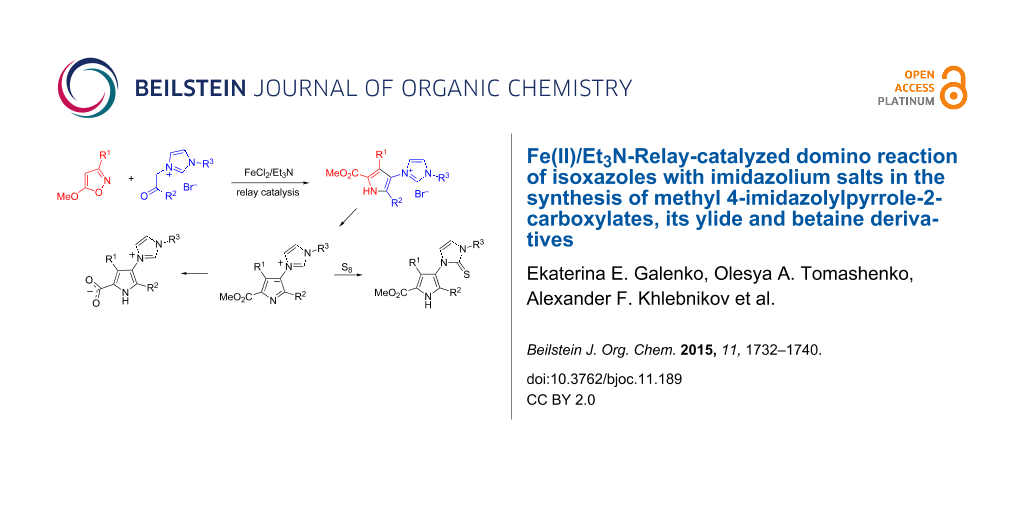
Abstract
A simple approach was developed for the synthesis of methyl 4-imidazolylpyrrole-2-carboxylates from easily available compounds, 5-methoxyisoxazoles and phenacylimidazolium salts under hybrid Fe(II)/Et3N relay catalysis. The products were easily transformed into the corresponding 3-(5-methoxycarbonyl-1H-imidazol-3-ium-3-yl)pyrrol-1-ides, which in turn can be hydrolyzed under basic conditions into the corresponding betaines. A carbene tautomeric form of the 4-methoxycarbonyl-substituted imidazolylpyrrolides was trapped by reaction with sulfur affording the corresponding imidazolethiones under very mild conditions.

Graphical Abstract
Introduction
Pyrrole-2-carboxyate and imidazole units are present in bioactive pyrrole-imidazole alkaloids and pyrrole-imidazole polyamides [1-5]. Derivatives of 4-imidazolylpyrrole-2-carboxylic acid are much less known, though some of these compounds showed various bioactivities [6-9] and were patented as inhibitors of c-Met protein kinase [8] and as anti-inflammatory agents [9]. Additionally, 5-alkoxycarbonylpyrrol-3-ylimidazolium salts 1 attracted our attention as the potential precursors of ylides 2, which in principle could be in equilibrium with N-heterocyclic carbenes (NHC) 3. Furthermore, hydrolysis of 2 could provide an easy access to unknown carboxy-substituted ylides 4, and then could potentially be in the equilibrium with N-heterocyclic carbenes 5 and betaine 6 (Scheme 1). Interplay between N-heterocyclic carbenes, heterocyclic betaines and ylides is currently intensively investigated as a promising route for tuning NHC for specific use. This topic was extensively reviewed [10-14] and many papers were published recently [15-22]. The 4–6 triads could particularly be interesting as new ligands for the preparation of mixed complexes: a chelate complex with the carboxypyrrole part and a monodentate NHC complex. Not so much is known about metal chelate complexes of pyrrole-2-carboxylic acid [23-25]. In some cases these complexes were prepared via dehydration of the corresponding proline complexes [25]. Substituted pyrrole-2-carboxylic acids as ligands of complexes are seldom used and are only exemplified by complexes of indole-2-carboxylic acid [26,27] and coenzyme pyrroloquinoline quinone [28].
![[1860-5397-11-189-i1]](/bjoc/content/inline/1860-5397-11-189-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The formation and possible tautomeric equilibria of 2-methoxycarbonyl- and 2-carboxy-3-(1H-imidazol-3-ium-3-yl)pyrrol-1-ides 2 and 4.
Scheme 1: The formation and possible tautomeric equilibria of 2-methoxycarbonyl- and 2-carboxy-3-(1H-imidazol...
The synthesis of 4-imidazolylpyrrole-2-carboxylic acid derivatives usually involves the corresponding pyrrole with a functional group allowing the formation of a imidazole ring [8,9]. Recently we developed a new approach to 3-(1H-pyrrol-3-yl)-1H-imidazoles based on the formation of a pyrrole ring via the reaction of 2H-azirines with 1-alkyl-3-phenacyl-1H-imidazolium bromides [29], in which one example of the synthesis of ethyl 4-imidazolylpyrrole-2-carboxylate from ethyl 3-methyl-2H-azirine-2-carboxylate was described. Earlier it was found that alkyl 2H-azirine-2-carboxylates can be prepared by isomerization of 5-alkoxyisoxazoles under Fe(II)-salt catalysis [30]. Quite recently this isomerization has been used for the preparation of substituted pyrrole-2-carboxylic acid derivatives by the domino reaction of 3-aryl-5-methoxyisoxazoles with 1,3-dicarbonyl compounds under relay catalysis [31]. Taking into account the facts discussed above, we envisioned that the synthesis of 5-alkoxycarbonylpyrrol-3-ylimidazolium salts 1 could be carried out starting from easily available 5-alkoxyisoxazoles 7 [32,33] and 1-alkyl-3-phenacyl-1H-imidazolium bromides 9 according to Scheme 2, whereby excluding the isolation of often unstable 2H-azirines [34].
![[1860-5397-11-189-i2]](/bjoc/content/inline/1860-5397-11-189-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: FeCl2/Et3N-catalyzed domino sequence leading to 5-alkoxycarbonylpyrrol-3-ylimidazolium salts 1.
