Abstract
The synthesis of zwitterionic dithiocarboxylate adducts was achieved by deprotonating various aldiminium or 1,2,3-triazolium salts with a strong base, followed by the nucleophilic addition of the in situ-generated cyclic (alkyl)(amino) or mesoionic carbenes (CAACs or MICs) onto carbon disulfide. Nine novel compounds were isolated and fully characterized by 1H and 13C NMR, FTIR, and HRMS techniques. Moreover, the molecular structures of two CAAC·CS2 and two MIC·CS2 betaines were determined by X-ray diffraction analysis. The analytical data recorded for all these adducts were compared with those reported previously for related NHC·CS2 betaines derived from imidazolinium or (benz)imidazolium salts. Due to the absence of electronic communication between the CS2 unit and the orthogonal heterocycle, all the CAAC·CS2, MIC·CS2, and NHC·CS2 zwitterions displayed similar electronic properties and featured the same bite angle. Yet, their steric properties are liable to ample modifications by varying the exact nature of their cationic heterocycle and its substituents.

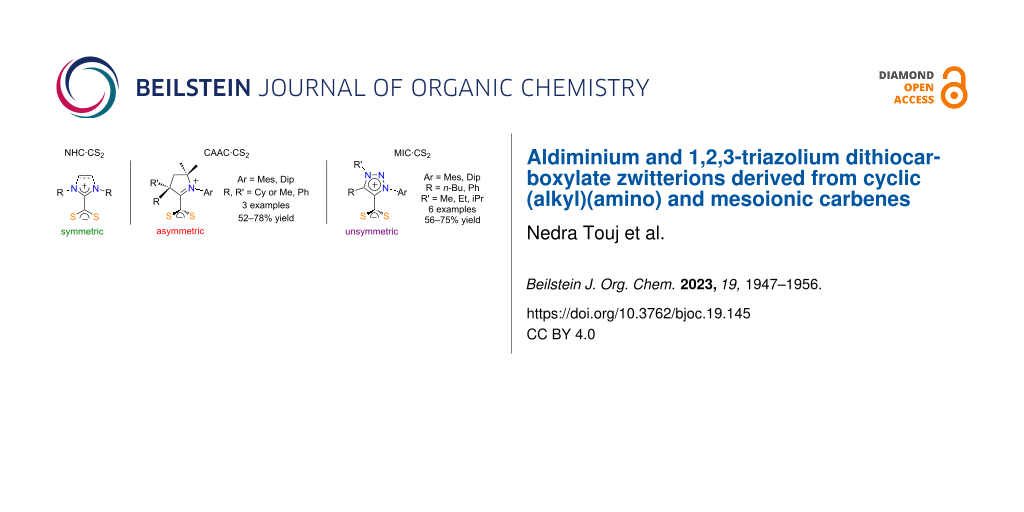
Graphical Abstract
Introduction
Following the seminal discovery from the group of Arduengo, who isolated and fully characterized 1,3-di(1-adamantyl)imidazol-2-ylidene in 1991 [1], stable divalent carbon species have evolved from fleeting intermediates to ubiquitous catalysts, ligands, and reagents in just three decades (Figure 1) [2]. In particular, cyclic diaminocarbenes based on the imidazoline, benzimidazole, or imidazole ring system (A–C) have led to a myriad of applications in organometallic chemistry, homogeneous catalysis, and materials science, to name just a few [3-5]. Due to their weaker basicity and greater modularity, the related 1,2,4-triazol-5-ylidene derivatives D have been mainly employed in organocatalysis [6]. Besides these four types of N-heterocyclic carbenes (NHCs), other families of cyclic compounds have been actively pursued to further expand the diversity of singlet carbenes available to the chemist [7]. Among them, the cyclic (alkyl)(amino)carbenes (CAACs, E) introduced by Bertrand et al. in 2005 [8] have attracted a great deal of attention, thanks to their remarkable nucleophilic (σ-donating) and electrophilic (π-accepting) properties, which allow them to activate a variety of small molecules and to bind strongly to metal centers, thereby affording very robust catalysts [9-12].
![[1860-5397-19-145-1]](/bjoc/content/figures/1860-5397-19-145-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Various types of stable singlet carbenes and their acronyms.
Figure 1: Various types of stable singlet carbenes and their acronyms.
Another category of stable carbenes that has emerged in the new millennium is made of mesoionic compounds, for which no reasonable canonical resonance form can be drawn in the absence of charges (Figure 1) [13-16]. Crabtree and co-workers first reported the abnormal binding of an imidazolium salt to an iridium hydride at the C4 carbon atom instead of C2 in 2001 [17,18]. Since then, many other metal complexes bearing imidazol-4-ylidene ligands (F) have been reported [7,19]. These mesoionic carbenes (MICs), together with their pyrazolin-4-ylidene [20] and 1,2-isoxazol-4-ylidene cousins [21], are the strongest donors among the various types of carbene ligands known thus far [22]. A distinct class of mesoionic or abnormal carbenes based on the 1,2,3-triazole ring system (G) was first investigated by Albrecht et al. in 2008 [23]. Because the heterocyclic precursors needed to prepare 1,2,3-triazol-5-ylidenes are readily available through the [3 + 2] cycloaddition of an azide and an alkyne, these compounds are currently the most popular MICs for catalytic and other applications [24-28].
