Abstract
Herein, we present the bottom-up, mechanochemical synthesis of phosphorus-bridged heptazine-based carbon nitrides (g-h-PCN). The structure of these materials was determined through a combination of powder X-ray diffraction (PXRD), X-ray photoelectron spectroscopy (XPS), 31P magic angle spinning nuclear magnetic resonance (MAS NMR), density functional theory (DFT) and electron energy loss spectroscopy (EELS). Compared to traditional furnace-based techniques, the presented method utilizes milder conditions, as well as shorter reaction times. Both samples of g-h-PCN directly after milling and aging and after an hour of annealing at 300 °C (g-h-PCN300) show a reduction in photoluminescent recombination, as well as a nearly two-time increase in photocurrent under broad spectrum irradiation, which are appealing properties for photocatalysis.

Graphical Abstract
Introduction
The development of heteroatom-doped graphitic carbon nitrides (g-CN) has been a rapidly growing area of research since their first report towards water splitting in 2009 [1]. Since that time, the addition of elements such as boron [2], phosphorus [3-5], sulfur and oxygen [6] have shown to help minimize the bandgap of these metal-free photocatalysts, as well as improve their overall stability. Traditional routes to incorporate phosphorus have relied on high-temperature [7] or microwave [8] syntheses, and often proceed through the introduction of a phosphorus atom within the heptazine ring, which constitutes the building block of g-CN, as opposed to in a linking position. Computational studies by Hartley and Martsinovich have investigated the influence of various linkers, including phosphorus atoms, on both the structure and optical behavior of heptazine-based graphitic carbon nitrides [3]. Yet, examples of carbon nitride materials linked together via phosphorus atoms are limited, likely due to challenges in controlling the insertion of phosphorus atoms as linkers under high energy conditions. Mechanochemistry [9-12] has proven to be effective for the synthesis of a variety of polymers [13-17], nanomaterials [18-22], in crystal engineering [23-25], as well as in the synthesis of inorganic materials [26-30] and organic small molecules [31-37]. The ability to avoid bulk solvent and mild reaction conditions allowed by such techniques are beneficial not only from a green chemistry perspective [11], but they also afford conditions conducive to new reactivities and the development of novel materials [9]. Previously, we have explored the synthesis of phosphorus-bridged g-CN-type materials produced from a triazine unit and found that the resulting material featured good photochemical properties (Scheme 1) [38]. Yet, conventional g-CN materials are not based on triazine units, but rather on heptazine ones, thus featuring more open structures. In an effort to replicate a structure closer to known g-CN systems, we explored herein the use of solvent-free, room temperature mechanochemistry to access phosphorus-linked carbon nitride with repeating heptazine units, which were found to show improved photochemistry over pristine graphitic carbon nitride (g-CN). Additionally, the effect of a 1-hour annealing period at 300 °C on the overall structure and photochemical properties of the material was investigated.
Results and Discussion
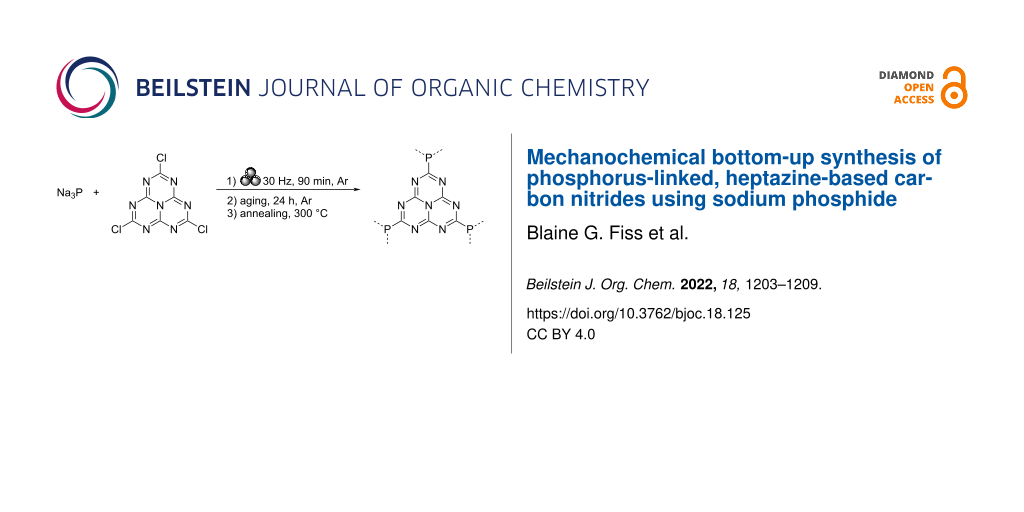
Employing a similar method to the one previously developed by our group (Scheme 1a) [38], equimolar amounts of sodium phosphide (Na3P) and trichloroheptazine were combined in a vibrational ball mill and milled at 30 Hz for 90 minutes under an argon atmosphere (Scheme 1b). As trichloroheptazine was not readily available commercially, it was synthesized from melem in three steps following a known procedure (see Supporting Information File 1 for the detailed procedure) [4]. The milled powder was then allowed to age under an argon atmosphere for 24 hours, prior to washing via centrifugation in a 3:1 by volume mixture of ethanol and deionized (DI) water. This afforded a material referred to below as g-h-PCN. Alternatively, the sample was annealed for 1 hour at 300 °C under a flow of argon gas, affording the material designated g-h-PCN300.