Scheme 2: FeCl2/Et3N-catalyzed domino sequence leading to 5-alkoxycarbonylpyrrol-3-ylimidazolium salts 1.
Results and Discussion
The synthetic scheme (Scheme 2) implies an implementation of all stages (1: generation of azirine 8 from isoxazole 7 under FeCl2 catalysis; 2: formation of phenacylimidazolium ylide 10 induced by Et3N; 3: activation of azirine 8 with Et3HN+Br−; 4: reaction of the activated azirine 11 with the imidazolium ylide 10) as a domino reaction under relay catalysis [35,36]. A simple procedure, consisting of stirring a mixture of isoxazole 7, phenacylimidazolium salt 9, FeCl2·4H2O and Et3N in MeCN for 6–7 h at 45 °C, gave 5-alkoxycarbonylpyrrol-3-ylimidazolium bromides 1 in reasonable yields (Table 1). All new compounds were characterized by 1H and 13C NMR, IR spectroscopy, and mass spectrometry.
Table 1: The synthesis of 5-alkoxycarbonylpyrrol-3-ylimidazolium salts 1 by the domino reaction of 5-methoxyisoxazoles 7 and phenacylimidazolium bromides 9 under FeCl2·4H2O/Et3N catalysis.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-189-i5.svg?max-width=637&scale=1.0)
|
||||||
| entry | R1 | R2 | R3 | 7 + 9 | 1 | yield, % |
|---|---|---|---|---|---|---|
| 1 | Ph | Ph | Me | 7a + 9a | 1a | 54 |
| 2 | Ph | 4-ClC6H4 | Me | 7a + 9b | 1b | 54 |
| 3 | Ph | 4-NO2C6H4 | Me | 7a + 9c | 1c | 63 |
| 4 | 4-BrC6H4 | 3-BrC6H4 | Me | 7b + 9d | 1d | 66 |
| 5 | Ph | Ph | Ph | 7a + 9e | 1e | 68 |
| 6 | Ph | 4-MeOC6H4 | Ph | 7a + 9f | 1f | 71 |
| 7 | Ph | 4-BrC6H4 | Ph | 7a + 9g | 1g | 57 |
| 8 | Me | Ph | Ph | 7c + 9e | 1h | 54 |
| 9 | Me | 4-MeOC6H4 | Ph | 7c + 9f | 1i | 59 |
| 10 | Ph | Ph | Bn | 7a + 9h | 1j | 51 |
| 11 | Ph | 4-MeOC6H4 | Bn | 7a + 9i | 1k | 51 |
| 12 | Ph | 4-FC6H4 | Bn | 7a + 9j | 1l | 55 |
| 13 | Ph | 4-ClC6H4 | Bn | 7a + 9k | 1m | 72 |
| 14 | Me | Ph | Bn | 7c + 9h | 1n, | 47 |
| 15 | Me | 4-ClC6H4 | Bn | 7c + 9k | 1o, | 69 |
1-Benzyl-3-(1H-pyrrol-3-yl)-1H-imidazol-3-ium bromides 1 can be easily debenzylated on Pd/C, with ammonium formate as a source of hydrogen, to give the corresponding methyl 4-(1H-imidazol-1-yl)-1H-pyrrole-2-carboxylates 12 in high yields (Scheme 3).
![[1860-5397-11-189-i3]](/bjoc/content/inline/1860-5397-11-189-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The synthesis of methyl 4-(1H-imidazol-1-yl)-1H-pyrrole-2-carboxylates 12.
Scheme 3: The synthesis of methyl 4-(1H-imidazol-1-yl)-1H-pyrrole-2-carboxylates 12.
The reaction of aq KOH with imidazolium bromides 1 at room temperature afforded the corresponding stable ylides 2 (Table 2) in high yields without hydrolyzing the ester group. Ylides 2 can also be debenzylated, affording the corresponding pyrrolyl imidazoles 12. Thus, ylide 2h was debenzylated on Pd/C with hydrogen to produce methyl 5-(4-fluorophenyl)-4-(1H-imidazol-1-yl)-3-phenyl-1H-pyrrole-2-carboxylate (12d) in quantitive yield.
Table 2: The preparation of ylides 2 and imidazolethiones 13.
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-189-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | R1 | R2 | R3 | 1 | 2, yield, % | 13, yield, % |
|---|---|---|---|---|---|---|
| 1 | Ph | Ph | Me | 1a | 2a, 71 | 13a, 80 |
| 2 | Ph | 4-ClC6H4 | Me | 1b | 2b, 86 | 13b, 81 |
| 3 | Ph | 4-NO2C6H4 | Me | 1c | 2c, 98 | 13c, 93 |
| 4 | 4-BrC6H4 | 3-BrC6H4 | Me | 1d | 2d, 80 | 13d, 92 |
| 5 | Ph | Ph | Ph | 1e | 2e, 91 | 13e, 91 |
| 6 | Ph | 4-BrC6H4 | Ph | 1g | 2f, 94 | 13f, 80 |
| 7 | Me | 4-MeOC6H4 | Ph | 1i | 2g, 82 | 13g, 90 |
| 8 | Ph | 4-FC6H4 | Bn | 1l | 2h, 88 | – |
| 9 | Ph | 4-ClC6H4 | Bn | 1m | 2i, 95 | 13h, 80 |
As mentioned above, ylides 2 can potentially be in tautomeric equilibrium with N-heterocyclic carbenes 3. No signals characteristic for carbenes 3a–i, however, were found in the NMR spectra of compound 2a–i. According to the DFT calculations mesomeric electron-donating substituents R2 in the pyrrole ring (Table 3, cf. entries 3 and 5) stabilize carbene tautomer 3 slightly. Changing N-alkyl for N-aryl substituents in the imidazolium ring has a relatively small effect on the tautomeric ratio (Table 3, entries 1 and 3). At the same time, the solvent has a dramatic effect on the equilibrium position. In the gas phase carbenes 3 are more thermodynamically stable than the corresponding ylides 2. However, as one can expect, the solvent stabilizes the zwitterion species much better than the uncharged ones. According to the DFT calculations in solution the equilibrium is shifted to the ylide side and the higher the polarity of the solvent the stronger the shift. Nevertheless, carbene tautomers 3 were trapped in THF by reaction with sulfur, leading to imidazolethiones 13, under unusually mild conditions (Table 2) [10,15,16].