N-Heterocyclic carbenes and their enetetramine dimers readily add to the central carbon atom of allenes and heteroallenes X=C=Y (X, Y = CR2, NR, O, S) to afford zwitterionic adducts [29]. In particular, their reaction with carbon disulfide affords stable azolium-2-dithiocarboxylate zwitterions (Figure 2) [30-43]. These 1,1-dithiolate inner salts strongly bind main group elements and transition metals through various coordination modes. Indeed, we and others have already reported the synthesis of diverse metallic complexes featuring monodentate [44,45], chelating bidentate [46-55], or bridging bidentate NHC·CS2 ligands [45,51,52]. Small bimetallic clusters [51,52,56], coordination polymers [57], self-assembled monolayers [58], and nanoparticles [45] based on these zwitterions were also prepared, while a few reports disclosed the formation of polynuclear clusters, in which the dithiocarboxylate unit underwent further chemical transformations [59-61].
![[1860-5397-19-145-2]](/bjoc/content/figures/1860-5397-19-145-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Various types of NHC·CS2 zwitterions and their coordination modes.
Figure 2: Various types of NHC·CS2 zwitterions and their coordination modes.
To the best of our knowledge, 1,2,3-triazolium-5-dithiocarboxylate species are hitherto unknown in the literature, and only a single report described the preparation of a CAAC·CS2 zwitterion. Thus, in 2009 Bertrand et al. obtained the betaine 2 by reacting the free carbene 1 with a slight excess of CS2 in THF at room temperature (Scheme 1) [62]. The starting material that featured a bulky and rigid spirocyclic alkyl group derived from (−)-menthone was obtained in a separate step by deprotonating the corresponding aldiminium triflate with lithium diisopropylamide (LDA) at −78 °C [8].
![[1860-5397-19-145-i1]](/bjoc/content/inline/1860-5397-19-145-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of CAAC·CS2 zwitterion 2 from its free carbene parent 1.
Scheme 1: Synthesis of CAAC·CS2 zwitterion 2 from its free carbene parent 1.
Herein, we disclose the synthesis of three CAAC·CS2 and six MIC·CS2 inner salts from the corresponding aldiminium or 1,2,3-triazolium salts and carbon disulfide. All these adducts were fully characterized by using various analytical techniques and their structural properties compared to those displayed by known, related imidazolinium and (benz)imidazolium-2-dithiocarboxylate betaines.
Results and Discussion
Currently, the most general strategy to prepare NHC·CS2 zwitterions relies on the deprotonation of an azolium salt with a strong base, typically potassium tert-butoxide or potassium bis(trimethylsilyl)amide (also known as potassium hexamethyldisilazide, KHMDS) followed by the addition of carbon disulfide either in one pot or after the isolation of the free carbene [39,41,42,58]. Hence, we decided to probe the feasibility of this approach for the synthesis of CAAC·CS2 and MIC·CS2 betaines from readily available aldiminium or 1,2,3-triazolium salts.
Synthesis of CAAC·CS2 zwitterions
To begin our study, we investigated the synthesis of CAAC·CS2 zwitterions starting from three commercially available aldiminium salts 3a–c (Scheme 2). These reagents were suspended in THF and cooled to 0 °C before a 1 M solution of KN(SiMe3)2 in THF was slowly added to release the free carbenes. Of note, compound 3b was purchased as a hydrogen dichloride salt and required the use of a double amount of base. The suspensions were brought back to room temperature and allowed to settle down to ease the filtration of the inorganic byproducts (KCl or KBF4), along with any unreacted starting materials. Adding an excess of CS2 to the free carbene solutions led to the instantaneous formation of the desired zwitterionic adducts, as evidenced by the appearance of an intense orange-red color. The solvent was removed and the residues were brought back to air, washed with n-pentane, and dried under high vacuum to afford pseudo-cross-conjugated mesomeric betaines 4a–c in high yields (ca. 80%). NMR analysis showed that compounds 4a and 4c required further purification. Thus, they were recrystallized from acetonitrile, which led to a non-negligible loss of materials, thereby leading to final yields in the 50–60% range.
![[1860-5397-19-145-i2]](/bjoc/content/inline/1860-5397-19-145-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of CAAC·CS2 zwitterions 4a–c with KN(SiMe3)2.
Scheme 2: Synthesis of CAAC·CS2 zwitterions 4a–c with KN(SiMe3)2.
Synthesis of MIC·CS2 zwitterions
The synthesis of 1,4-disubstituted-1,2,3-triazole derivatives is readily achieved via the copper(I)-catalyzed [3 + 2] cycloaddition of an azide and a terminal alkyne (CuAAC) [63-65]. A further alkylation of the N3 position with an alkyl halide is an equally straightforward procedure that ultimately affords a large assortment of MIC precursors [24-28]. By analogy with the archetypical NHCs bearing mesityl (Mes) or 2,6-diisopropylphenyl (Dipp) substituents on their nitrogen atoms, we have prepared three triazole derivatives with mixed Mes/Ph, Mes/Bu, or Dipp/Ph substituents on N1 and C4, respectively (Scheme 3). The active catalytic species for the CuAAC reaction were generated by reducing copper(II) sulfate with sodium ascorbate according to literature procedures [66,67]. 2-Azido-1,3,5-trimethylbenzene (mesityl azide) was easily synthesized in a distinct, preliminary step through the Sandmeyer reaction of mesitylamine with sodium nitrite and acetic acid followed by a substitution of the intermediate diazonium salt with sodium azide [68]. All our attempts to prepare 2-azido-1,3-diisopropylbenzene along the same lines failed. Nevertheless, its in situ formation in the presence of phenylacetylene led to the desired cycloadduct, although in a modest 30% yield [69]. The subsequent alkylation of N3 with methyl, ethyl, or isopropyl iodide afforded triazolium salts 5a–f in satisfactory to excellent yields. It should be pointed out that compounds 5c–f had never been described before (see the Supporting Information File 1 for experimental details).