![[1860-5397-18-125-i1]](/bjoc/content/inline/1860-5397-18-125-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: a) Mechanochemical synthesis of g-PCN from sodium phosphide and trichlorotriazine (previous work [38]) and b) g-h-PCN from sodium phosphide and trichloroheptazine (this work).
Scheme 1: a) Mechanochemical synthesis of g-PCN from sodium phosphide and trichlorotriazine (previous work [38]) ...
Powder X-ray diffraction (PXRD)
To confirm the formation of a layered structure, powder X-ray diffraction (PXRD) was performed on g-h-PCN and g-h-PCN300 (Figure 1, green and teal). Both g-h-PCN and g-h-PCN300 were largely amorphous but showed two broad Bragg reflections at 2θ = 16° and 28°. This suggests a high thermal stability of the g-h-PCN structure, being formed during the mild milling and aging conditions, with no need for annealing.
![[1860-5397-18-125-1]](/bjoc/content/figures/1860-5397-18-125-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: PXRD patterns of g-h-PCN (green) and g-h-PCN300 (teal).
Figure 1: PXRD patterns of g-h-PCN (green) and g-h-PCN300 (teal).
X-ray photoelectron spectroscopy (XPS)
To gain insight into the atomic speciation within the structure and establish phosphorus atoms are linkers between heptazine units, X-ray photoelectron spectroscopy (XPS) was used to probe the surface. In g-h-PCN, XPS scans focused on carbon 1s showed three major peaks at 284.7, 286.4, and 288.6 eV, corresponding to C=N, C–OH and C=O signals, respectively (Figure 2a), as well as a peak centered on 292.1 eV due to charging effects [39]. The presence of C=N bonds established by XPS indicates that the heptazine structure was preserved during milling and aging with Na3P. Nitrogen 1s focused scans of g-h-PCN showed a 62% to 38% ratio of pyridinic to pyridonic-N type nitrogen environments, centered on 399.0 eV and 400.7 eV, respectively [16,40] (Figure 2b). This suggests that while the majority of the heptazine ring remained pyridinic in nature, partial reduction of the ring structure, due to the reductive nature of Na3P and mild oxidation during ambient workup following milling and aging, can also have occurred. Additionally, the phosphorus 2p signal in g-h-PCN showed the majority of phosphorus exists as a mixture of P=O and P–O species, with a major peak centered at 133.6 eV (69%, Figure 2c). These species are formed by oxidation with air and hydrolysis upon quenching in water and ethanol at the end of the aging step. g-h-PCN300 featured the same three major carbon 1s peaks at 284.8, 286.2 and 288.2 eV for C=N/C=C, C=O and C–OH species, respectively, as well as the charging peak seen in g-h-PCN. In g-h-PCN300, a reduction of the C=N ratio to 37%, reduction of C=O character from 21% to 18% compared to g-h-PCN and an increase in C–OH character from 35% to 43% suggests mild hydrolysis, likely of terminal trichloroheptazine, during the annealing step at 300 °C, even under a flow of argon gas (Figure 2d). The nitrogen 1s scans show a similar trend, with the ratio of pyridinic and pyridone nitrogen being 68% and 29%, respectively, compared to 62% and 38% in g-h-PCN (Figure 2e). Phosphorus 2p focused scans of g-h-PCN300 showed a slight reduction in P–O and P=O bond character from g-h-PCN to g-h-PCN300 down from 69% to 66% (Figure 2f).
![[1860-5397-18-125-2]](/bjoc/content/figures/1860-5397-18-125-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: XPS scans of a) C 1s, b) N 1s and c) P 2p for the pre-annealed g-h-PCN and d) C 1s, e) N 1s and f) P 2p focused scans of g-h-PCN300 after annealing at 300 °C for 1 hour.
Figure 2: XPS scans of a) C 1s, b) N 1s and c) P 2p for the pre-annealed g-h-PCN and d) C 1s, e) N 1s and f) ...
Analysis by infrared spectroscopy
The structural motifs seen through XPS were further validated by Fourier-transform infrared attenuated total reflectance (FTIR-ATR) spectroscopy. For g-h-PCN, the sharp absorption band at 800 cm−1 was indicative of the heptazine breathing mode, typically seen for nitrogen- and phosphorus-linked C3N4 materials (Supporting Information File 1, Figure S3, purple) [41]. The retention of C=N bonds was also further supported by the observation of a series of bands in the range of 1300–1800 cm−1. While the retention of the heptazine ring structure was evident by FTIR-ATR, a low-intensity additional signal at ≈950 cm−1 is also seen, indicative of the formation of P–C bonds [42], consistent with the results of XPS analysis (Supporting Information File 1, Figure S3, teal). For the g-h-PCN300 material, the overall spectrum showed similar features to that of g-h-PCN, notably retaining the sharp absorption band corresponding to the heptazine breathing mode at 800 cm−1, while also retaining the characteristic P–C vibration at 950 cm−1 (Supporting Information File 1, Figure S3, green).