Table 3: Relative free energies (carbene 3/ylide 2, ylide 4/betaine 6 and carbene 5/betaine 6) computed at the DFT B3LYP/6-31+G(d,p) level in the gas phase or with the PCM model for the corresponding solvent at 298 K. Calculated data are based on the most stable conformer of 2−6.
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-189-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | equilibrium system | ΔG3-2, kcal·mol−1 | |||
|---|---|---|---|---|---|
| gas phase | DCM | THF | DMSO | ||
equilibrium 2 ![[Graphic 4]](/bjoc/content/inline/1860-5397-11-189-i8.svg?max-width=637&scale=1.0) 3 (ΔG3-2, kcal·mol−1) 3 (ΔG3-2, kcal·mol−1)
|
|||||
| 1 | a | –1.5 | 9.6 | – | 12.3 |
| 2 | b | –0.5 | 10.2 | 9.5 | 12.8 |
| 3 | e | –1.3 | 7.9 | – | 10.1 |
| 4 | i | –0.8 | 9.6 | 9.3 | 12.4 |
| 5 | j | –2.7 | 6.8 | – | 9.5 |
|
equilibrium 4 5 6 (ΔG4-6/ΔG5-6, kcal·mol−1)
|
|||||
| 6 | a | –11.4/–8.6 | 1.5/16.0 | – | 4.0/20.0 |
| 7 | b | –12.4/–9.0 | 1.2/15.7 | 0.5/14.6 | 4.0/20.5 |
| 8 | e | –11.6/–8.8 | 1.9/14.4 | – | 4.0/17.9 |
| 9 | i | –10.9/–7.0 | 1.8/16.3 | – | 3.8/20.1 |
| 10 | j | –11.3/–9.1 | 2.1/14.1 | – | 4.3/18.0 |
Hydrolysis of the ester group in compounds 1 or 2 needs much harsher conditions. Reflux of 1b in a NaOH solution in methanol/water 2:1 or in a LiOH solution in THF/water 9:1 leaved the ester group unchanged and only by refluxing 1b in a LiOH solution in dioxane/water 9:1 the Li salt 14b was obtained. Betaine 6b was isolated in quantitative yield after treatment of 14b with 1 equiv of CCl3CO2H (Scheme 4). Ylide 2a was hydrolyzed with a lower yield due to the high solubility of the salt 14a in water.
![[1860-5397-11-189-i4]](/bjoc/content/inline/1860-5397-11-189-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The hydrolysis of ylide 2a and bromide 1b.
Scheme 4: The hydrolysis of ylide 2a and bromide 1b.
Betaines 6 can, in principle, exist in tautomeric equilibrium with the corresponding carboxy-substituted ylides 4 and N-heterocyclic carbenes 5. The results of the DFT study of the relative thermodynamic stability of tautomers 4–6 revealed (Table 3, entries 6–10) that solvent has a crucial impact on the equilibrium position. Betaines 6 are the most unstable species in the gas phase, whereas in solvents they become the most stable species and therefore dominate in solution. It is worth noting that the concentration of carbene 5 in solution is negligible and much less than carbene 3 in equilibrium 2/3. It is therefore not surprising that the corresponding imidazolethiones were not formed from 6a with sulfur either in THF (rt or reflux) or even in refluxing dioxane.
The structure of the crystalline compound 6b was also analyzed by single X-ray diffraction (Figure 1). X-ray analysis cannot give preference to one of the three possible structures with the same positions of heavy atoms: ylide 4b, carbene 5b and betaine 6b. A comparison, however, of the carbon–oxygen bond lengths of the carboxy group obtained from X-ray analysis with bond lengths calculated at the B3LYP/6-31+G(d,p) level of theory for structures mentioned above shows that betaine 6b is the correct structure (for calculated geometries of ylide 4b, carbene 5b and betaine 6b see Supporting Information File 1). It can therefore be concluded that betaine 6b is thermodynamically much more stable than the corresponding ylide 4b and carbene 5b both in solution and in the solid state.
![[1860-5397-11-189-1]](/bjoc/content/figures/1860-5397-11-189-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of compound 6b (CCDC 1406417). Carbon, nitrogen and oxygen atoms are grey, blue and red, respectively. Chlorine is dark-green. Thermal ellipsoids are drawn at the 50% probability level.
Figure 1: Molecular structure of compound 6b (CCDC 1406417). Carbon, nitrogen and oxygen atoms are grey, blue...
Conclusion
A convenient approach was developed for the synthesis of derivatives of methyl 4-imidazolylpyrrole-2-carboxylates from easily available compounds, 5-methoxyisoxazoles and phenacylimidazolium salts, under hybrid Fe(II)/Et3N relay catalysis. 3-(5-Methoxycarbonyl-1H-pyrrol-3-yl)-1H-imidazol-3-ium bromides were easily transformed into the corresponding 3-(5-methoxycarbonyl-1H-imidazol-3-ium-3-yl)pyrrol-1-ides. The carbene form of the latter were trapped by reaction with sulfur with formation of the corresponding imidazolethiones under very mild conditions. Hydrolysis of 3-(5-methoxycarbonyl-1H-pyrrol-3-yl)-1H-imidazol-3-ium bromides under harsh conditions leads to (1H-imidazol-3-ium-3-yl)-1H-pyrrole-2-carboxylates which are potential ligands for hybrid chelate/NHC complexes.
