet.2007.07.066
Return to citation in text: [1] [2] [3] -
Hewlett, N. M.; Tepe, J. J. Org. Lett. 2011, 13, 4550–4553. doi:10.1021/ol201741r
Return to citation in text: [1] -
Bailey, D. M.; Johnson, R. E.; Albertson, N. F. Org. Synth. 1971, 51, 100–102. doi:10.15227/orgsyn.051.0100
Return to citation in text: [1] -
Menges, N.; Sari, O.; Abdullayev, Y.; Erdem, S. S.; Balci, M. J. Org. Chem. 2013, 78, 5184–5195. doi:10.1021/jo4001228
Return to citation in text: [1] -
Nguyen, T. M.; Guzei, I. A.; Lee, D. J. Org. Chem. 2002, 67, 6553–6556. doi:10.1021/jo025630t
Return to citation in text: [1] -
Nguyen, T. M.; Lee, D. Org. Lett. 2001, 3, 3161–3163. doi:10.1021/ol016462e
Return to citation in text: [1] -
Blanco Jaimes, M. C.; Ahrens, A.; Pflästerer, D.; Rudolph, M.; Hashmi, A. S. K. Chem. – Eur. J. 2015, 21, 427–433. doi:10.1002/chem.201402524
Return to citation in text: [1] -
Ueda, H.; Yamaguchi, M.; Kameya, H.; Sugimoto, K.; Tokuyama, H. Org. Lett. 2014, 16, 4948–4951. doi:10.1021/ol5024695
Return to citation in text: [1] -
Sánchez-Larios, E.; Giacometti, R. D.; Hanessian, S. Eur. J. Org. Chem. 2014, 5664–5669. doi:10.1002/ejoc.201402803
Return to citation in text: [1] -
Kim, C.; Kang, S.; Rhee, Y. H. J. Org. Chem. 2014, 79, 11119–11124. doi:10.1021/jo502103p
Return to citation in text: [1] -
Tokimizu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Org. Lett. 2014, 16, 3138–3141. doi:10.1021/ol5012604
Return to citation in text: [1] -
Vaidya, T.; Cheng, R.; Carlsen, P. N.; Frontier, A. J.; Eisenberg, R. Org. Lett. 2014, 16, 800–803. doi:10.1021/ol403542k
Return to citation in text: [1] -
Hashmi, A. S. K. Acc. Chem. Res. 2014, 47, 864–876. doi:10.1021/ar500015k
Return to citation in text: [1] -
Corma, A.; Ruiz, V. R.; Leyva-Pérez, A.; Sabater, M. J. Adv. Synth. Catal. 2010, 352, 1701–1710. doi:10.1002/adsc.201000094
Return to citation in text: [1] -
Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136
Return to citation in text: [1] -
Molteni, G. Heterocycles 2004, 63, 1423–1428. doi:10.3987/COM-04-10050
Return to citation in text: [1] [2] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] -
Doucet, H.; Hierso, J.-C. Angew. Chem., Int. Ed. 2007, 46, 834–871. doi:10.1002/anie.200602761
Return to citation in text: [1] -
Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/S0040-4039(00)91094-3
Return to citation in text: [1] -
Kost, A. N.; Grandberg, I. I. Adv. Heterocycl. Chem. 1966, 6, 347–429. doi:10.1016/S0065-2725(08)60579-6
Return to citation in text: [1] -
Caporale, A.; Tartaggia, S.; Castellin, A.; de Lucchi, O. Beilstein J. Org. Chem. 2014, 10, 384–393. doi:10.3762/bjoc.10.36
Return to citation in text: [1] -
Meng, G.; Zheng, M.-L.; Wang, M. Org. Prep. Proced. Int. 2011, 43, 308–311. doi:10.1080/00304948.2011.582005
-
Fumie, S. PCT Int. Appl. WO 9628250 A1, Sept 19, 1996; pp 232 ff.