STEM-EELS analysis
The composition and particle morphology were investigated further using scanning tunneling electron microscopy-electron energy loss spectroscopy (STEM-EELS). The STEM-EELS data for a g-h-PCN sample prior to annealing showed equal distribution of carbon and nitrogen with minimal phosphorus present, and particles roughly 400 nm in length (Supporting Information File 1, Figure S4a). Upon annealing at 300 °C for 1 hour under argon, the phosphorus content is shown to increase, while still remaining partially localized, with the particles retaining their size and morphology (Supporting Information File 1, Figure S4b).
31P magic angle spinning (MAS) NMR
Bulk solid-state analysis of the heptazine-based materials showed similar resonances to previous work by our group on phosphorus-linked triazine networks [38]. The 31P MAS NMR of g-h-PCN showed a broad resonance centered around −8.9 ppm, with a sharp residual phosphate resonance at 0.9 ppm (Figure 3a). NMR analysis of similar materials, by our group as well as others, suggest that the broad resonance corresponds to a largely amorphous phase with predominately phosphate and phosphite-like environments, with the broad resonance at −8.9 ppm possibly corresponding to hydrated sodium phosphate byproducts [43,44]. The NMR spectrum of the g-h-PCN300 material showed an up-field shift of all main resonances towards −14.4 ppm and −20.6 ppm (Figure 3b). As previously shown by our group [38], such a shift in main resonance positioning is indicative of the organization of the formed sheets, indicating a layered structure.
![[1860-5397-18-125-3]](/bjoc/content/figures/1860-5397-18-125-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 31P MAS NMR of a) g-h-PCN and b) g-h-PCN300. Asterisks denote spinning sidebands.
Figure 3: 31P MAS NMR of a) g-h-PCN and b) g-h-PCN300. Asterisks denote spinning sidebands.
Computational analysis
The 31P NMR chemical shifts were calculated using the plane-wave density function theory (DFT) code CASTEP v20.11 (see Supporting Information File 1 for full computational details) [45]. In the absence of an experimentally resolved crystal structure for g-h-PCN, we followed a similar methodology to our previous work [38] of substituting bridging nitrogen atoms for phosphorus in previously reported heptazine-based graphitic carbon nitrides. We adapted the ab initio predicted structures for a network of corrugated sheets [46] (Figure 4a and 4b) and planar sheets (Figure 4c) [47]. Additionally, we modelled a chlorine terminated monomeric unit based on an experimentally resolved, nitrogen-bridged, paddlewheel structure (Figure 4d) [48]. Calculations resulted in a single chemical environment for the phosphorus atoms in the three model structures. The corrugated and planar network structures have calculated 31P chemical shifts at 37.7 ppm (P1) and 23.3 ppm (P2), respectively, while that of the paddlewheel monomer is at 11.2 ppm (P3). The calculated shifts demonstrate phosphorus environments in the mechanochemically synthesized material are distinct to those calculated in ab initio predicted models of the pure reaction product. The variations may be ascribed to the oxygen content found by XPS (Figure 2) or that the mechanochemically synthesized material exists in a different spatial configuration to those previously predicted [46,47].
![[1860-5397-18-125-4]](/bjoc/content/figures/1860-5397-18-125-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Calculated structures for a) corrugated (edge facing), b) corrugated (single layer), c) layered g-h-PCN and d) phosphine paddlewheel.
Figure 4: Calculated structures for a) corrugated (edge facing), b) corrugated (single layer), c) layered g-h...
Thermal stability
Prior to annealing, g-h-PCN was found to show thermal stability upwards of 200 °C through TGA in nitrogen and air (Supporting Information File 1, Figures S6 and S7). The initial loss seen for both samples is attributed to surface-bound water and carbon dioxide. The g-h-PCN300 retains more mass to upwards of 400 °C in both air and nitrogen, however, at higher temperature (>500 °C), g-h-PCN shows an additional loss of ≈5 wt % compared to g-h-PCN. Overall, under both air and nitrogen, the g-h-PCN retains between 25–35% of its relative mass, up to 800 °C.
Photochemical activity
The photochemical behavior of both g-h-PCN (Supporting Information File 1, Figure S5a, blue trace) and g-h-PCN300 (Figure S5a, green) was investigated by diffuse reflectance spectroscopy (DRS), and compared to that of a pure g-CN sample made by annealing melamine in a loosely capped alumina crucible at 550 °C for 4 hours, with a ramp rate of 5 °C min−1 [49]. The g-CN material was found to exhibit an absorption edge at ≈425 nm (Figure S5a, purple), typical for polymerized and graphitic heptazine materials [50,51]. Both phosphorus-containing structures featured broadened absorption ranges compared to g-CN, with g-h-PCN showing a red-shifted maximum at ≈572 nm (Figure S5a, blue) and g-h-PCN300 (Figure S5a, teal) showing a similar, also red-shifted maximum at 525 nm. Additionally, photoluminescence (PL) measurements showed an initially reduced absorption intensity for g-h-PCN (Figure S5b, blue) compared to that of g-CN (Figure S5b, purple) with further reduction notable in absorption for g-h-PCN300 (Figure S5b, teal). This reduction in photoluminescence has previously been reported for phosphorus-doped carbons and carbon nitrides [52], as the addition of Lewis basic heteroatoms improves the stability of excitons, slowing the rate of recombination. Time-resolved lifetimes showed a marked increase upon replacement of the nitrogen linker for phosphorus. Nitrogen-linked g-CN showed exciton lifetimes of 4.2 µs, while the introduction of a phosphorus linker in g-h-PCN increases the lifetime to 67 µs, with g-h-PCN300 showing lifetimes of 42 µs. We have also observed a similar effect in triazine-based phosphorus-linked graphitic CN structures, with lifetimes of 4.7 and 39 µs seen for g-PCN and g-PCN300, respectively [38].
