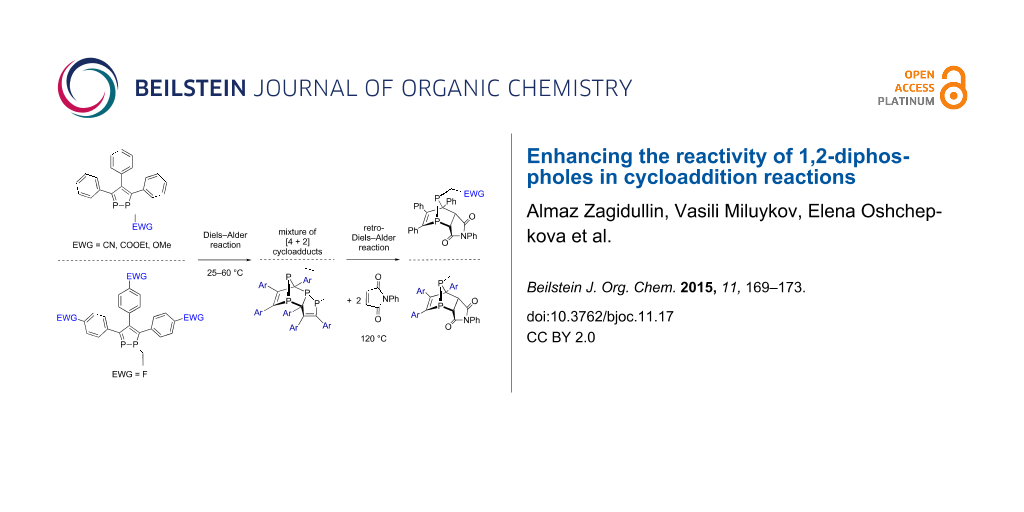
Abstract
Two different approaches have been employed to enhance the reactivity of 1-alkyl-1,2-diphospholes – the introduction of electron-withdrawing groups either at the phosphorus atoms or in the para-position of the arene ring. The alkylation of sodium 1,2-diphospha-3,4,5-triphenylcyclopentadienide with alkyl halides Hal-CH2-R (R = CN, COOEt, OMe, CH2OEt) results in corresponding 1-alkyl-3,4,5-triphenyl-1,2-diphospholes (alkyl = CH2CN (1a), CH2COOEt (1b), CH2OMe (1c), and (CH2)2OEt (1d)), which spontaneously undergo the intermolecular [4 + 2] cycloaddition reactions at room temperature to form the mixture of the cycloadducts, 2a–c, respectively. However the alkylation of sodium 1,2-diphospha-3,4,5-tri(p-fluorophenyl)cyclopentadienide with ethyl iodide leads to stable 1-ethyl-3,4,5-tris(p-fluorophenyl)-1,2-diphosphole (1e), which forms the [4 + 2] cycloadduct 2,3,4,4a,5,6-hexa(p-fluorophenyl)-1-ethyl-1,7,7a-triphospha-4,7-(ethylphosphinidene)indene (2e) only upon heating up to 60 °C. With further heating to 120 °C with N-phenylmaleimide, the cycloadducts 2a–c and 2e undergo the retro-Diels–Alder reaction and form only one product of the [4 + 2] cycloaddition reaction 3a–с, 3e with good yields up to 65%.

Graphical Abstract
Introduction
Phospholes are weakly aromatic heterocycles and demonstrate rather different properties from those of their S, N and O counterparts [1,2]. Due to low their aromaticity, phospholes are of significant interest for the preparation of highly effective catalysts, materials for light-emitting diodes and nonlinear optics [3,4]. In contrast to furans, thiophenes and pyrroles, phospholes display cycloaddition and complexation reactions and can be used as starting materials for caged phosphines, phosphinidenes, etc. [2]. At the same time, the presence of electron-withdrawing substituents (cyano-, alkoxy-, or halo-) at the phosphorus atom reduces the aromaticity of the monophosphole ring and facilitates cycloaddition reactions resulting in novel 7-phosphanorbornenes [5,6], which was verified by theoretical calculations and experimental work [7,8].