-
Verniest, G.; Padwa, A. Org. Lett. 2008, 10, 4379–4382. doi:10.1021/ol801847j
-
Defossa, E.; Heinelt, U.; Klingler, O.; Zoller, G.; Al-Obeidi, F.; Walser, A.; Wildgoose, P.; Matter, H. Patent W 9933800 A1, July 8, 1999.
(Chem. Abstr. 1999, 131, 87814).
-
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2009.
Return to citation in text: [1] -
Wang, D.; Cai, R.; Sharma, S.; Jirak, J.; Thummanapelli, S. K.; Akhmedov, N. G.; Zhang, H.; Liu, X.; Petersen, J. L.; Shi, X. J. Am. Chem. Soc. 2012, 134, 9012–9019. doi:10.1021/ja303862z
Return to citation in text: [1] -
Patrick, S. R.; Boogaerts, I. I. F.; Gaillard, S.; Slawin, A. M. Z.; Nolan, S. P. Beilstein J. Org. Chem. 2011, 7, 892–896. doi:10.3762/bjoc.7.102
Return to citation in text: [1] -
Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e
Return to citation in text: [1] -
Genin, E.; Toullec, P. Y.; Antoniotti, S.; Brancour, C.; Genêt, J.-P.; Michelet, V. J. Am. Chem. Soc. 2006, 128, 3112–3113. doi:10.1021/ja056857j
Return to citation in text: [1] -
Chernyak, N.; Gevorgyan, V. J. Am. Chem. Soc. 2008, 130, 5636–5637. doi:10.1021/ja8006534
Return to citation in text: [1] -
Yamada, W.; Sugawara, Y.; Cheng, H. M.; Ikeno, T.; Yamada, T. Eur. J. Org. Chem. 2007, 2604–2607. doi:10.1002/ejoc.200700169
Return to citation in text: [1]
| 54. | Corma, A.; Ruiz, V. R.; Leyva-Pérez, A.; Sabater, M. J. Adv. Synth. Catal. 2010, 352, 1701–1710. doi:10.1002/adsc.201000094 |
| 55. | Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136 |
| 1. |
Gossauer, A. Pyrrole. Houben-Weyl – Methods of Organic Chemistry; Thieme: Stuttgart, 1994; Vol. E6a/1, pp 556 ff.
See for a review. |
| 11. | Manzanaro, S.; Salvá, J.; de la Fuente, J. Á. J. Nat. Prod. 2006, 69, 1485–1487. doi:10.1021/np0503698 |
| 44. | Menges, N.; Sari, O.; Abdullayev, Y.; Erdem, S. S.; Balci, M. J. Org. Chem. 2013, 78, 5184–5195. doi:10.1021/jo4001228 |
| 45. | Nguyen, T. M.; Guzei, I. A.; Lee, D. J. Org. Chem. 2002, 67, 6553–6556. doi:10.1021/jo025630t |
| 46. | Nguyen, T. M.; Lee, D. Org. Lett. 2001, 3, 3161–3163. doi:10.1021/ol016462e |
| 8. | Yoshida, W. Y.; Lee, K. K.; Carrol, A. R.; Scheuer, P. J. Helv. Chim. Acta 1992, 75, 1721–1725. doi:10.1002/hlca.19920750525 |
| 9. | Lu, Y.; Arndtsen, B. A. Org. Lett. 2009, 11, 1369–1372. doi:10.1021/ol900185n |
| 10. | Takamura, K.; Matsuo, H.; Tanaka, A.; Tanaka, J.; Fukuda, T.; Ishibashi, F.; Iwao, M. Tetrahedron 2013, 69, 2782–2788. doi:10.1016/j.tet.2013.01.077 |