Improved charge transfer was further confirmed through photocurrent and Nyquist measurements, comparing to pristine g-CN. Photocurrent measurements showed an initial decrease from 10 to ≈5 µA for g-h-PCN (Supporting Information File 1, Figure S4c, blue) compared to pristine g-CN (Figure S4c, purple) respectively after 250 s. The g-h-PCN300 material showed increased photocurrent behavior to both g-CN and g-h-PCN, with photocurrent values of ≈16 µA (Figure S4c, teal), further demonstrating the benefit of both the presence of phosphorus linkages, as well as thermal annealing for the photoactivity. Finally, Nyquist plots (see Supporting Information File 1, Table S1 for details) of g-h-PCN (Figure S4d, blue) and g-h-PCN300 (Figure S4d, teal) showed lower resistivity compared to pure g-CN (Figure S4d, purple), further supporting the idea that the g-h-PCN series enables better charge mobility due to phosphorus linkages.
Conclusion
Mechanochemistry provided modular, room temperature access to phosphorus-linked carbon nitrides based on heptazine units, through the combination of sodium phosphide and trichloroheptazine. A combination of experimental PXRD, XPS, MAS NMR, as well as theoretical (DFT) approaches confirmed the formation of P–C linkages between repeating heptazine units in the mechanochemically prepared material, with the retention of the heptazine subunits. The introduction of phosphorus linkages reduced photoluminescent recombination and improved exciton lifetimes, when compared to nitrogen-linked g-CN. Overall, this supports future investigations of the room-temperature mechanochemical synthesis of heteroatom containing carbons as well as the benefit of pairing DFT calculations to experimental, structural studies.
Supporting Information
| Supporting Information File 1: General methods and materials as well as additional spectra. | ||
| Format: PDF | Size: 887.2 KB | Download |
Acknowledgements
We thank the McGill Institute for Advanced Materials, the Facility for Electron Microscopy Research (FEMR), the MC2 facility, McGill University Institute for Advanced Material (MIAM) for the use of their microscopy and spectroscopy equipment.
Funding
We are grateful for the support of the Natural Science and Engineering Research Council of Canada (NSERC) Discovery Grant and Discovery Accelerator Supplement, the Canada Foundation for Innovation (CFI), the McGill Sustainability Systems Initiative (MSSI), the Fonds de Recherche du Québec – Nature et Technologies (FRQNT) – Centre for Green Chemistry and Catalysis (CGCC), the Walter C. Sumner Memorial Fellowship (B. G. F.), McGill University and Université de Laval. This research was enabled in part by support provided by Calcul Québec (https://www.calculquebec.ca) and Compute Canada (https://www.computecanada.ca).
References
-
Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J. M.; Domen, K.; Antonietti, M. Nat. Mater. 2009, 8, 76–80. doi:10.1038/nmat2317
Return to citation in text: [1] -
Guo, Y.; Wang, R.; Wang, P.; Rao, L.; Wang, C. ACS Sustainable Chem. Eng. 2019, 7, 5727–5741. doi:10.1021/acssuschemeng.8b05150
Return to citation in text: [1] -
Hartley, G. O.; Martsinovich, N. Faraday Discuss. 2021, 227, 341–358. doi:10.1039/c9fd00147f
Return to citation in text: [1] [2] -
Kroke, E.; Schwarz, M.; Horath-Bordon, E.; Kroll, P.; Noll, B.; Norman, A. D. New J. Chem. 2002, 26, 508–512. doi:10.1039/b111062b
Return to citation in text: [1] [2] -
Zhang, Y.; Mori, T.; Ye, J.; Antonietti, M. J. Am. Chem. Soc. 2010, 132, 6294–6295. doi:10.1021/ja101749y
Return to citation in text: [1] -
Liu, M.; Yang, K.; Li, Z.; Fan, E.; Fu, H.; Zhang, L.; Zhang, Y.; Zheng, Z. Chem. Commun. 2022, 58, 92–95. doi:10.1039/d1cc05619k
Return to citation in text: [1] -