![[1860-5397-19-145-i3]](/bjoc/content/inline/1860-5397-19-145-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1,2,3-triazolium iodides 5a–f.
Scheme 3: Synthesis of 1,2,3-triazolium iodides 5a–f.
We initially carried out the synthesis of MIC·CS2 zwitterions from the set of six triazolium iodides in our hands according to the experimental procedure described above for CAAC·CS2 betaines (cf. Scheme 2). Thus, the salts 5a–f were deprotonated with KN(SiMe3)2 (1.2 equiv) in THF at 0 °C. The potassium iodide byproduct was filtered off and carbon disulfide (3.3 equiv) was added to the carbene solution leading to an immediate color change. After 30 min at room temperature, the solvent was evaporated under vacuum. The residue was washed with petroleum ether and dried under high vacuum. 1H NMR analysis revealed that a significant amount of starting material was still present in most cases. Moreover, unidentified byproducts were also detected in some instances. Products 6a and 6b could be isolated in pure form and satisfactory yields upon recrystallization from acetonitrile (Scheme 4). However, all our attempts to purify compounds 6c–f by recrystallization or column chromatography remained unsuccessful.
![[1860-5397-19-145-i4]](/bjoc/content/inline/1860-5397-19-145-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of MIC·CS2 zwitterions 6a and 6b with KN(SiMe3)2.
Scheme 4: Synthesis of MIC·CS2 zwitterions 6a and 6b with KN(SiMe3)2.
Several studies have shown that metal alkoxides, such as potassium tert-butoxide (pKa = 22), were basic enough to deprotonate 1,2,3-triazolium salts (pKa ≈ 22–23) depending on the nature of their aliphatic or aromatic substituents, and that the use of the stronger base KN(SiMe3)2 (pKa = 26) was not always mandatory [25,69,70]. Grubbs, Bertrand et al. also noticed that treating 1-benzyl-3-methyl-4-phenyl-1H-1,2,3-triazolium iodide with KOt-Bu did not afford the desired MIC but led to a debenzylated triazole instead [71]. Based on these observations, we decided to revise our experimental procedure for the synthesis of MIC·CS2 zwitterions by using a mixture of NaOt-Bu and CS2 from the onset of the reaction in THF at 60 °C. These two reagents were added in large excess to compensate for the possible formation of sodium O-tert-butyl xanthate [72-74]. We reasoned that these conditions should favor a quantitative deprotonation of the starting triazolium salts and the concomitant trapping of the free carbenes by CS2 prior to their potential decomposition. Gratifyingly, this method allowed us to isolate the cross-conjugated mesomeric betaines 6c–f in good yields (Scheme 5). Clean NMR spectra were recorded in all cases, although elemental analysis revealed that the products were not entirely homogeneous. This might be due to the presence of inorganic impurities. Yet, we did not try to purify them any further.
![[1860-5397-19-145-i5]](/bjoc/content/inline/1860-5397-19-145-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of MIC·CS2 zwitterions 6c–f with NaOt-Bu.
Scheme 5: Synthesis of MIC·CS2 zwitterions 6c–f with NaOt-Bu.
Before closing this section, it should be stressed that we were not able to isolate any dithiocarboxylate betaines when aldiminium salt 3b or triazolium salt 5a were treated with cesium carbonate and carbon disulfide in acetonitrile in the presence of air and moisture, followed by an aqueous work-up. These results sharply contrasted with those obtained previously with a range of imidazolinium, benzimidazolium, or imidazolium salts, which were successfully converted into NHC·CS2 zwitterions under mild aerobic conditions [75]. Yet, the greater proton affinity and the higher basicity of CAACs and MICs vs NHCs [25,69-71] did not prevent the formation of the desired adducts using Cs2CO3 instead of KN(SiMe3)2 or NaOt-Bu, as evidenced by the appearance of revealing orange-red colors and by the emergence of a characteristic resonance for the CS2− unit on 13C NMR spectroscopy (see below). We suspect that deleterious hydrophilic effects caused the subsequent decomposition of the CAAC·CS2 and MIC·CS2 zwitterions when an aqueous work-up was applied.
Structural analysis
Several analytical techniques were employed to characterize the nine aldiminium and 1,2,3-triazolium dithiocarboxylate betaines under investigation. 1H NMR spectra of compounds 4a–c and 6a–f were rather similar to those of their precursors 3a–c and 5a–f, respectively, with only one less resonance due to the replacement of their acidic proton with a CS2 group (Table 1). Because the vanishing signal was always the most deshielded singlet in the spectra of the reagents, it was a very convenient probe to monitor the success of the deprotonation step. Concomitantly, the incorporation of CS2 in products 4a–c and 6a–f led to the emergence of an equally characteristic resonance in the 13C NMR spectra. Indeed, with values higher than 220 ppm, the chemical shift of a dithiocarboxylate unit is located in a spectral region where it can hardly be mistaken for anything else.