| 41. | Marchal, E.; Uriac, P.; Legouin, B.; Toupet, L.; van de Weghe, P. Tetrahedron 2007, 63, 9979–9990. doi:10.1016/j.tet.2007.07.066 |
| 47. | Blanco Jaimes, M. C.; Ahrens, A.; Pflästerer, D.; Rudolph, M.; Hashmi, A. S. K. Chem. – Eur. J. 2015, 21, 427–433. doi:10.1002/chem.201402524 |
| 48. | Ueda, H.; Yamaguchi, M.; Kameya, H.; Sugimoto, K.; Tokuyama, H. Org. Lett. 2014, 16, 4948–4951. doi:10.1021/ol5024695 |
| 49. | Sánchez-Larios, E.; Giacometti, R. D.; Hanessian, S. Eur. J. Org. Chem. 2014, 5664–5669. doi:10.1002/ejoc.201402803 |
| 50. | Kim, C.; Kang, S.; Rhee, Y. H. J. Org. Chem. 2014, 79, 11119–11124. doi:10.1021/jo502103p |
| 51. | Tokimizu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Org. Lett. 2014, 16, 3138–3141. doi:10.1021/ol5012604 |
| 52. | Vaidya, T.; Cheng, R.; Carlsen, P. N.; Frontier, A. J.; Eisenberg, R. Org. Lett. 2014, 16, 800–803. doi:10.1021/ol403542k |
| 53. | Hashmi, A. S. K. Acc. Chem. Res. 2014, 47, 864–876. doi:10.1021/ar500015k |
| 6. | Benaamane, N.; Nedjar-Kolli, B.; Bentarzi, Y.; Hammal, L.; Geronikaki, A.; Eleftheriou, P.; Lagunin, A. Bioorg. Med. Chem. 2008, 16, 3059–3066. doi:10.1016/j.bmc.2007.12.033 |
| 7. | Kern, J. C.; Terefenko, E. A.; Fensome, A.; Unwallla, R.; Wrobel, J.; Zhu, Y.; Cohen, J.; Winneker, R.; Zhang, Z.; Zhang, P. Bioorg. Med. Chem. Lett. 2007, 17, 189–192. doi:10.1016/j.bmcl.2006.09.060 |
| 41. | Marchal, E.; Uriac, P.; Legouin, B.; Toupet, L.; van de Weghe, P. Tetrahedron 2007, 63, 9979–9990. doi:10.1016/j.tet.2007.07.066 |
| 41. | Marchal, E.; Uriac, P.; Legouin, B.; Toupet, L.; van de Weghe, P. Tetrahedron 2007, 63, 9979–9990. doi:10.1016/j.tet.2007.07.066 |
| 71. | Chernyak, N.; Gevorgyan, V. J. Am. Chem. Soc. 2008, 130, 5636–5637. doi:10.1021/ja8006534 |
| 72. | Yamada, W.; Sugawara, Y.; Cheng, H. M.; Ikeno, T.; Yamada, T. Eur. J. Org. Chem. 2007, 2604–2607. doi:10.1002/ejoc.200700169 |
| 2. | Meng, Y.; Wang, G.; Li, Y.; Hou, K.; Yuan, Y.; Zhang, L.-J.; Song, H.-R.; Shi, W. Chin. Chem. Lett. 2013, 24, 619–621. doi:10.1016/j.cclet.2013.04.020 |
| 3. | Han, S.; Siegel, D. S.; Morrison, K. C.; Hergenrother, P. J.; Movassaghi, M. J. Org. Chem. 2013, 78, 11970–11984. doi:10.1021/jo4020112 |
| 4. | Krishnaiah, P.; Reddy, V. L. N.; Venkataramana, G.; Ravinder, K.; Srinivasulu, M.; Raju, T. V.; Ravikumar, K.; Chandrasekar, D.; Ramakrishna, S.; Venkateswarlu, Y. J. Nat. Prod. 2004, 67, 1168–1171. doi:10.1021/np030503t |
| 5. | Rochais, C.; Lisowski, V.; Dallemagne, P.; Rault, S. Bioorg. Med. Chem. 2006, 14, 8162–8175. doi:10.1016/j.bmc.2006.09.022 |