Wang, Q.; Gou, H.; Zhu, L.; Huang, H.-T.; Biswas, A.; Chaloux, B. L.; Epshteyn, A.; Yesinowski, J. P.; Liu, Z.; Cody, G.; Ma, M.; Zhao, Z.; Fei, Y.; Prescher, C.; Greenberg, E.; Prakapenka, V. B.; Strobel, T. A. ACS Mater. Lett. 2019, 1, 14–19. doi:10.1021/acsmaterialslett.9b00010
Return to citation in text: [1] -
Zou, J.; Yu, Y.; Qiao, K.; Wu, S.; Yan, W.; Cheng, S.; Jiang, N.; Wang, J. J. Mater. Sci. 2020, 55, 13618–13633. doi:10.1007/s10853-020-04862-6
Return to citation in text: [1] -
Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277
Return to citation in text: [1] [2] -
Friščić, T.; Mottillo, C.; Titi, H. M. Angew. Chem., Int. Ed. 2020, 59, 1018–1029. doi:10.1002/anie.201906755
Return to citation in text: [1] -
Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478
Return to citation in text: [1] [2] -
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a
Return to citation in text: [1] -
Ohn, N.; Kim, J. G. ACS Macro Lett. 2018, 7, 561–565. doi:10.1021/acsmacrolett.8b00171
Return to citation in text: [1] -
Troschke, E.; Grätz, S.; Lübken, T.; Borchardt, L. Angew. Chem., Int. Ed. 2017, 56, 6859–6863. doi:10.1002/anie.201702303
Return to citation in text: [1] -
Casco, M. E.; Kirchhoff, S.; Leistenschneider, D.; Rauche, M.; Brunner, E.; Borchardt, L. Nanoscale 2019, 11, 4712–4718. doi:10.1039/c9nr01019j
Return to citation in text: [1] -
Schneidermann, C.; Kensy, C.; Otto, P.; Oswald, S.; Giebeler, L.; Leistenschneider, D.; Grätz, S.; Dörfler, S.; Kaskel, S.; Borchardt, L. ChemSusChem 2019, 12, 310–319. doi:10.1002/cssc.201801997
Return to citation in text: [1] [2] -
Fiss, B. G.; Hatherly, L.; Stein, R. S.; Friščić, T.; Moores, A. ACS Sustainable Chem. Eng. 2019, 7, 7951–7959. doi:10.1021/acssuschemeng.9b00764
Return to citation in text: [1] -
Moores, A. Curr. Opin. Green Sustainable Chem. 2018, 12, 33–37. doi:10.1016/j.cogsc.2018.05.004
Return to citation in text: [1] -
Rak, M. J.; Saadé, N. K.; Friščić, T.; Moores, A. Green Chem. 2014, 16, 86–89. doi:10.1039/c3gc41827h
Return to citation in text: [1] -
Malca, M. Y.; Bao, H.; Bastaille, T.; Saadé, N. K.; Kinsella, J. M.; Friščić, T.; Moores, A. Chem. Mater. 2017, 29, 7766–7773. doi:10.1021/acs.chemmater.7b02134
Return to citation in text: [1] -
Xu, C.; De, S.; Balu, A. M.; Ojeda, M.; Luque, R. Chem. Commun. 2015, 51, 6698–6713. doi:10.1039/c4cc09876e
Return to citation in text: [1] -
Fiss, B. G.; Vu, N.-N.; Douglas, G.; Do, T.-O.; Friščić, T.; Moores, A. ACS Sustainable Chem. Eng. 2020, 8, 12014–12024. doi:10.1021/acssuschemeng.0c02762
Return to citation in text: [1] -
Braga, D.; Dichiarante, E.; Grepioni, F.; Lampronti, G. I.; Maini, L.; Mazzeo, P. P.; D'Agostino, S. Mechanical Preparation of Crystalline Materials. An Oxymoron?. In Supramolecular Chemistry: From Molecules to Nanomaterials; Steed, J. W.; Gale, P. A., Eds.; Supramolecular Materials Chemistry, Vol. 6; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp 2993–3007. doi:10.1002/9780470661345.smc115
Return to citation in text: [1] -
Toda, F.; Tanaka, K.; Sekikawa, A. J. Chem. Soc., Chem. Commun. 1987, 279–280. doi:10.1039/c39870000279
Return to citation in text: [1] -
Friščić, T.; Trask, A. V.; Jones, W.; Motherwell, W. D. S. Angew. Chem., Int. Ed. 2006, 45, 7546–7550. doi:10.1002/anie.200603235
Return to citation in text: [1] -
Braga, D.; Giaffreda, S. L.; Grepioni, F.; Pettersen, A.; Maini, L.; Curzi, M.; Polito, M. Dalton Trans. 2006, 1249–1263. doi:10.1039/b516165g
Return to citation in text: [1] -
Boldyrev, V. V.; Avvakumov, E. G. Russ. Chem. Rev. 1971, 40, 847–859. doi:10.1070/rc1971v040n10abeh001977
Return to citation in text: [1] -
Beillard, A.; Bantreil, X.; Métro, T.-X.; Martinez, J.; Lamaty, F. Chem. Rev. 2019, 119, 7529–7609. doi:10.1021/acs.chemrev.8b00479
Return to citation in text: [1] -
Rightmire, N. R.; Hanusa, T. P. Dalton Trans. 2016, 45, 2352–2362. doi:10.1039/c5dt03866a
Return to citation in text: [1] -