Table 1: Characteristic 1H and 13C NMR chemical shifts recorded for CAAC·CS2 and MIC·CS2 zwitterions 4a–c and 6a–f and their precursors 3a–c and 5a–f (data recorded in CDCl3 at 298 K).
| Reagent | δ NCH (ppm) | δ NCH (ppm) | Product | δ NCCS2 (ppm) | δ CS2 (ppm) |
| 3a | 8.78 | 191.3 | 4a | 188.7a | 230.5a |
| 3b | 10.7 | 193.0 | 4b | 189.0a | 227.7a |
| 3c | 9.53 | 189.7 | 4c | 188.2a | 228.8a |
| 5a | 9.03 | 130.3 | 6a | 150.5a | 225.6a |
| 5b | 8.77 | 130.6 | 6b | 150.5 | 225.0 |
| 5c | 8.90 | 130.7 | 6c | 150.2 | 225.0 |
| 5d | 9.43 | 130.0 | 6d | 150.3 | 224.9 |
| 5e | 8.46 | 130.3 | 6e | 150.3 | 225.5 |
| 5f | 8.55 | 130.5 | 6f | 150.4a | 226.3a |
aData recorded in CD2Cl2.
On average, the δ CS2 value recorded for aldiminium inner salts 4a–c (229 ppm) was slightly higher than for triazolium derivatives 6a–f (225 ppm). Previously, we had reported chemical shifts in the 220–226 ppm range for a series of imidazolinium, benzimidazolium, or imidazolium-2-dithiocarboxylate zwitterions with various aliphatic or aromatic substituents on their nitrogen atoms [40,75]. Hence, the CS2 resonance is not significantly altered by the nature of the adjacent heterocycle, in line with a lack of electronic communication between these two moieties, as further discussed below. Contrastingly, the 13C NMR resonance for the carbenoid center of all the reagents and products used in this study was clearly affected by the type of heterocycle it belonged to (Table 1). The average chemical shift for C2 was 191 ppm in aldiminium salts 3a–c and 189 ppm in inner salts 4a–c. These values are significantly higher than those recorded for NHC·CS2 zwitterions based on imidazolinium (165 ppm), benzimidazolium (152 ppm), or imidazolium (149 ppm) derivatives [75], which are surrounded by two nitrogen atoms instead of one. It is noteworthy that the C2 resonance found at ca. 130 ppm in triazolium salts 5a–f underwent a significant downfield shift to about 150 ppm in inner salts 6a–f. This is the largest variation of chemical shift observed for C2 when replacing its acidic proton with a CS2 group among all the nucleophilic carbene precursors that we have investigated so far [40,75]. Yet, we do not have an explanation for it.
On IR spectroscopy, the most intense absorption in the ATR spectra of compounds 4a–c and 6a–f was always due to the asymmetric stretching of the CS2 group (Table 2). This band was observed at wavenumbers ranging from 1037 to 1050 cm−1, down from 1052–1080 cm−1 for common imidazol(in)ium-2-dithiocarboxylate inner salts bearing aliphatic or aromatic substituents on their nitrogen atoms [40]. This shift to lower energies is a likely consequence of the greater donicity of CAACs and MICs vs NHCs. Hence, the ν̃ CS2 values recorded on IR spectroscopy constitute a more sensitive probe than the δ CS2 values obtained from 13C NMR spectroscopy to help discriminate the various types of dithiocarboxylate adducts derived from nucleophilic carbenes. More sophisticated methods, such as the determination of the Huynh electronic parameter, should, however, be better suited to evaluate more precisely the influence of substituents on the donating ability of carbene ligands [22,76,77].
Table 2: IR stretching vibrations recorded for CAAC·CS2 and MIC·CS2 zwitterions 4a–c and 6a–f in the ATR mode.
| Compound | ν̃ C=S (cm−1) | ν̃ C=C (cm−1) | ν̃ C=N (cm−1) |
| 4a | 1037 | 1424 | 1552 |
| 4b | 1050 | 1446 | 1536 |
| 4c | 1040 | 1456 | 1554 |
| 6a | 1043 | 1482, 1456 | |
| 6b | 1044 | 1465, 1440 | |
| 6c | 1045 | 1481, 1448 | |
| 6d | 1047 | 1480, 1448 | |
| 6e | 1044 | 1454 | |
| 6f | 1042 | 1455 | |
Apart from the asymmetric stretching vibration of the S=C–S− group, another strong absorption was clearly visible in the IR spectra of CAAC·CS2 betaines 4a–c. This second most intense band was observed around 1550 cm−1 (Table 2). It probably originated from the asymmetric stretching of the aldiminium group, in line with similar high intensity bands previously observed at ca. 1528 and 1477 cm−1, respectively, in the IR spectra of imidazolinium and imidazolium inner salts [40]. Contrastingly, no remarkable absorption was detected in the IR spectra of triazolium derivatives 6a–f for the CNN or NNN motifs. Yet, in all the cases, medium bands were observed in the 1400–1500 cm−1 region (Table 2). These patterns, often a doublet, were tentatively assigned to skeletal vibrations involving C=C stretching of the aromatic rings, although the intervention of asymmetrical CH deformation modes (e.g., bending or scissoring) could not be excluded.
Crystallography
Crystals of CAAC·CS2 zwitterions 4a and 4c suitable for X-ray diffraction (XRD) analysis were grown by slow diffusion of cyclohexane in a THF solution at 6 °C. Their molecular structures are depicted in Figure 3. The orange-red needles of compound 4a belonged to the trigonal ![[Graphic 1]](/bjoc/content/inline/1860-5397-19-145-i6.svg?max-width=637&scale=1.18182) space group, while the orange plates of compound 4c belonged to the monoclinic P21/c group. Due to the asymmetry of the quaternary carbon atom adjacent to the carbene center, the latter compound crystallized as a racemic mixture.
space group, while the orange plates of compound 4c belonged to the monoclinic P21/c group. Due to the asymmetry of the quaternary carbon atom adjacent to the carbene center, the latter compound crystallized as a racemic mixture.