| 42. | Hewlett, N. M.; Tepe, J. J. Org. Lett. 2011, 13, 4550–4553. doi:10.1021/ol201741r |
| 43. | Bailey, D. M.; Johnson, R. E.; Albertson, N. F. Org. Synth. 1971, 51, 100–102. doi:10.15227/orgsyn.051.0100 |
| 2. | Meng, Y.; Wang, G.; Li, Y.; Hou, K.; Yuan, Y.; Zhang, L.-J.; Song, H.-R.; Shi, W. Chin. Chem. Lett. 2013, 24, 619–621. doi:10.1016/j.cclet.2013.04.020 |
| 27. | Dumas, D. J. J. Org. Chem. 1988, 53, 4650–4653. doi:10.1021/jo00255a002 |
| 28. | Khan, S. T.; Yu, P.; Hua, E.; Ali, S. N.; Nisa, M. Acta Crystallogr., Sect. E 2010, 66, o/711. doi:10.1107/S1600536810006951 |
| 29. | Reddy, G. S.; Rajasekhar, K.; Praveen, C.; Mukkanti, K.; Reddy, P. P. Synth. Commun. 2008, 38, 3884–3893. doi:10.1080/00397910802238742 |
| 30. | Vaillard, V. A.; Rossi, R. A.; Martín, S. E. Org. Biomol. Chem. 2011, 9, 4927–4935. doi:10.1039/c1ob05269a |
| 32. | Vandavasi, J. K.; Hu, W.-P.; Senadi, G. C.; Boominathan, S. S. K.; Chen, H.-Y.; Wang, J.-J. Eur. J. Org. Chem. 2014, 6219–6226. doi:10.1002/ejoc.201402818 |
| 67. | Wang, D.; Cai, R.; Sharma, S.; Jirak, J.; Thummanapelli, S. K.; Akhmedov, N. G.; Zhang, H.; Liu, X.; Petersen, J. L.; Shi, X. J. Am. Chem. Soc. 2012, 134, 9012–9019. doi:10.1021/ja303862z |
| 68. | Patrick, S. R.; Boogaerts, I. I. F.; Gaillard, S.; Slawin, A. M. Z.; Nolan, S. P. Beilstein J. Org. Chem. 2011, 7, 892–896. doi:10.3762/bjoc.7.102 |
| 69. | Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e |
| 21. | Mathew, P.; Asokan, C. W. Tetrahedron Lett. 2005, 46, 475–478. doi:10.1016/j.tetlet.2004.11.076 |
| 22. | Fürstner, A.; Weintritt, H.; Hupperts, A. J. Org. Chem. 1995, 60, 6637–6641. doi:10.1021/jo00125a068 |
| 23. | Boger, D. L.; Boyce, C. W.; Labroli, M. A.; Sehon, C. A.; Jin, Q. J. Am. Chem. Soc. 1999, 121, 54–62. doi:10.1021/ja982078+ |
| 24. | Banwell, M. G.; Flynn, B. L.; Hamel, E.; Hockles, D. C. R. Chem. Commun. 1997, 207–208. doi:10.1039/a606793j |
| 25. | Hinze, C.; Kreipl, A.; Terpin, A.; Steglich, W. Synthesis 2007, 608–612. doi:10.1055/s-2007-965876 |
| 26. | Peschko, C.; Winklhofer, C.; Terpin, A.; Steglich, W. Synthesis 2006, 3048–3057. doi:10.1055/s-2006-950191 |
| 33. | Hou, Q.; Zhang, Z.; Kong, F.; Wang, S.; Wang, H.; Yao, Z.-J. Chem. Commun. 2013, 49, 695–697. doi:10.1039/C2CC36245G |
| 34. | Hoffmann, M.; Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. Beilstein J. Org. Chem. 2013, 9, 1774–1780. doi:10.3762/bjoc.9.206 |
| 35. | Seo, H.; Jun, M. E.; Egorova, O. A.; Lee, K.-H.; Kim, K.-T.; Ahn, K. H. Org. Lett. 2012, 14, 5062–5065. doi:10.1021/ol302291c |