Tan, D.; García, F. Chem. Soc. Rev. 2019, 48, 2274–2292. doi:10.1039/c7cs00813a
Return to citation in text: [1] -
Tanaka, K. Solvent-free organic synthesis; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/3527601821
Return to citation in text: [1] -
Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h
Return to citation in text: [1] -
Tan, D.; Friščić, T. Eur. J. Org. Chem. 2018, 18–33. doi:10.1002/ejoc.201700961
Return to citation in text: [1] -
Wang, G.-W. Chin. J. Chem. 2021, 39, 1797–1803. doi:10.1002/cjoc.202100085
Return to citation in text: [1] -
Zhu, S.-E.; Li, F.; Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7535–7570. doi:10.1039/c3cs35494f
Return to citation in text: [1] -
Mack, J.; Fulmer, D.; Stofel, S.; Santos, N. Green Chem. 2007, 9, 1041–1043. doi:10.1039/b706167f
Return to citation in text: [1] -
Fiss, B. G.; Richard, A. J.; Friščić, T.; Moores, A. Can. J. Chem. 2021, 99, 93–112. doi:10.1139/cjc-2020-0408
Return to citation in text: [1] -
Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Ronning, C.; Feldermann, H.; Merk, R.; Hofsäss, H.; Reinke, P.; Thiele, J.-U. Phys. Rev. B 1998, 58, 2207–2215. doi:10.1103/physrevb.58.2207
Return to citation in text: [1] -
Wang, J.; Senkovska, I.; Oschatz, M.; Lohe, M. R.; Borchardt, L.; Heerwig, A.; Liu, Q.; Kaskel, S. J. Mater. Chem. A 2013, 1, 10951–10961. doi:10.1039/c3ta11995e
Return to citation in text: [1] -
Bojdys, M. J.; Müller, J.-O.; Antonietti, M.; Thomas, A. Chem. – Eur. J. 2008, 14, 8177–8182. doi:10.1002/chem.200800190
Return to citation in text: [1] -
Kumar, A.; Kumar, P.; Joshi, C.; Manchanda, M.; Boukherroub, R.; Jain, S. L. Nanomaterials 2016, 6, 59. doi:10.3390/nano6040059
Return to citation in text: [1] -
Turner, G. L.; Smith, K. A.; Kirkpatrick, R. J.; Oldfieldt, E. J. Magn. Reson. (1969-1992) 1986, 70, 408–415. doi:10.1016/0022-2364(86)90129-0
Return to citation in text: [1] -
MacKenzie, K. J.; Smith, M. E. NMR of Other Commonly Studied Nuclei. Multinuclear solid-state nuclear magnetic resonance of inorganic materials; Elsevier: New York, NY, USA, 2002. doi:10.1016/s1470-1804(02)80008-6
Return to citation in text: [1] -
Clark, S. J.; Segall, M. D.; Pickard, C. J.; Hasnip, P. J.; Probert, M. I. J.; Refson, K.; Payne, M. C. Z. Kristallogr. 2005, 220, 567–570. doi:10.1524/zkri.220.5.567.65075
Return to citation in text: [1] -
Gracia, J.; Kroll, P. J. Mater. Chem. 2009, 19, 3013–3019. doi:10.1039/b821568e
Return to citation in text: [1] [2] -
Wang, J.; Hao, D.; Ye, J.; Umezawa, N. Chem. Mater. 2017, 29, 2694–2707. doi:10.1021/acs.chemmater.6b02969
Return to citation in text: [1] [2] -
Tragl, S.; Gibson, K.; Glaser, J.; Heydenrych, G.; Frenking, G.; Duppel, V.; Simon, A.; Meyer, H.-J. Z. Anorg. Allg. Chem. 2008, 634, 2754–2760. doi:10.1002/zaac.200800092
Return to citation in text: [1] -
Yan, S. C.; Li, Z. S.; Zou, Z. G. Langmuir 2009, 25, 10397–10401. doi:10.1021/la900923z
Return to citation in text: [1] -
Lin, L.; Ou, H.; Zhang, Y.; Wang, X. ACS Catal. 2016, 6, 3921–3931. doi:10.1021/acscatal.6b00922
Return to citation in text: [1] -
Wang, Y.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2012, 51, 68–89. doi:10.1002/anie.201101182
Return to citation in text: [1] -
Ma, X.; Lv, Y.; Xu, J.; Liu, Y.; Zhang, R.; Zhu, Y. J. Phys. Chem. C 2012, 116, 23485–23493. doi:10.1021/jp308334x
Return to citation in text: [1]
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 46. | Gracia, J.; Kroll, P. J. Mater. Chem. 2009, 19, 3013–3019. doi:10.1039/b821568e |
| 47. | Wang, J.; Hao, D.; Ye, J.; Umezawa, N. Chem. Mater. 2017, 29, 2694–2707. doi:10.1021/acs.chemmater.6b02969 |
| 1. | Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J. M.; Domen, K.; Antonietti, M. Nat. Mater. 2009, 8, 76–80. doi:10.1038/nmat2317 |