At the same time both the presence of the P=C bond in phospholes as well as the transient 2H-phospholes [3] increase the cycloaddition reactivity. It was previously demonstrated that 1-alkyl-1,2-diphospholes combine the properties of both 1H-phospholes (with thermal stability up to 190 °C) and 2H-phospholes (exhibiting high reactivity in the cycloaddition reaction at 25 °C) [9-11]. In the present work, attempts to increase the reactivity of the dienic system of 1,2-diphospholes using two different approaches are described: (a) by the introduction of electron-withdrawing groups (EWGs) at the phosphorus atom or (b) the introduction of EWGs to the carbon atoms of aryl substituents. This work will provide access to new polycyclic, organophosphorus compounds having significant potential as weak, bulky ligands in homogeneous catalysis [12-14].
Results and Discussion
The 1-alkyl-1,2-diphospholes 1a–e, incorporating EWGs at the phosphorus atom or in aromatic fragments, are easily accessible by the alkylation of sodium 1,2-diphospha-3,4,5-triarylcyclopentadienide with alkyl halides. The reactions were carried out in THF at −80 °C with yields of 55–60% for 1a–e (Scheme 1).
![[1860-5397-11-17-i1]](/bjoc/content/inline/1860-5397-11-17-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 1-alkyl-1,2-diphospholes 1a–e.
Scheme 1: Synthesis of 1-alkyl-1,2-diphospholes 1a–e.
The structures of the obtained compounds, 1a–e, were unambiguously confirmed by 31Р, 1Н and 13С NMR spectroscopy. The 31P NMR spectra of 1a–e (Table 1) showed two doublets in the range of 30–60 and 210–225 ppm, corresponding to three- and two-coordinated phosphorus atoms, respectively, with a large coupling constant 1JPP ≈ 365–410 Hz, which is typical for 1-alkyl-1,2-diphospholes [15].
Table 1: 31Р {1H} NMR spectral data of new 1-R-1,2-diphospholes 1a–e.
| δP, ppm | ΔδP, ppm | 1JPР, Hz | R | X | |
|---|---|---|---|---|---|
| 1a | 225.7 (P1=C) and 30.8 (P2) | 194.9 | 363.1 | CN | H |
| 1b | 223.3 (P1=C) and 40.5 (P2) | 182.8 | 389.0 | COOEt | H |
| 1c | 209.3 (P1=C) and 61.2 (P2) | 148.1 | 404.0 | OMe | H |
| 1d | 214.1 (P1=C) and 51.9 (P2) | 162.2 | 407.3 | CH2OEt | H |
| 1e | 218.5 (P1=C) and 73.4 (P2) | 145.1 | 407.8 | Me | F |
Remarkably, the coupling constants 1JPР increase with the decrease of the electron-withdrawing properties of the substitutents in the diphosphole ring (CN > COOEt > OMe > CH2OEt) in the series 1a–1d.
A large phosphorus–phosphorus coupling constant, 1JPР, usually indicates significant σ–π delocalization of the lone pair of the tricoordinated phosphorus atom into the diphosphole ring system. Thus, the 1JPP coupling constant for a non-aromatic 1,2-diphosphacyclopentene is observed at around 220 Hz [16], although both phosphorus atoms of the highly aromatic 1-(2,4,6-tri-tert-butylphenyl)-1H-1,2-diphosphole are coupled with a larger phosphorus–phosphorus constant (1JPР = 528.2 Hz) [17]. The same phenomena was noted for the 1,2,4-triphosphole with the planar tricoordinated phosphorus [18]. Thus, the increase of 1JPР in a sequence from 1a to 1d could imply the increasing delocalization of the RP-fragment within the diphosphole system that reflects the stability and the reactivity of 1,2-diphosphole.