Experimental
General methods
Melting points were determined on a capillary melting point apparatus Stuart® SMP30. 1H (400 MHz) and 13C (100 MHz) NMR spectra were determined in CDCl3 and DMSO-d6 with a Bruker AVANCE III 400 spectrometer. Chemical shifts (δ) are reported in parts per million downfield from tetramethylsilane (TMS δ = 0.00). 1H NMR spectra were calibrated according to the residual peak of CDCl3 (7.26 ppm) or DMSO-d6 (2.50 ppm). For all new compounds 13C{1H} and 13C DEPT135 were recorded and calibrated according to the peak of CDCl3 (77.00 ppm) or DMSO-d6 (39.51 ppm). Mass spectra were recorded on a Bruker maXis HRMS–ESI–QTOF, with electrospray ionization in positive mode. IR spectra were recorded on a Bruker FTIR spectrometer Tensor 27 for tablets in KBr, only characteristic absorption is indicated. The single crystal X-ray diffraction experiment was performed on Agilent Technologies SuperNova diffractometer at 100 K using monochromated Cu Kα radiation. Thin-layer chromatography (TLC) was conducted on aluminium sheets with 0.2 mm silica gel (fluorescent indicator, Macherey-Nagel). The isoxazoles 7 [37,38] and imidazolium salts 9 [29] were synthesized by known literature procedures.
General procedure for the synthesis of 5-methoxycarbonylpyrrol-3-ylimidazolium bromides 1a–o from isoxazoles 7a–c and imidazolium bromides 9a–k. Isoxazole 7 (1.2–1.5 mmol) and imidazolium bromide 9 (1.0 mmol) were suspended in MeCN (4 mL), FeCl2·4H2O (0.06–0.08 mmol, 5 mol % calcd on isoxazole) and Et3N (3.0 mmol, 3 equiv) were added and the mixture was stirred at 45 °C for 6–7 h (monitored by TLC). Reaction mixture was evaporated to dryness, ethyl acetate was added and the precipitate formed was filtered off and washed with ethyl acetate or an ethyl acetate/CH2Cl2 mixture. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 12:1), additionally washed with ethyl acetate or an ethyl acetate/CH2Cl2 mixture and dried to give the analytically pure compound.
3-(2-(4-Chlorophenyl)-5-methoxycarbonyl-4-phenyl-1H-pyrrol-3-yl)-1-methyl-1H-imidazol-3-ium bromide (1b): colorless solid, mp 242–243 °C (dec, ethyl acetate), yield 204 mg, 54%, obtained from 5-methoxy-3-phenylisoxazole (7a, 175 mg, 1 mmol), 3-(2-(4-chlorophenyl)-2-oxoethyl)-1-methyl-1H-imidazol-3-ium bromide (9b, 253 mg, 0.8 mmol), FeCl2·4H2O (10 mg, 0.05 mmol, 5 mol %) and Et3N (242 mg, 2.4 mmol) according to the general procedure. 1Н NMR (DMSO-d6) δ 3.68 (s, 3H), 3.83 (s, 3H), 7.25–7.28 (m, 2H), 7.31–7.32 (m, 3H), 7.37–7.40 (m, 2H), 7.48–7.50 (m, 2H), 7.81–7.82 (m, 1H), 7.83–7.84 (m, 1H), 9.35-9.36 (m, 1H), 13.03 (br s, 1H); 13С NMR (DMSO-d6) δ 36.2 (CH3), 51.5 (CH3), 116.5 (C), 118.1(C), 124.2 (CH), 125.6 (CH), 126.6 (C), 127.8 (CH), 127.9 (CH), 128.0 (C), 128.9 (CH), 129.5 (CH), 129.7 (CH), 130.4 (C), 130.6 (C), 133.8 (C), 138.8 (CH), 160.2 (C); HRMS–ESI (m/z): [M – Br]+ calcd for C22Н19ClN3O2, 392.1160; found, 392.1168; IR (KBr, cm−1) ν: 3389, 3042, 1706.
General procedure for debenzylation of 1-benzyl-3-pyrrol-3-yl-1H-imidazol-3-ium bromides 1j,k,n. 1-Benzyl-1H-imidazol-3-ium bromide 1 (100 mg) was dissolved in MeOH (10 mL), Pd/C (10 mg, 10 wt %) and ammonium formate (10 equiv) were added. The suspension was stirred under reflux for 1 h (monitored by TLC). The reaction mixture was filtered to remove Pd/C, MeOH was evaporated under reduced pressure, water was added to the residue and the product was filtered, washed with water and dried to give the analytically pure compound.
Methyl 4-(1H-imidazol-1-yl)-3,5-diphenyl-1H-pyrrole-2-carboxylate (12a): colorless solid, mp 239–241 °C (dec., water), yield 128 mg, 90%, obtained from 1-benzyl-3-(5-(methoxycarbonyl)-2,4-diphenyl-1H-pyrrol-3-yl)-1H-imidazol-3-ium bromide (1j, 220 mg, 0.43 mmol), Pd/C (22 mg, 10 wt %) and ammonium formate (270 mg, 4.3 mmol) according to the general procedure. 1Н NMR (DMSO-d6) δ 3.66 (s, 3H), 6.89–6.90 (m, 1H), 7.10–7.11 (m, 1H), 7.21–7.26 (m, 7H), 7.30–7.33 (m, 3H), 7.51–7.52 (m, 1H), 12.49 (br s, 1H); 13С NMR (DMSO-d6) δ 51.2 (CH3), 117.3 (C), 119.1 (C), 122.5 (CH), 126.9 (CH), 127.1 (CH), 127.4 (CH), 128.2 (CH), 128.5 (CH), 128.69 (CH), 128.70 (C), 128.9 (C), 129.7 (CH), 131.1 (C), 131.8 (C), 139.1 (CH), 160.5 (C); HRMS–ESI (m/z): [M + H]+ calcd for C21Н18N3O2, 344.1394; found, 344.1401; IR (KBr, cm–1) ν: 3124, 2951, 1688.
General procedure for the synthesis of pyrrolydes 2 from 5-methoxycarbonylpyrrol-3-ylimidazolium bromides 1. A suspension of 3-(1H-pyrrol)-1H-imidazol-3-ium bromide 1 (1 mmol) in aqueous solution of KOH (2 mmol, 2 equiv, 5 mL H2O) was sonicated for 5 min and then vigorously stirred for 12 h. The precipitate was filtered, washed with water (2–3 mL) and dried to give analytically pure compound.