| 36. | Yoshikawa, T.; Shindo, M. Org. Lett. 2009, 11, 5378–5381. doi:10.1021/ol902086t |
| 37. | Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Org. Lett. 2006, 8, 5517–5520. doi:10.1021/ol062190+ |
| 38. | Harkat, H.; Weibel, J.-M.; Pale, P. Tetrahedron Lett. 2006, 47, 6273–6276. doi:10.1016/j.tetlet.2006.06.129 |
| 39. | Yao, T.; Larock, R. C. J. Org. Chem. 2003, 68, 5936–5942. doi:10.1021/jo034308v |
| 40. | Biagetti, M.; Bellina, F.; Carpita, A.; Stabile, P.; Rossi, R. Tetrahedron 2002, 58, 5023–5038. doi:10.1016/S0040-4020(02)00469-6 |
| 70. | Genin, E.; Toullec, P. Y.; Antoniotti, S.; Brancour, C.; Genêt, J.-P.; Michelet, V. J. Am. Chem. Soc. 2006, 128, 3112–3113. doi:10.1021/ja056857j |
| 15. | Andersen, R. J.; Faulkner, D. J.; Cun-heng, H.; Van Duyne, G. D.; Clardy, J. J. Am. Chem. Soc. 1985, 107, 5492–5495. doi:10.1021/ja00305a027 |
| 16. | Carroll, A. R.; Bowden, B. F.; Coll, J. C. Aust. J. Chem. 1993, 46, 489–501. doi:10.1071/CH9930489 |
| 17. | Urban, S.; Butler, M. S.; Capon, R. J. Aust. J. Chem. 1994, 47, 1919–1924. doi:10.1071/CH9941919 |
| 18. | Fan, H.; Peng, J.; Hamann, M. T.; Hu, J.-F. Chem. Rev. 2008, 108, 264–287. doi:10.1021/cr078199m |
| 19. | Chittchang, M.; Paul, G. M.; Ploypradith, P.; Ruchirawat, S. Eur. J. Med. Chem. 2010, 45, 2165–2172. doi:10.1016/j.ejmech.2010.01.053 |
| 20. | Ueda, K.; Amaike, K.; Maceiczyk, R. M.; Itami, K.; Yamaguchi, J. J. Am. Chem. Soc. 2014, 136, 13226–13232. doi:10.1021/ja508449y |
| 56. | Molteni, G. Heterocycles 2004, 63, 1423–1428. doi:10.3987/COM-04-10050 |
| 57. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 58. | Doucet, H.; Hierso, J.-C. Angew. Chem., Int. Ed. 2007, 46, 834–871. doi:10.1002/anie.200602761 |
| 59. | Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/S0040-4039(00)91094-3 |
| 60. | Kost, A. N.; Grandberg, I. I. Adv. Heterocycl. Chem. 1966, 6, 347–429. doi:10.1016/S0065-2725(08)60579-6 |
| 61. | Caporale, A.; Tartaggia, S.; Castellin, A.; de Lucchi, O. Beilstein J. Org. Chem. 2014, 10, 384–393. doi:10.3762/bjoc.10.36 |
| 12. | Kang, H.; Fenical, W. J. Org. Chem. 1997, 62, 3254–3262. doi:10.1021/jo962132+ |
| 13. | Hasse, K.; Willis, A. C.; Banwell, M. G. Aust. J. Chem. 2009, 62, 683–691. doi:10.1071/CH09158 |
| 14. | Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Org. Lett. 2011, 13, 312–315. doi:10.1021/ol1027877 |
| 31. | Çetinkaya, Y.; Balci, M. Tetrahedron Lett. 2014, 55, 6698–6702. doi:10.1016/j.tetlet.2014.10.044 |
© 2015 Taskaya et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)