Indeed, the compounds 1a,b are stable only at temperatures below +5 °C, while 1c is stable at room temperature for a few hours. 1,2-Diphosphole 1d is more stable and no cycloaddition was observed upon heating in toluene. Upon standing, the diphospholes 1a–c undergo spontaneous [4 + 2] cycloaddition reactions leading to a mixture of cycloadducts (Scheme 2). The 31P NMR spectra of the reaction mixtures showed many multiplets at 80 and −40 ppm with a coupling constant 1JPP ca. 200–210 Hz characteristic for the products of [4 + 2] cycloaddition reaction – 1,7-diphosphanorbornadienes [9,11]. Remarkably, 1-alkyl-1,2-diphospholes without EWGs reveal significant thermal stability and dimerization was observed only upon heating to 190 °C leading to the product of the [2 + 2] cycloaddition reaction [9]. At the same time, 3,4,5-tri(p-fluorophenyl)-1-ethyl-1,2-diphosphole (1e) is stable at room temperature and undergoes the [4 + 2] cycloaddition reaction only upon heating at 60 °C resulting in only one product, 2e (Scheme 2).
![[1860-5397-11-17-i2]](/bjoc/content/inline/1860-5397-11-17-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The cycloaddition reactions of 1-alkyl-1,2-diphospholes 1a–e.
Scheme 2: The cycloaddition reactions of 1-alkyl-1,2-diphospholes 1a–e.
The molecular structure of 2e (Figure 1) was verified by X-ray crystallography. The crystal structure analysis of 2e showed that only the endo isomer was formed with the alkyl group in anti-orientation with respect to the double bond of the ring.
![[1860-5397-11-17-1]](/bjoc/content/figures/1860-5397-11-17-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP view of 2,3,4,4a,5,6-hexa(p-fluorophenyl)-1-ethyl-1,7,7a-triphospha-4,7-(ethylphosphinidene)indene (2e). Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–C1 1.850(5); P1–С4 1.923(6); P1–Р2 2.213(2); P2–С43 1.843(5); P2–С3 1.896(5); C1–С2 1.360(7); C2–С3 1.540(7); P3–С3 1.924(6); P3–С4 1.902(6); P3–P4 2.193(2); P4–С6 1.832(5); C5–С6 1.340(7); C5–С4 1.539(7); C1–P1–C4 97.9(2); C1–P1–P2 85.79(18); C4–P1–P2 99.14(17); C43–P2–C3 105.5(2); C43–P2–P1 109.9(2); C3–P2–P1 87.07(17); C4–P3–C3 100.1(2); C4–P3–P4 95.44(17); C3–P3–P4 101.35(17); C6–P4–C45 101.6(3);C6–P4–P3 93.40(18); C45–P4–P3 100.8(2).
Figure 1: ORTEP view of 2,3,4,4a,5,6-hexa(p-fluorophenyl)-1-ethyl-1,7,7a-triphospha-4,7-(ethylphosphinidene)i...
In the case of 2a–c, it would be assumed that similar [4 + 2] cycloadducts are formed according to the range of signals in the 31P NMR spectra. However, in this case, several stereoisomers are formed due to the high reactivity of the 1,2-diphospholes containing EWGs on the phosphorus atom. It should be noted that this is the first example of [4 + 2] cycloaddition between two diphosphole molecules where 1,2-diphosphole acts as a diene and a dienophile in one reaction. Therefore, these isomeric cycloadducts, 2a–с, can be a source of reactive 1,2-diphospholes containing EWGs 1a–с in the retro-Diels–Alder reaction [19].
Indeed, upon further heating up to 120 °C, the mixture of the cycloadducts 2a–c as well the cycloadduct 2e underwent the retro-Diels–Alder reaction to form monomeric 1,2-diphospholes, 1a–c, 1e, which were trapped by N-phenylmaleimide to form the compounds 3a–c, 3e with yields of 55–65% (Scheme 3).
![[1860-5397-11-17-i3]](/bjoc/content/inline/1860-5397-11-17-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The retro-Diels–Alder reactions of the cycloadducts 2a–с, and 2e.
Scheme 3: The retro-Diels–Alder reactions of the cycloadducts 2a–с, and 2e.
After heating for 25–30 hours at 120 °C in toluene, the 31Р NMR spectra of the reaction mixture displayed only two doublets at around −25 and 60 ppm with 1JPP = 200 Hz. This indicates the high stereoselectivity of this reaction. The 1H NMR spectra of the reaction mixtures show only two doublets for protons of the N-phenylmaleimide fragment in the range of 4.6–4.8 ppm. Based on our previous results [20], we can conclude that only an anti-endo-isomer was formed in each case.