2-Methoxycarbonyl-4-(1-methyl-1H-imidazol-3-ium-3-yl)-3,5-diphenylpyrrol-1-ide (2a): colorless solid, mp 237–238 °C (dec.), yield 188 mg, 71%, obtained from 3-(5-(methoxycarbonyl)-2,4-diphenyl-1H-pyrrol-3-yl)-1-methyl-1H-imidazol-3-ium bromide (1a, 323 mg, 0.737 mmol) and a aqueous solution of KOH (83 mg, 1.482 mmol, 4 mL H2O) according to the general procedure. 1Н NMR (DMSO-d6) δ 3.51 (s, 3H), 3.81 (s, 3H), 7.03–7.21 (m, 10H), 7.69–7.72 (m. 2H), 9.11 (s, 1H); 13С NMR (DMSO-d6) δ 35.7 (CH3), 49.4 (CH3), 115.2 (C), 123.3 (CH), 124.6 (CH), 124.9 (CH), 125.2 (CH), 125.8 (C), 126.3 (CH), 127.0 (CH), 128.0 (CH), 128.0 (C), 129.6 (CH), 135.5 (C), 135.8 (C), 137.1 (C), 137.9 (CH), 165.0 (C); HRMS–ESI (m/z): [M + H]+ calcd for C22H20N3O2, 358.1550; found, 358.1566; IR (KBr, cm–1) ν: 3528, 3144, 3059, 1676.
General procedure for the synthesis of 4-(2-thioxo-2,3-dihydro-1H-imidazol-1-yl)-1H-pyrrol-2-carboxylates 13 from pyrrolides 2. A suspension of 3-(1H-imidazol-3-ium-3-yl)-5-(methoxycarbonyl)-pyrrol-1-ide (2, 1 mmol) and sulfur (2 mmol, 2 equiv) in dry THF was stirred at rt for 1–2 hours (monitored by TLC). Then the reaction mixture was evaporated to dryness and the residue was purified by column chromatography on silica gel (hexane/ethyl acetate from 1:1 to 0:1) to give the analytically pure compound.
Methyl 4-(3-methyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)-3,5-diphenyl-1H-pyrrole-2-carboxylate (13a): colorless solid, mp 261–262 °C, yield 43 mg, 80%, obtained from 2-(methoxycarbonyl)-4-(1-methyl-1H-imidazol-3-ium-3-yl)-3,5-diphenylpyrrol-1-ide (2a, 50 mg, 0.140 mmol) and sulfur (9 mg, 0.280 mmol) according to the general procedure. 1Н NMR (DMSO-d6) δ 3.46 (s, 3H), 3.67 (s, 3H), 6.84 (d, J = 2.3 Hz, 1H), 7.03 (d, J = 2.3 Hz, 1H), 7.17–7.42 (m, 8H), 7.52 (d, J = 7.0 Hz, 2H), 12.41 (s, 1H); 13С NMR (DMSO-d6) δ 34.9 (CH3), 51.1 (CH3), 117.1 (C), 118.9 (CH), 119.6 (CH), 119.7 (C), 127.0 (CH), 127.3 (CH), 127.5 (CH), 128.1 (CH), 128.3 (CH), 129.4 (C), 129.7 (C), 130.1 (CH), 132.1 (C), 132.4 (C), 160.6 (C), 165.4 (C); HRMS–ESI (m/z): [M + Na]+ calcd for C22H19N3O2SNa, 412.1090; found 412.1112; IR (KBr, cm−1) ν: 3304, 1662, 1454, 1375.
5-(4-Chlorophenyl)-4-(1-methyl-1H-imidazol-3-ium-3-yl)-3-phenyl-1H-pyrrole-2-carboxylate (6b). A suspension of 3-(2-(4-chlorophenyl)-5-(methoxycarbonyl)-4-phenyl-1H-pyrrol-3-yl)-1-methyl-1H-imidazol-3-ium bromide 1b (100 mg, 0.212 mmol) and LiOH (253 mg, 10.6 mmol, 50 equiv) in dioxane (30 mL) and water (3 mL) was stirred at 110 °C for 24 h. Then reaction mixture was evaporated to dryness and water (5 mL) was added. The suspension was filtered, the solid was washed with water (2 × 5 mL) and thoroughly dried to obtain lithium salt 14b in quantitive yield. To convert the lithium salt 14b into 6b trichloroacetic acid (35 mg, 0.212 mmol, 1 equiv) was added to a suspension of the lithium salt in water (5 mL). The suspension was sonicated for 5 min, stirred for 1 h and filtered. The solid was washed with water and dried to obtain 6b as a colorless solid, mp 205–207 °C, yield 79 mg, 98%. 1Н NMR (DMSO-d6) δ 3.82 (s, 3H), 7.11–7.26 (m, 5H), 7.29 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 7.77 (s, 1H), 7.80 (s, 1H), 9.26 (s, 1H); 13С NMR (DMSO-d6) δ 36.0 (CH3), 115.1 (C), 120.8 (C), 123.9 (CH), 124.0 (C), 124.2 (C), 125.9 (CH), 126.0 (CH), 127.1 (CH), 128.4 (CH), 128.42 (C), 128.6 (CH), 130.1 (CH), 131.8 (C), 133.1 (C), 138.6 (CH), 162.2 (C); HRMS–ESI (m/z): [M + H]+ calcd for C21H17ClN3O2, 378.1004; found, 378.1009; IR (KBr, cm−1) ν: 3498, 3033, 1694. Carboxylate 6a (CCDC 1406417) was analyzed by single X-ray diffraction. It is triclinic P−1, a = 9.2999(5) Å, b = 9.5698(4) Å, c = 15.3875(5) Å, α = 72.782(3)°, β = 78.712(4)°, γ = 66.401(4)°, V = 1194.02(9) Å3, Z = 2, 4960 unique reflections were measured, which were used in all calculations. The final R1 was 0.0561 and wR2 was 0.1681 (all data) (>2sigma(I)) (see Supporting Information File 1 for details).