![[1860-5397-19-145-3]](/bjoc/content/figures/1860-5397-19-145-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP representations of zwitterions 4a (CAAC-Mes-Cy·CS2, top) and 4c (CAAC-Die-MePh·CS2, bottom) with thermal ellipsoids drawn at the 50% probability level.
Figure 3: ORTEP representations of zwitterions 4a (CAAC-Mes-Cy·CS2, top) and 4c (CAAC-Die-MePh·CS2, bottom) w...
Solutions of MIC·CS2 zwitterions 6a–f in CD2Cl2 or CDCl3 employed for NMR analyses were layered with petroleum ether or n-hexane and kept at −18 °C for a few weeks. This procedure successfully afforded single crystals of products 6b and 6e suitable for XRD analysis (Figure 4). Orange prisms of zwitterion 6b belonged to the monoclinic P21/n space group, while the dark red-brown blocks of compound 6e belonged to the P21/c space group.
![[1860-5397-19-145-4]](/bjoc/content/figures/1860-5397-19-145-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP representations of zwitterions 6b (MIC-Dip-Ph-Me·CS2, top) and 6e (MIC-Mes-Bu-Me·CS2, bottom) with thermal ellipsoids drawn at the 50% probability level.
Figure 4: ORTEP representations of zwitterions 6b (MIC-Dip-Ph-Me·CS2, top) and 6e (MIC-Mes-Bu-Me·CS2, bottom)...
A comparison of the C1–S1 and C1–S2 distances recorded in the four crystal structures under scrutiny and those determined previously for 1,3-dimesitylimidazolium-2-dithiocarboxylate (IMes·CS2) and its more saturated analogue SIMes·CS2 showed that the negative charge of the CS2− unit was uniformly delocalized between the two sulfur atoms (Table 3). Moreover, the average length of 1.67 Å recorded for all the C–S bonds matched the typical distance compiled for double rather than single CS bonds (1.67 vs 1.75 Å) [78]. At (130 ± 1)°, the S1–C1–S2 bite angle of the various 1,1-dithiolate ligands was also remarkably constant. With values comprised between 87 and 126°, the S1–C1–C2–N1 dihedral angle between the CS2− moiety and the heterocyclic ring was more fluctuant, but a nearly orthogonal rather than planar disposition was maintained in all cases. Through-space attractive coulombic interactions between the opposite charges were held responsible for this orientation [79]. In addition, the presence of bulky aryl substituents in the vicinity of the sulfur atoms should further restrict their conformational freedom. As a matter of fact, the largest deviation from perpendicularity was observed in compound 6e, which featured a flexible n-butyl chain rather than a more rigid cycloalkyl or aryl group next to the attachment point of the CS2− group. The twisted nature of the zwitterions prevented any electronic communication between their anionic and cationic parts in the solid state, in line with their cross-conjugated or pseudo-cross-conjugated nature [80,81]. Indeed, an average C1–C2 distance of 1.49 Å indicated that the adducts were essentially assembled via the formation of a single rather than a double C–C bond (1.51 vs 1.34 Å) [78]. These data are in line with the trends observed on 13C NMR and IR spectroscopies for δ CS2 and ν̃ CS2 and support the hypothesis that the perpendicular arrangement between the CS2− and CCN+ units is retained in solution. Likewise, all the exocyclic C–C and C–N bond lengths in the crystal structures under examination were typical of single rather than double bonds (see for instance the N–Mes distances N1–C8 or N1–C4 in Table 3). This observation, combined with the orthogonal disposition of all the aryl substituents relative to the central heterocycle evidenced by C2–N1–C8–C9 or C2–N1–C4–C5 dihedral angles close to 90°, evidenced the lack of conjugation between the heterocyclic core of the molecules and their peripherical decorations.
Table 3: Selected bond lengths (Å) and angles (°) derived from the molecular structures of various CAAC·CS2, MIC·CS2, and NHC·CS2 zwitterions.a
| compound | C1–S1 | C1–S2 | C1–C2 |
C2–C3
(or C2–C5) |
C2–N1 |
N1–C8
(or N1–C4) |
| 4a | 1.664(2) | 1.671(2) | 1.483(3) | 1.520(2) | 1.302(3) | 1.456(2) |
| 4c | 1.675(1) | 1.661(2) | 1.487(2) | 1.529(2) | 1.302(2) | 1.465(2) |
| 6b | 1.661(1) | 1.672(1) | 1.491(2) | 1.384(2) | 1.367(1) | 1.446(1) |
| 6e | 1.680(2) | 1.674(2) | 1.486(3) | 1.383(2) | 1.365(2) | 1.449(2) |
| IMes·CS2b,c | 1.667(3) | 1.667(3) | 1.489(7) | / | 1.336(5) | 1.461(6) |
| SIMes·CS2b,d | 1.662(2) | 1.662(2) | 1.502(6) | / | 1.315(4) | 1.446(4) |
| compound | S1–C1–S2 |
N1–C2–C5
(or N1–C2–C3) |
S1–C1–C2–N1 |
N1–C3–C4–C5
(or N1–N2–N3–C3) |
C2–N1–C8–C9
(or C2–N1–C4–C5) |
|
| 4a | 131.4(2) | 112.2(1) | 86.9(2) | −23.0(2) | 85.6(2) | |
| 4c | 131.17(9) | 112.4(1) | 91.6(2) | 12.2(2) | 90.1(2) | |
| 6b | 129.00(7) | 104.57(9) | 87.7(1) | 0.3(1) | 94.2(1) | |
| 6e | 128.5(1) | 104.9(1) | 126.3(2) | 0.8(2) | 100.1(2) | |
| IMes·CS2b,c | 129.1(4) | 107.2(4) | 114.7(2) | 0.5(5) | 104.9(6) | |
| SIMes·CS2b,d | 130.3(3) | 112.0(4) | 92.4(2) | 9.5(5) | 94.4(5) | |
aSee Figure 3 and Figure 4 for atom labeling. bData from ref. [40]. cIMes·CS2 crystallized with two molecules in the asymmetric unit. dOnly half of the molecule of SIMes·CS2 formed the asymmetric unit.