Conclusion
In summary, we have demonstrated the prospect of increasing the reactivity of 1,2-diphospholes using two different approaches: (a) introduction of EWGs at the phosphorus atom and (b) introduction of EWGs at the carbon atoms of aryl substituents. New 1-alkyl-1,2-diphospholes, 1a–c, 1e, containing EWGs demonstrated high reactivity and underwent intermolecular [4 + 2] cycloaddition reactions at 25–60 °C leading to a single product, 2e, or a mixture of [4 + 2] cycloadducts 2a–c. Additionally, upon further heating up to 120 °C with N-phenylmaleimide, the mixture of isomeric cycloadducts 2a–c, 2e underwent the retro-Diels–Alder reaction, yielding only one product of [4 + 2] cycloaddition 3a–c, and 3e. The same chemical behavior was observed for 1-alkyl-1,2-diphosphole-1-oxides, which underwent [4 + 2] cycloaddition at 25 °C and the retro-Diels–Alder reaction at 100 °C [21]. Compared with 1-alkyl-1,2-diphospholes, the new 1-R-1,2-diphospholes 1a–c, 1e containing EWGs were less stable. Given that they are more reactive in cycloaddition reactions, this work presents the opportunity for new polycyclic phosphines.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 390.6 KB | Download |
References
-
von Ragué Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N. J. R. J. Am. Chem. Soc. 1996, 118, 6317–6318. doi:10.1021/ja960582d
Return to citation in text: [1] -
Zagidullin, A. A.; Bezkishko, I. A.; Miluykov, V. A.; Sinyashin, O. G. Mendeleev Commun. 2013, 23, 117–130. doi:10.1016/j.mencom.2013.05.001
Return to citation in text: [1] [2] -
Mathey, F. Acc. Chem. Res. 2004, 37, 954–960. doi:10.1021/ar030118v
Return to citation in text: [1] [2] -
Quin, L. D. Curr. Org. Chem. 2006, 10, 43–78. doi:10.2174/138527206775192997
Return to citation in text: [1] -
Mattmann, E.; Mathey, F.; Sevin, A.; Frison, G. J. Org. Chem. 2002, 67, 1208–1213. doi:10.1021/jo0108156
Return to citation in text: [1] -
Mattmann, E.; Simonutti, D.; Ricard, L.; Mercier, F.; Mathey, F. J. Org. Chem. 2001, 66, 755–758. doi:10.1021/jo001096i
Return to citation in text: [1] -
Westerhausen, M.; Stein, B.; Ossberger, M.; Görls, H.; Ruiz, J.; Nöth, H.; Mayer, P. ARKIVOC 2007, No. iii, 46–59.
Return to citation in text: [1] -
Mattmann, E.; Mercier, F.; Ricard, L.; Mathey, F. J. Org. Chem. 2002, 67, 5422–5425. doi:10.1021/jo025713+
Return to citation in text: [1] -
Miluykov, V.; Bezkishko, I.; Zagidullin, A.; Sinyashin, O.; Lönnecke, P.; Hey-Hawkins, E. Eur. J. Org. Chem. 2009, 1269–1274. doi:10.1002/ejoc.200801181
Return to citation in text: [1] [2] [3] -
Zagidullin, A. A.; Miluykov, V. A.; Krivolapov, D. B.; Kharlamov, S. V.; Latypov, S. K.; Sinyashin, O. G.; Hey-Hawkins, E. Eur. J. Org. Chem. 2011, 4910–4918. doi:10.1002/ejoc.201100615
Return to citation in text: [1] -