Supporting Information
| Supporting Information File 1: Detailed experimental procedures including characterization data for all synthesized compounds, 1H and 13C NMR spectra for all new compounds. Computational details: energies of molecules, transition states and their Cartesian coordinates of atoms. X-ray details. | ||
| Format: PDF | Size: 7.8 MB | Download |
Acknowledgements
We gratefully acknowledge the financial support of the Russian Foundation for Basic Research (Grant No. 14-03-00187) and Saint Petersburg State University (Grant No. 12.50.1565.2013, 12.38.239.2014, 12.38.217.2015). This research was carried out using resources of the X-ray Diffraction Centre, the Centre for Magnetic Resonance, the Computer Center and the Centre for Chemical Analysis and Materials of St. Petersburg State University.
References
-
Forte, B.; Malgesini, B.; Piutti, C.; Quartieri, F.; Scolaro, A.; Papeo, G. Mar. Drugs 2009, 7, 705–753. doi:10.3390/md7040705
Return to citation in text: [1] -
Zhang, L.; Peng, X.-M.; Damu, G. L. V.; Geng, R.-X.; Zhou, C.-H. Med. Res. Rev. 2014, 34, 340–437. doi:10.1002/med.21290
Return to citation in text: [1] -
Shinohara, K.-I.; Bando, T.; Sugiyama, H. Anti-Cancer Drugs 2010, 21, 228–242. doi:10.1097/CAD.0b013e328334d8f9
Return to citation in text: [1] -
Yanga, F.; Nickols, N. G.; Li, B. C.; Marinov, G. K.; Said, J. W.; Dervan, P. B. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1863–1868. doi:10.1073/pnas.1222035110
Return to citation in text: [1] -
Edwards, T. G.; Koeller, K. J.; Slomczynska, U.; Fok, K.; Helmus, M.; Bashkin, J. K.; Fisher, C. Antiviral Res. 2011, 91, 177–186. doi:10.1016/j.antiviral.2011.05.014
Return to citation in text: [1] -
Reger, T. S.; Zunic, J.; Stock, N.; Wang, B.; Smith, N. D.; Munoz, B.; Green, M. D.; Gardner, M. F.; James, J. P.; Chen, W.; Alves, K.; Si, Q.; Treonze, K. M.; Lingham, R. B.; Mumford, R. A. Bioorg. Med. Chem. Lett. 2010, 20, 1173–1176. doi:10.1016/j.bmcl.2009.12.009
Return to citation in text: [1] -
Natchus, M. G.; Bookland, R. G.; De, B.; Almstead, N. G.; Pikul, S.; Janusz, M. J.; Heitmeyer, S. A.; Hookfin, E. B.; Hsieh, L. C.; Dowty, M. E.; Dietsch, C. R.; Patel, V. S.; Garver, S. M.; Gu, F.; Pokross, M. E.; Mieling, G. E.; Baker, T. R.; Foltz, D. J.; Peng, S. X.; Bornes, D. M.; Strojnowski, M. J.; Taiwo, Y. O. J. Med. Chem. 2000, 43, 4948–4963. doi:10.1021/jm000246e
Return to citation in text: [1] -
Aronov, A.; Bandarage, U. K.; Lauffer, D. J. Pyrrole compositions useful as inhibitors of c-met. PCT Int. Appl. WO2005/016920A1, Feb 24, 2005.
Return to citation in text: [1] [2] [3] -
Klose, W.; Boettcher, I. Antiinflammatory and antiallergic imidazole derivatives. U.S. Patent US4466976, Aug 21, 1984.
Return to citation in text: [1] [2] [3] -
Schmidt, A.; Wiechmann, S.; Freese, T. ARKIVOC 2013, No. i, 424–469. doi:10.3998/ark.5550190.p008.251
Return to citation in text: [1] [2] -
Ramsden, C. A. Tetrahedron 2013, 69, 4146–4159. doi:10.1016/j.tet.2013.02.081
Return to citation in text: [1] -
Schmidt, A.; Guan, Z. Synthesis 2012, 44, 3251–3268. doi:10.1055/s-0032-1316787
Return to citation in text: [1] -
Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 2897–2970. doi:10.2174/138527211796378497
Return to citation in text: [1] -
Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263
Return to citation in text: [1] -
Liu, M.; Nieger, M.; Schmidt, A. Chem. Commun. 2015, 51, 477–479. doi:10.1039/C4CC08032G
Return to citation in text: [1] [2] -
Pidlypnyi, N.; Wolf, S.; Liu, M.; Rissanen, K.; Nieger, M.; Schmidt, A. Tetrahedron 2014, 70, 8672–8680. doi:10.1016/j.tet.2014.09.035
Return to citation in text: [1] [2] -
Benhamou, L.; Bastin, S.; Lugan, N.; Lavigne, G.; César, V. Dalton Trans. 2014, 43, 4474–4482. doi:10.1039/C3DT53089B
Return to citation in text: [1] -
Danopoulos, A. A.; Monakhov, K. Yu.; Braunstein, P. Chem. – Eur. J. 2013, 19, 450–455. doi:10.1002/chem.201203488
Return to citation in text: [1] -
Pidlypnyi, N.; Uhrner, F.; Nieger, M.; Drafz, M. H. H.; Hübner, E. G.; Namyslo, J. C.; Schmidt, A. Eur. J. Org. Chem. 2013, 7739–7748. doi:10.1002/ejoc.201300728
Return to citation in text: [1] -
Pidlypnyi, N.; Namyslo, J. C.; Drafz, M. H. H.; Nieger, M.; Schmidt, A. J. Org. Chem. 2013, 78, 1070–1079. doi:10.1021/jo302479p
Return to citation in text: [1] -
Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/C1CC16460K
Return to citation in text: [1] -
César, V.; Tourneux, J.-C.; Vujkovic, N.; Brousses, R.; Lugan, N.; Lavigne, G. Chem. Commun. 2012, 48, 2349–2351. doi:10.1039/c2cc17870b
Return to citation in text: [1] -
Singh, K. S.; Kaminsky, W. Polyhedron 2014, 68, 279–286. doi:10.1016/j.poly.2013.10.032
Return to citation in text: [1] -
Kemmitt, R. D. W.; Mason, S.; Fawcett, J.; Russell, D. R. J. Chem. Soc., Dalton Trans. 1992, 1165–1176. doi:10.1039/DT9920001165
Return to citation in text: [1] -
Hammershøi, A.; Hartshorn, R. M.; Sargeson, A. M. J. Chem. Soc., Chem. Commun. 1988, 1267–1269. doi:10.1039/c39880001267
Return to citation in text: [1] [2] -
Ahmed, I. T. Spectrochim. Acta, Part A 2006, 63, 416–422. doi:10.1016/j.saa.2005.05.028
Return to citation in text: [1] -
Taubmann, S.; Alt, H. G. J. Organomet. Chem. 2008, 693, 1808–1814. doi:10.1016/j.jorganchem.2008.02.003
Return to citation in text: [1] -
Nakamura, N.; Kohzuma, T.; Kuma, H.; Suzuki, S. Inorg. Chem. 1994, 33, 1594–1599. doi:10.1021/ic00086a007
Return to citation in text: [1] -
Khlebnikov, A. F.; Tomashenko, O. A.; Funt, L. D.; Novikov, M. S. Org. Biomol. Chem. 2014, 12, 6598–6609. doi:10.1039/C4OB00865K
Return to citation in text: [1] [2] -
Auricchio, S.; Bini, A.; Pastormerlo, E.; Truscello, A. M. Tetrahedron 1997, 53, 10911–10920. doi:10.1016/S0040-4020(97)00696-0
Return to citation in text: [1] -
Galenko, E. E.; Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S. RSC Adv. 2015, 5, 18172–18176. doi:10.1039/C5RA01889G
Return to citation in text: [1] -
Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/RCR4503
Return to citation in text: [1] -
Pinho e Melo, T. M. V. D. Curr. Org. Chem. 2005, 9, 925–958. doi:10.2174/1385272054368420
Return to citation in text: [1] -
Khlebnikov, A. F.; Novikov, M. S. Tetrahedron 2013, 69, 3363–3401. doi:10.1016/j.tet.2013.02.020
Return to citation in text: [1] -
Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337–1378. doi:10.1039/C2CS35258C
Return to citation in text: [1] -
Wende, R. C.; Schreiner, P. R. Green Chem. 2012, 14, 1821–1849. doi:10.1039/c2gc35160a
Return to citation in text: [1] -
Katritzky, A. R.; Øksne, S.; Boulton, A. J. Tetrahedron 1962, 18, 777–790. doi:10.1016/S0040-4020(01)92728-0
Return to citation in text: [1] -
Micetich, R. G.; Chin, C. G. Can. J. Chem. 1970, 48, 1371–1376. doi:10.1139/v70-226
Return to citation in text: [1]
| 37. | Katritzky, A. R.; Øksne, S.; Boulton, A. J. Tetrahedron 1962, 18, 777–790. doi:10.1016/S0040-4020(01)92728-0 |
| 38. | Micetich, R. G.; Chin, C. G. Can. J. Chem. 1970, 48, 1371–1376. doi:10.1139/v70-226 |
| 35. | Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337–1378. doi:10.1039/C2CS35258C |
| 36. | Wende, R. C.; Schreiner, P. R. Green Chem. 2012, 14, 1821–1849. doi:10.1039/c2gc35160a |
| 10. | Schmidt, A.; Wiechmann, S.; Freese, T. ARKIVOC 2013, No. i, 424–469. doi:10.3998/ark.5550190.p008.251 |
| 15. | Liu, M.; Nieger, M.; Schmidt, A. Chem. Commun. 2015, 51, 477–479. doi:10.1039/C4CC08032G |
| 16. | Pidlypnyi, N.; Wolf, S.; Liu, M.; Rissanen, K.; Nieger, M.; Schmidt, A. Tetrahedron 2014, 70, 8672–8680. doi:10.1016/j.tet.2014.09.035 |
| 1. | Forte, B.; Malgesini, B.; Piutti, C.; Quartieri, F.; Scolaro, A.; Papeo, G. Mar. Drugs 2009, 7, 705–753. doi:10.3390/md7040705 |
| 2. | Zhang, L.; Peng, X.-M.; Damu, G. L. V.; Geng, R.-X.; Zhou, C.-H. Med. Res. Rev. 2014, 34, 340–437. doi:10.1002/med.21290 |
| 3. | Shinohara, K.-I.; Bando, T.; Sugiyama, H. Anti-Cancer Drugs 2010, 21, 228–242. doi:10.1097/CAD.0b013e328334d8f9 |
| 4. | Yanga, F.; Nickols, N. G.; Li, B. C.; Marinov, G. K.; Said, J. W.; Dervan, P. B. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1863–1868. doi:10.1073/pnas.1222035110 |
| 5. | Edwards, T. G.; Koeller, K. J.; Slomczynska, U.; Fok, K.; Helmus, M.; Bashkin, J. K.; Fisher, C. Antiviral Res. 2011, 91, 177–186. doi:10.1016/j.antiviral.2011.05.014 |
| 10. | Schmidt, A.; Wiechmann, S.; Freese, T. ARKIVOC 2013, No. i, 424–469. doi:10.3998/ark.5550190.p008.251 |
| 11. | Ramsden, C. A. Tetrahedron 2013, 69, 4146–4159. doi:10.1016/j.tet.2013.02.081 |
| 12. | Schmidt, A.; Guan, Z. Synthesis 2012, 44, 3251–3268. doi:10.1055/s-0032-1316787 |
| 13. | Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 2897–2970. doi:10.2174/138527211796378497 |