Conclusion
The synthesis of three CAAC·CS2 and six MIC·CS2 zwitterions derived from aldiminium or 1,2,3-triazolium salts was achieved via a two-step procedure involving the in situ generation of free carbenes with a strong base, followed by their nucleophilic addition onto carbon disulfide. The nine products obtained were characterized by 1H and 13C NMR spectroscopy, FTIR spectroscopy, HR–ESI mass spectrometry, and elemental analysis. Moreover, the molecular structures of two CAAC·CS2 and two MIC·CS2 betaines were determined by X-ray diffraction analysis. The various analytical data recorded for all these compounds were compared with those reported previously for related NHC·CS2 zwitterions derived from imidazolinium or (benz)imidazolium salts.
Due to the absence of electronic communication between the CS2 unit and the orthogonal heterocycle, all the CAAC·CS2, MIC·CS2, and NHC·CS2 zwitterions under scrutiny displayed rather similar electronic properties and featured the same bite angle. Yet, their steric properties are liable to ample modifications by varying the nature of the cationic heterocycle and its substituents. The synthesis of 1,2,3-triazolium salts via a “click” reaction is a particularly attractive and straightforward strategy to prepare dithiocarboxylate zwitterions with two different alkyl or aryl groups flanking the carbenoid center and the adjacent CS2 unit. This is in sharp contrast with the most common cyclization processes leading to imidazol(in)ium derivatives, which afford symmetrical products with identical substituents on both nitrogen atoms [82]. Although cyclic aldiminium salts are less readily available, they feature a quaternary carbon atom next to the carbenoid center that may act as a source of chirality. Thus, the novel compounds reported in this study represent a valuable addition to the family of neutral dithiolate ligands derived from stable nucleophilic carbenes, and we are currently investigating their coordination chemistry toward various transition metals. Details of these studies will be disclosed in a forthcoming publication.
Supporting Information
| Supporting Information File 1: Experimental procedures, X-ray crystal structure determinations, copies of 1H NMR, 13C NMR, and FTIR spectra. | ||
| Format: PDF | Size: 3.5 MB | Download |
Acknowledgements
The authors would like to thank Apeiron Synthesis for a gift of aldiminium tetrafluoroborate salts 3a and 3c, Mr. Lucas Langue and Dr. Jan Lorkowski for their help with the synthesis of triazolium iodides 5a–f, Ms. Patricia Mestdag for her involvement in this work, Mr. Stéphane Luts and Dr. Cédric Malherbe for the FTIR analyses, and RIAIDT-USC for the use of its analytical facilities.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Arduengo, A. J., III; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361–363. doi:10.1021/ja00001a054
Return to citation in text: [1] -
Bertrand, G., Ed. Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents; Marcel Dekker: New York, NY, USA, 2002.