Zagidullin, A.; Ganushevich, Y.; Miluykov, V.; Sinyashin, O.; Hey-Hawkins, E. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 238–242. doi:10.1080/10426507.2012.744017
Return to citation in text: [1] [2] -
Robin, F.; Mercier, F.; Ricard, L.; Mathey, F.; Spagnol, M. Chem. – Eur. J. 1997, 3, 1365–1369. doi:10.1002/chem.19970030824
Return to citation in text: [1] -
Clochard, M.; Mattmann, E.; Mercier, F.; Ricard, L.; Mathey, F. Org. Lett. 2003, 5, 3093–3094. doi:10.1021/ol035067g
Return to citation in text: [1] -
Germoni, A.; Deschamps, B.; Ricard, L.; Mercier, F.; Mathey, F. J. Organomet. Chem. 2005, 690, 1133–1139. doi:10.1016/j.jorganchem.2004.11.018
Return to citation in text: [1] -
Milyukov, V. A.; Bezkishko, I. A.; Zagidullin, A. A.; Sinyashin, O. G.; Hey-Hawkins, E. Russ. Chem. Bull. 2010, 59, 1232–1236. doi:10.1007/s11172-010-0226-9
Return to citation in text: [1] -
Doxsee, K. M.; Wood, N. P.; Hanawalt, E. M.; Weakley, T. J. R. Heteroat. Chem. 1996, 7, 383–389. doi:10.1002/(SICI)1098-1071(199610)7:5<383::AID-HC16>3.0.CO;2-M
Return to citation in text: [1] -
Ionkin, A. S.; Marshall, W. J.; Fish, B. M.; Schiffhauer, M. F.; Davidson, F.; McEwen, C. N.; Keys, D. E. Organometallics 2007, 26, 5050–5058. doi:10.1021/om7005084
Return to citation in text: [1] -
Cloke, F. G. N.; Hitchcock, P. B.; Hunnable, P.; Nixon, J. F.; Nyulászi, L.; Niecke, E.; Thelen, V. Angew. Chem., Int. Ed. 1998, 37, 1083–1086. doi:10.1002/(SICI)1521-3773(19980504)37:8<1083::AID-ANIE1083>3.0.CO;2-C
Return to citation in text: [1] -
Kotha, S.; Banerjee, S. RSC Adv. 2013, 3, 7642–7666. doi:10.1039/c3ra22762f
Return to citation in text: [1] -
Zagidullin, A.; Miluykov, V.; Sinyashin, O.; Lönnecke, P.; Hey-Hawkins, E. Heteroat. Chem. 2014, 25, 28–34. doi:10.1002/hc.21132
Return to citation in text: [1] -
Zagidullin, A.; Ganushevich, Y.; Miluykov, V.; Krivolapov, D.; Kataeva, O.; Sinyashin, O.; Hey-Hawkins, E. Org. Biomol. Chem. 2012, 10, 5298–5306. doi:10.1039/c2ob25532d
Return to citation in text: [1]
| 21. | Zagidullin, A.; Ganushevich, Y.; Miluykov, V.; Krivolapov, D.; Kataeva, O.; Sinyashin, O.; Hey-Hawkins, E. Org. Biomol. Chem. 2012, 10, 5298–5306. doi:10.1039/c2ob25532d |
| 1. | von Ragué Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N. J. R. J. Am. Chem. Soc. 1996, 118, 6317–6318. doi:10.1021/ja960582d |
| 2. | Zagidullin, A. A.; Bezkishko, I. A.; Miluykov, V. A.; Sinyashin, O. G. Mendeleev Commun. 2013, 23, 117–130. doi:10.1016/j.mencom.2013.05.001 |
| 7. | Westerhausen, M.; Stein, B.; Ossberger, M.; Görls, H.; Ruiz, J.; Nöth, H.; Mayer, P. ARKIVOC 2007, No. iii, 46–59. |
| 8. | Mattmann, E.; Mercier, F.; Ricard, L.; Mathey, F. J. Org. Chem. 2002, 67, 5422–5425. doi:10.1021/jo025713+ |
| 5. | Mattmann, E.; Mathey, F.; Sevin, A.; Frison, G. J. Org. Chem. 2002, 67, 1208–1213. doi:10.1021/jo0108156 |