| 7. | Wang, Q.; Gou, H.; Zhu, L.; Huang, H.-T.; Biswas, A.; Chaloux, B. L.; Epshteyn, A.; Yesinowski, J. P.; Liu, Z.; Cody, G.; Ma, M.; Zhao, Z.; Fei, Y.; Prescher, C.; Greenberg, E.; Prakapenka, V. B.; Strobel, T. A. ACS Mater. Lett. 2019, 1, 14–19. doi:10.1021/acsmaterialslett.9b00010 |
| 9. | Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277 |
| 6. | Liu, M.; Yang, K.; Li, Z.; Fan, E.; Fu, H.; Zhang, L.; Zhang, Y.; Zheng, Z. Chem. Commun. 2022, 58, 92–95. doi:10.1039/d1cc05619k |
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 3. | Hartley, G. O.; Martsinovich, N. Faraday Discuss. 2021, 227, 341–358. doi:10.1039/c9fd00147f |
| 4. | Kroke, E.; Schwarz, M.; Horath-Bordon, E.; Kroll, P.; Noll, B.; Norman, A. D. New J. Chem. 2002, 26, 508–512. doi:10.1039/b111062b |
| 5. | Zhang, Y.; Mori, T.; Ye, J.; Antonietti, M. J. Am. Chem. Soc. 2010, 132, 6294–6295. doi:10.1021/ja101749y |
| 31. | Tanaka, K. Solvent-free organic synthesis; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/3527601821 |
| 32. | Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h |
| 33. | Tan, D.; Friščić, T. Eur. J. Org. Chem. 2018, 18–33. doi:10.1002/ejoc.201700961 |
| 34. | Wang, G.-W. Chin. J. Chem. 2021, 39, 1797–1803. doi:10.1002/cjoc.202100085 |
| 35. | Zhu, S.-E.; Li, F.; Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7535–7570. doi:10.1039/c3cs35494f |
| 36. | Mack, J.; Fulmer, D.; Stofel, S.; Santos, N. Green Chem. 2007, 9, 1041–1043. doi:10.1039/b706167f |
| 37. | Fiss, B. G.; Richard, A. J.; Friščić, T.; Moores, A. Can. J. Chem. 2021, 99, 93–112. doi:10.1139/cjc-2020-0408 |
| 52. | Ma, X.; Lv, Y.; Xu, J.; Liu, Y.; Zhang, R.; Zhu, Y. J. Phys. Chem. C 2012, 116, 23485–23493. doi:10.1021/jp308334x |
| 2. | Guo, Y.; Wang, R.; Wang, P.; Rao, L.; Wang, C. ACS Sustainable Chem. Eng. 2019, 7, 5727–5741. doi:10.1021/acssuschemeng.8b05150 |
| 11. | Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478 |
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 13. | Ohn, N.; Kim, J. G. ACS Macro Lett. 2018, 7, 561–565. doi:10.1021/acsmacrolett.8b00171 |
| 14. | Troschke, E.; Grätz, S.; Lübken, T.; Borchardt, L. Angew. Chem., Int. Ed. 2017, 56, 6859–6863. doi:10.1002/anie.201702303 |
| 15. | Casco, M. E.; Kirchhoff, S.; Leistenschneider, D.; Rauche, M.; Brunner, E.; Borchardt, L. Nanoscale 2019, 11, 4712–4718. doi:10.1039/c9nr01019j |
| 16. | Schneidermann, C.; Kensy, C.; Otto, P.; Oswald, S.; Giebeler, L.; Leistenschneider, D.; Grätz, S.; Dörfler, S.; Kaskel, S.; Borchardt, L. ChemSusChem 2019, 12, 310–319. doi:10.1002/cssc.201801997 |
| 17. | Fiss, B. G.; Hatherly, L.; Stein, R. S.; Friščić, T.; Moores, A. ACS Sustainable Chem. Eng. 2019, 7, 7951–7959. doi:10.1021/acssuschemeng.9b00764 |
| 23. | Braga, D.; Dichiarante, E.; Grepioni, F.; Lampronti, G. I.; Maini, L.; Mazzeo, P. P.; D'Agostino, S. Mechanical Preparation of Crystalline Materials. An Oxymoron?. In Supramolecular Chemistry: From Molecules to Nanomaterials; Steed, J. W.; Gale, P. A., Eds.; Supramolecular Materials Chemistry, Vol. 6; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp 2993–3007. doi:10.1002/9780470661345.smc115 |
| 24. | Toda, F.; Tanaka, K.; Sekikawa, A. J. Chem. Soc., Chem. Commun. 1987, 279–280. doi:10.1039/c39870000279 |
| 25. | Friščić, T.; Trask, A. V.; Jones, W.; Motherwell, W. D. S. Angew. Chem., Int. Ed. 2006, 45, 7546–7550. doi:10.1002/anie.200603235 |
| 49. | Yan, S. C.; Li, Z. S.; Zou, Z. G. Langmuir 2009, 25, 10397–10401. doi:10.1021/la900923z |
| 9. | Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277 |
| 10. | Friščić, T.; Mottillo, C.; Titi, H. M. Angew. Chem., Int. Ed. 2020, 59, 1018–1029. doi:10.1002/anie.201906755 |
| 11. | Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478 |
| 12. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a |
| 26. | Braga, D.; Giaffreda, S. L.; Grepioni, F.; Pettersen, A.; Maini, L.; Curzi, M.; Polito, M. Dalton Trans. 2006, 1249–1263. doi:10.1039/b516165g |