Return to citation in text: [1] -
Nolan, S. P., Ed. N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527671229
Return to citation in text: [1] -
Díez-González, S., Ed. N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, 2nd ed.; RSC Catalysis Series, Vol. 27; Royal Society of Chemistry: Cambridge, UK, 2017. doi:10.1039/9781782626817
Return to citation in text: [1] -
Nolan, S. P.; Cazin, C. S. J., Eds. N-Heterocyclic Carbenes in Catalytic Organic Synthesis; Science of Synthesis; Thieme: Stuttgart, Germany, 2017. doi:10.1055/b-004-132254
Return to citation in text: [1] -
Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z
Return to citation in text: [1] -
Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165
Return to citation in text: [1] [2] -
Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. doi:10.1002/anie.200501841
Return to citation in text: [1] [2] -
Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2015, 48, 256–266. doi:10.1021/ar5003494
Return to citation in text: [1] -
Melaimi, M.; Jazzar, R.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2017, 56, 10046–10068. doi:10.1002/anie.201702148
Return to citation in text: [1] -
Morvan, J.; Mauduit, M.; Bertrand, G.; Jazzar, R. ACS Catal. 2021, 11, 1714–1748. doi:10.1021/acscatal.0c05508
Return to citation in text: [1] -
Singh, R. K.; Khan, T. K.; Misra, S.; Singh, A. K. J. Organomet. Chem. 2021, 956, 122133. doi:10.1016/j.jorganchem.2021.122133
Return to citation in text: [1] -
Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/c1cc16460k
Return to citation in text: [1] -
César, V.; Tourneux, J.-C.; Vujkovic, N.; Brousses, R.; Lugan, N.; Lavigne, G. Chem. Commun. 2012, 48, 2349–2351. doi:10.1039/c2cc17870b
Return to citation in text: [1] -
Mummel, S.; Lederle, F.; Hübner, E. G.; Namyslo, J. C.; Nieger, M.; Schmidt, A. Angew. Chem., Int. Ed. 2021, 60, 18882–18887. doi:10.1002/anie.202107495
Return to citation in text: [1] -
Mummel, S.; Lederle, F.; Hübner, E. G.; Namyslo, J. C.; Nieger, M.; Schmidt, A. Eur. J. Org. Chem. 2023, 26, e202300216. doi:10.1002/ejoc.202300216
Return to citation in text: [1] -
Gründemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J. W.; Crabtree, R. H. Chem. Commun. 2001, 2274–2275. doi:10.1039/b107881j
Return to citation in text: [1] -
Gründemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J. W.; Crabtree, R. H. J. Am. Chem. Soc. 2002, 124, 10473–10481. doi:10.1021/ja026735g
Return to citation in text: [1] -
Crabtree, R. H. Coord. Chem. Rev. 2013, 257, 755–766. doi:10.1016/j.ccr.2012.09.006
Return to citation in text: [1] -
Han, Y.; Huynh, H. V. Dalton Trans. 2011, 40, 2141–2147. doi:10.1039/c0dt01037e
Return to citation in text: [1] -
Iglesias, M.; Albrecht, M. Dalton Trans. 2010, 39, 5213–5215. doi:10.1039/c0dt00027b
Return to citation in text: [1] -
Teng, Q.; Huynh, H. V. Dalton Trans. 2017, 46, 614–627. doi:10.1039/c6dt04222h
Return to citation in text: [1] [2] -
Mathew, P.; Neels, A.; Albrecht, M. J. Am. Chem. Soc. 2008, 130, 13534–13535. doi:10.1021/ja805781s
Return to citation in text: [1] -
Crowley, J. D.; Lee, A.-L.; Kilpin, K. J. Aust. J. Chem. 2011, 64, 1118–1132. doi:10.1071/ch11185
Return to citation in text: [1] [2] -
Donnelly, K. F.; Petronilho, A.; Albrecht, M. Chem. Commun. 2013, 49, 1145–1159. doi:10.1039/c2cc37881g
Return to citation in text: [1] [2] [3] [4] -
Marichev, K. O.; Patil, S. A.; Bugarin, A. Tetrahedron 2018, 74, 2523–2546. doi:10.1016/j.tet.2018.04.013
Return to citation in text: [1] [2] -
Vivancos, Á.; Segarra, C.; Albrecht, M. Chem. Rev. 2018, 118, 9493–9586. doi:10.1021/acs.chemrev.8b00148
Return to citation in text: [1] [2] -
Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2018, 51, 3236–3244. doi:10.1021/acs.accounts.8b00480
Return to citation in text: [1] [2] -
Delaude, L. Eur. J. Inorg. Chem. 2009, 1681–1699. doi:10.1002/ejic.200801227
Return to citation in text: [1] -
Sheldrick, W. S.; Schönberg, A.; Singer, E.; Eckert, P. Chem. Ber. 1980, 113, 3605–3609. doi:10.1002/cber.19801131118
Return to citation in text: [1] -
Krasuski, W.; Nikolaus, D.; Regitz, M. Liebigs Ann. Chem. 1982, 1451–1465. doi:10.1002/jlac.198219820805
Return to citation in text: [1] -
Kuhn, N.; Bohnen, H.; Henkel, G. Z. Naturforsch., B: J. Chem. Sci. 1994, 49, 1473–1480. doi:10.1515/znb-1994-1105
Return to citation in text: [1] -
Küçükbay, H.; Çetinkaya, E.; Durmaz, R. Arzneim. Forsch. 1995, 45, 1331–1334.