| 14. | Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263 |
| 32. | Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/RCR4503 |
| 33. | Pinho e Melo, T. M. V. D. Curr. Org. Chem. 2005, 9, 925–958. doi:10.2174/1385272054368420 |
| 9. | Klose, W.; Boettcher, I. Antiinflammatory and antiallergic imidazole derivatives. U.S. Patent US4466976, Aug 21, 1984. |
| 34. | Khlebnikov, A. F.; Novikov, M. S. Tetrahedron 2013, 69, 3363–3401. doi:10.1016/j.tet.2013.02.020 |
| 8. | Aronov, A.; Bandarage, U. K.; Lauffer, D. J. Pyrrole compositions useful as inhibitors of c-met. PCT Int. Appl. WO2005/016920A1, Feb 24, 2005. |
| 30. | Auricchio, S.; Bini, A.; Pastormerlo, E.; Truscello, A. M. Tetrahedron 1997, 53, 10911–10920. doi:10.1016/S0040-4020(97)00696-0 |
| 6. | Reger, T. S.; Zunic, J.; Stock, N.; Wang, B.; Smith, N. D.; Munoz, B.; Green, M. D.; Gardner, M. F.; James, J. P.; Chen, W.; Alves, K.; Si, Q.; Treonze, K. M.; Lingham, R. B.; Mumford, R. A. Bioorg. Med. Chem. Lett. 2010, 20, 1173–1176. doi:10.1016/j.bmcl.2009.12.009 |
| 7. | Natchus, M. G.; Bookland, R. G.; De, B.; Almstead, N. G.; Pikul, S.; Janusz, M. J.; Heitmeyer, S. A.; Hookfin, E. B.; Hsieh, L. C.; Dowty, M. E.; Dietsch, C. R.; Patel, V. S.; Garver, S. M.; Gu, F.; Pokross, M. E.; Mieling, G. E.; Baker, T. R.; Foltz, D. J.; Peng, S. X.; Bornes, D. M.; Strojnowski, M. J.; Taiwo, Y. O. J. Med. Chem. 2000, 43, 4948–4963. doi:10.1021/jm000246e |
| 8. | Aronov, A.; Bandarage, U. K.; Lauffer, D. J. Pyrrole compositions useful as inhibitors of c-met. PCT Int. Appl. WO2005/016920A1, Feb 24, 2005. |
| 9. | Klose, W.; Boettcher, I. Antiinflammatory and antiallergic imidazole derivatives. U.S. Patent US4466976, Aug 21, 1984. |
| 31. | Galenko, E. E.; Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S. RSC Adv. 2015, 5, 18172–18176. doi:10.1039/C5RA01889G |
| 26. | Ahmed, I. T. Spectrochim. Acta, Part A 2006, 63, 416–422. doi:10.1016/j.saa.2005.05.028 |
| 27. | Taubmann, S.; Alt, H. G. J. Organomet. Chem. 2008, 693, 1808–1814. doi:10.1016/j.jorganchem.2008.02.003 |
| 8. | Aronov, A.; Bandarage, U. K.; Lauffer, D. J. Pyrrole compositions useful as inhibitors of c-met. PCT Int. Appl. WO2005/016920A1, Feb 24, 2005. |
| 9. | Klose, W.; Boettcher, I. Antiinflammatory and antiallergic imidazole derivatives. U.S. Patent US4466976, Aug 21, 1984. |
| 25. | Hammershøi, A.; Hartshorn, R. M.; Sargeson, A. M. J. Chem. Soc., Chem. Commun. 1988, 1267–1269. doi:10.1039/c39880001267 |
| 29. | Khlebnikov, A. F.; Tomashenko, O. A.; Funt, L. D.; Novikov, M. S. Org. Biomol. Chem. 2014, 12, 6598–6609. doi:10.1039/C4OB00865K |
| 23. | Singh, K. S.; Kaminsky, W. Polyhedron 2014, 68, 279–286. doi:10.1016/j.poly.2013.10.032 |
| 24. | Kemmitt, R. D. W.; Mason, S.; Fawcett, J.; Russell, D. R. J. Chem. Soc., Dalton Trans. 1992, 1165–1176. doi:10.1039/DT9920001165 |
| 25. | Hammershøi, A.; Hartshorn, R. M.; Sargeson, A. M. J. Chem. Soc., Chem. Commun. 1988, 1267–1269. doi:10.1039/c39880001267 |
| 29. | Khlebnikov, A. F.; Tomashenko, O. A.; Funt, L. D.; Novikov, M. S. Org. Biomol. Chem. 2014, 12, 6598–6609. doi:10.1039/C4OB00865K |
| 15. | Liu, M.; Nieger, M.; Schmidt, A. Chem. Commun. 2015, 51, 477–479. doi:10.1039/C4CC08032G |
| 16. | Pidlypnyi, N.; Wolf, S.; Liu, M.; Rissanen, K.; Nieger, M.; Schmidt, A. Tetrahedron 2014, 70, 8672–8680. doi:10.1016/j.tet.2014.09.035 |
| 17. | Benhamou, L.; Bastin, S.; Lugan, N.; Lavigne, G.; César, V. Dalton Trans. 2014, 43, 4474–4482. doi:10.1039/C3DT53089B |
| 18. | Danopoulos, A. A.; Monakhov, K. Yu.; Braunstein, P. Chem. – Eur. J. 2013, 19, 450–455. doi:10.1002/chem.201203488 |
| 19. | Pidlypnyi, N.; Uhrner, F.; Nieger, M.; Drafz, M. H. H.; Hübner, E. G.; Namyslo, J. C.; Schmidt, A. Eur. J. Org. Chem. 2013, 7739–7748. doi:10.1002/ejoc.201300728 |
| 20. | Pidlypnyi, N.; Namyslo, J. C.; Drafz, M. H. H.; Nieger, M.; Schmidt, A. J. Org. Chem. 2013, 78, 1070–1079. doi:10.1021/jo302479p |
| 21. | Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/C1CC16460K |
| 22. | César, V.; Tourneux, J.-C.; Vujkovic, N.; Brousses, R.; Lugan, N.; Lavigne, G. Chem. Commun. 2012, 48, 2349–2351. doi:10.1039/c2cc17870b |
| 28. | Nakamura, N.; Kohzuma, T.; Kuma, H.; Suzuki, S. Inorg. Chem. 1994, 33, 1594–1599. doi:10.1021/ic00086a007 |
© 2015 Galenko et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)