| 27. | Boldyrev, V. V.; Avvakumov, E. G. Russ. Chem. Rev. 1971, 40, 847–859. doi:10.1070/rc1971v040n10abeh001977 |
| 28. | Beillard, A.; Bantreil, X.; Métro, T.-X.; Martinez, J.; Lamaty, F. Chem. Rev. 2019, 119, 7529–7609. doi:10.1021/acs.chemrev.8b00479 |
| 29. | Rightmire, N. R.; Hanusa, T. P. Dalton Trans. 2016, 45, 2352–2362. doi:10.1039/c5dt03866a |
| 30. | Tan, D.; García, F. Chem. Soc. Rev. 2019, 48, 2274–2292. doi:10.1039/c7cs00813a |
| 50. | Lin, L.; Ou, H.; Zhang, Y.; Wang, X. ACS Catal. 2016, 6, 3921–3931. doi:10.1021/acscatal.6b00922 |
| 51. | Wang, Y.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2012, 51, 68–89. doi:10.1002/anie.201101182 |
| 3. | Hartley, G. O.; Martsinovich, N. Faraday Discuss. 2021, 227, 341–358. doi:10.1039/c9fd00147f |
| 48. | Tragl, S.; Gibson, K.; Glaser, J.; Heydenrych, G.; Frenking, G.; Duppel, V.; Simon, A.; Meyer, H.-J. Z. Anorg. Allg. Chem. 2008, 634, 2754–2760. doi:10.1002/zaac.200800092 |
| 8. | Zou, J.; Yu, Y.; Qiao, K.; Wu, S.; Yan, W.; Cheng, S.; Jiang, N.; Wang, J. J. Mater. Sci. 2020, 55, 13618–13633. doi:10.1007/s10853-020-04862-6 |
| 18. | Moores, A. Curr. Opin. Green Sustainable Chem. 2018, 12, 33–37. doi:10.1016/j.cogsc.2018.05.004 |
| 19. | Rak, M. J.; Saadé, N. K.; Friščić, T.; Moores, A. Green Chem. 2014, 16, 86–89. doi:10.1039/c3gc41827h |
| 20. | Malca, M. Y.; Bao, H.; Bastaille, T.; Saadé, N. K.; Kinsella, J. M.; Friščić, T.; Moores, A. Chem. Mater. 2017, 29, 7766–7773. doi:10.1021/acs.chemmater.7b02134 |
| 21. | Xu, C.; De, S.; Balu, A. M.; Ojeda, M.; Luque, R. Chem. Commun. 2015, 51, 6698–6713. doi:10.1039/c4cc09876e |
| 22. | Fiss, B. G.; Vu, N.-N.; Douglas, G.; Do, T.-O.; Friščić, T.; Moores, A. ACS Sustainable Chem. Eng. 2020, 8, 12014–12024. doi:10.1021/acssuschemeng.0c02762 |
| 46. | Gracia, J.; Kroll, P. J. Mater. Chem. 2009, 19, 3013–3019. doi:10.1039/b821568e |
| 47. | Wang, J.; Hao, D.; Ye, J.; Umezawa, N. Chem. Mater. 2017, 29, 2694–2707. doi:10.1021/acs.chemmater.6b02969 |
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 4. | Kroke, E.; Schwarz, M.; Horath-Bordon, E.; Kroll, P.; Noll, B.; Norman, A. D. New J. Chem. 2002, 26, 508–512. doi:10.1039/b111062b |
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 45. | Clark, S. J.; Segall, M. D.; Pickard, C. J.; Hasnip, P. J.; Probert, M. I. J.; Refson, K.; Payne, M. C. Z. Kristallogr. 2005, 220, 567–570. doi:10.1524/zkri.220.5.567.65075 |
| 38. | Fiss, B.; Douglas, G.; Ferguson, M.; Becerra, J.; Valdez, J.; Do, T.-O.; Friščić, T.; Moores, A. ChemRxiv 2022. doi:10.26434/chemrxiv-2022-15cwv-v2 |
| 43. | Turner, G. L.; Smith, K. A.; Kirkpatrick, R. J.; Oldfieldt, E. J. Magn. Reson. (1969-1992) 1986, 70, 408–415. doi:10.1016/0022-2364(86)90129-0 |
| 44. | MacKenzie, K. J.; Smith, M. E. NMR of Other Commonly Studied Nuclei. Multinuclear solid-state nuclear magnetic resonance of inorganic materials; Elsevier: New York, NY, USA, 2002. doi:10.1016/s1470-1804(02)80008-6 |
| 41. | Bojdys, M. J.; Müller, J.-O.; Antonietti, M.; Thomas, A. Chem. – Eur. J. 2008, 14, 8177–8182. doi:10.1002/chem.200800190 |
| 42. | Kumar, A.; Kumar, P.; Joshi, C.; Manchanda, M.; Boukherroub, R.; Jain, S. L. Nanomaterials 2016, 6, 59. doi:10.3390/nano6040059 |
| 39. | Ronning, C.; Feldermann, H.; Merk, R.; Hofsäss, H.; Reinke, P.; Thiele, J.-U. Phys. Rev. B 1998, 58, 2207–2215. doi:10.1103/physrevb.58.2207 |
| 16. | Schneidermann, C.; Kensy, C.; Otto, P.; Oswald, S.; Giebeler, L.; Leistenschneider, D.; Grätz, S.; Dörfler, S.; Kaskel, S.; Borchardt, L. ChemSusChem 2019, 12, 310–319. doi:10.1002/cssc.201801997 |
| 40. | Wang, J.; Senkovska, I.; Oschatz, M.; Lohe, M. R.; Borchardt, L.; Heerwig, A.; Liu, Q.; Kaskel, S. J. Mater. Chem. A 2013, 1, 10951–10961. doi:10.1039/c3ta11995e |
© 2022 Fiss et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.