| 6. | Mattmann, E.; Simonutti, D.; Ricard, L.; Mercier, F.; Mathey, F. J. Org. Chem. 2001, 66, 755–758. doi:10.1021/jo001096i |
| 20. | Zagidullin, A.; Miluykov, V.; Sinyashin, O.; Lönnecke, P.; Hey-Hawkins, E. Heteroat. Chem. 2014, 25, 28–34. doi:10.1002/hc.21132 |
| 2. | Zagidullin, A. A.; Bezkishko, I. A.; Miluykov, V. A.; Sinyashin, O. G. Mendeleev Commun. 2013, 23, 117–130. doi:10.1016/j.mencom.2013.05.001 |
| 9. | Miluykov, V.; Bezkishko, I.; Zagidullin, A.; Sinyashin, O.; Lönnecke, P.; Hey-Hawkins, E. Eur. J. Org. Chem. 2009, 1269–1274. doi:10.1002/ejoc.200801181 |
| 11. | Zagidullin, A.; Ganushevich, Y.; Miluykov, V.; Sinyashin, O.; Hey-Hawkins, E. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 238–242. doi:10.1080/10426507.2012.744017 |
| 3. | Mathey, F. Acc. Chem. Res. 2004, 37, 954–960. doi:10.1021/ar030118v |
| 4. | Quin, L. D. Curr. Org. Chem. 2006, 10, 43–78. doi:10.2174/138527206775192997 |
| 9. | Miluykov, V.; Bezkishko, I.; Zagidullin, A.; Sinyashin, O.; Lönnecke, P.; Hey-Hawkins, E. Eur. J. Org. Chem. 2009, 1269–1274. doi:10.1002/ejoc.200801181 |
| 15. | Milyukov, V. A.; Bezkishko, I. A.; Zagidullin, A. A.; Sinyashin, O. G.; Hey-Hawkins, E. Russ. Chem. Bull. 2010, 59, 1232–1236. doi:10.1007/s11172-010-0226-9 |
| 17. | Ionkin, A. S.; Marshall, W. J.; Fish, B. M.; Schiffhauer, M. F.; Davidson, F.; McEwen, C. N.; Keys, D. E. Organometallics 2007, 26, 5050–5058. doi:10.1021/om7005084 |
| 12. | Robin, F.; Mercier, F.; Ricard, L.; Mathey, F.; Spagnol, M. Chem. – Eur. J. 1997, 3, 1365–1369. doi:10.1002/chem.19970030824 |
| 13. | Clochard, M.; Mattmann, E.; Mercier, F.; Ricard, L.; Mathey, F. Org. Lett. 2003, 5, 3093–3094. doi:10.1021/ol035067g |
| 14. | Germoni, A.; Deschamps, B.; Ricard, L.; Mercier, F.; Mathey, F. J. Organomet. Chem. 2005, 690, 1133–1139. doi:10.1016/j.jorganchem.2004.11.018 |
| 18. | Cloke, F. G. N.; Hitchcock, P. B.; Hunnable, P.; Nixon, J. F.; Nyulászi, L.; Niecke, E.; Thelen, V. Angew. Chem., Int. Ed. 1998, 37, 1083–1086. doi:10.1002/(SICI)1521-3773(19980504)37:8<1083::AID-ANIE1083>3.0.CO;2-C |
| 9. | Miluykov, V.; Bezkishko, I.; Zagidullin, A.; Sinyashin, O.; Lönnecke, P.; Hey-Hawkins, E. Eur. J. Org. Chem. 2009, 1269–1274. doi:10.1002/ejoc.200801181 |
| 10. | Zagidullin, A. A.; Miluykov, V. A.; Krivolapov, D. B.; Kharlamov, S. V.; Latypov, S. K.; Sinyashin, O. G.; Hey-Hawkins, E. Eur. J. Org. Chem. 2011, 4910–4918. doi:10.1002/ejoc.201100615 |
| 11. | Zagidullin, A.; Ganushevich, Y.; Miluykov, V.; Sinyashin, O.; Hey-Hawkins, E. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 238–242. doi:10.1080/10426507.2012.744017 |
| 16. | Doxsee, K. M.; Wood, N. P.; Hanawalt, E. M.; Weakley, T. J. R. Heteroat. Chem. 1996, 7, 383–389. doi:10.1002/(SICI)1098-1071(199610)7:5<383::AID-HC16>3.0.CO;2-M |
© 2015 Zagidullin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)