Return to citation in text: [1] -
Enders, D.; Breuer, K.; Runsink, J.; Teles, J. H. Liebigs Ann. 1996, 2019–2028. doi:10.1002/jlac.199619961212
Return to citation in text: [1] -
Kuhn, N.; Niquet, E.; Steimann, M.; Walker, I. Z. Naturforsch., B: J. Chem. Sci. 1999, 54, 1181–1187. doi:10.1515/znb-1999-0915
Return to citation in text: [1] -
Küçükbay, H.; Durmaz, R.; Orhan, E.; Günal, S. Farmaco 2003, 58, 431–437. doi:10.1016/s0014-827x(03)00068-5
Return to citation in text: [1] -
Akkurt, M.; Öztürk, S.; Küçükbay, H.; Orhan, E.; Büyükgüngör, O. Acta Crystallogr., Sect. E: Struct. Rep. Online 2004, 60, o219–o221. doi:10.1107/s160053680400073x
Return to citation in text: [1] -
Nyce, G. W.; Csihony, S.; Waymouth, R. M.; Hedrick, J. L. Chem. – Eur. J. 2004, 10, 4073–4079. doi:10.1002/chem.200400196
Return to citation in text: [1] -
Sereda, O.; Blanrue, A.; Wilhelm, R. Chem. Commun. 2009, 1040–1042. doi:10.1039/b817991c
Return to citation in text: [1] [2] -
Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Dagmara Konieczna, D.; Blanrue, A.; Wilhelm, R. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 596–604. doi:10.5560/znb.2014-4014
Return to citation in text: [1] [2] -
Aydogan Gokturk, P.; Donmez, S. E.; Ulgut, B.; Türkmen, Y. E.; Suzer, S. New J. Chem. 2017, 41, 10299–10304. doi:10.1039/c7nj01996c
Return to citation in text: [1] [2] -
Yılmaz, Ü.; Küçükbay, H. J. Turk. Chem. Soc., Sect. A 2018, 5, 1037–1042. doi:10.18596/jotcsa.447056
Return to citation in text: [1] -
Fujihara, T.; Sugaya, T.; Nagasawa, A.; Nakayama, J. Acta Crystallogr., Sect. E: Struct. Rep. Online 2004, 60, m282–m284. doi:10.1107/s160053680400217x
Return to citation in text: [1] -
Naeem, S.; Delaude, L.; White, A. J. P.; Wilton-Ely, J. D. E. T. Inorg. Chem. 2010, 49, 1784–1793. doi:10.1021/ic9021504
Return to citation in text: [1] [2] [3] -
Borer, L. L.; Kong, J.; Sinn, E. Inorg. Chim. Acta 1986, 122, 145–148. doi:10.1016/s0020-1693(00)81631-x
Return to citation in text: [1] -
Delaude, L.; Sauvage, X.; Demonceau, A.; Wouters, J. Organometallics 2009, 28, 4056–4064. doi:10.1021/om9002363
Return to citation in text: [1] -
Naeem, S.; Thompson, A. L.; Delaude, L.; Wilton‐Ely, J. D. E. T. Chem. – Eur. J. 2010, 16, 10971–10974. doi:10.1002/chem.201001235
Return to citation in text: [1] -
Naeem, S.; Thompson, A. L.; White, A. J. P.; Delaude, L.; Wilton-Ely, J. D. E. T. Dalton Trans. 2011, 40, 3737–3747. doi:10.1039/c1dt10048c
Return to citation in text: [1] -
Champion, M. J. D.; Solanki, R.; Delaude, L.; White, A. J. P.; Wilton-Ely, J. D. E. T. Dalton Trans. 2012, 41, 12386–12394. doi:10.1039/c2dt31413d
Return to citation in text: [1] -
Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2016, 45, 18346–18355. doi:10.1039/c6dt03428d
Return to citation in text: [1] [2] [3] -
Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 1779–1788. doi:10.1039/c6dt04780g
Return to citation in text: [1] [2] [3] -
Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 9036–9048. doi:10.1039/c7dt01889d
Return to citation in text: [1] -
Beltrán, T. F.; Zaragoza, G.; Delaude, L. Polyhedron 2021, 197, 115055. doi:10.1016/j.poly.2021.115055
Return to citation in text: [1] -
Zain Aldin, M.; Zaragoza, G.; Deschamps, W.; Tomani, J.-C. D.; Souopgui, J.; Delaude, L. Inorg. Chem. 2021, 60, 16769–16781. doi:10.1021/acs.inorgchem.1c02648
Return to citation in text: [1] -
Ortmeyer, J.; Flörke, U.; Henkel, G.; Wilhelm, R.; Neuba, A. Eur. J. Inorg. Chem. 2017, 3191–3197. doi:10.1002/ejic.201700328
Return to citation in text: [1] -
Neuba, A.; Ortmeyer, J.; Konieczna, D. D.; Weigel, G.; Flörke, U.; Henkel, G.; Wilhelm, R. RSC Adv. 2015, 5, 9217–9220. doi:10.1039/c4ra09033k
Return to citation in text: [1] -
Siemeling, U.; Memczak, H.; Bruhn, C.; Vogel, F.; Träger, F.; Baio, J. E.; Weidner, T. Dalton Trans. 2012, 41, 2986–2994. doi:10.1039/c2dt11976e
Return to citation in text: [1] [2] -
Cabeza, J. A.; García‐Álvarez, P.; Guadalupe Hernández‐Cruz, M. Eur. J. Inorg. Chem. 2012, 2928–2932. doi:10.1002/ejic.201200245
Return to citation in text: [1] -
Shi, Y.-C.; Shi, Y. Inorg. Chim. Acta 2015, 434, 92–96. doi:10.1016/j.ica.2015.04.024
Return to citation in text: [1] -
Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 13002–13009. doi:10.1039/c7dt03202a
Return to citation in text: [1] -
Kuchenbeiser, G.; Soleilhavoup, M.; Donnadieu, B.; Bertrand, G. Chem. – Asian J. 2009, 4, 1745–1750. doi:10.1002/asia.200900338
Return to citation in text: [1] -
Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s
Return to citation in text: [1] -
Bock, V. D.; Hiemstra, H.; van Maarseveen, J. H. Eur. J. Org. Chem. 2006, 51–68. doi:10.1002/ejoc.200500483
Return to citation in text: [1] -
Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479
Return to citation in text: [1] -
Barral, K.; Moorhouse, A. D.; Moses, J. E. Org. Lett. 2007, 9, 1809–1811. doi:10.1021/ol070527h
Return to citation in text: [1] -
Lorkowski, J.; Żak, P.; Kubicki, M.; Pietraszuk, C.; Jędrzkiewicz, D.; Ejfler, J. New J. Chem. 2018, 42, 10134–10141. doi:10.1039/c8nj00981c
Return to citation in text: [1] -
Brown, D. G.; Sanguantrakun, N.; Schulze, B.; Schubert, U. S.; Berlinguette, C. P. J. Am. Chem. Soc. 2012, 134, 12354–12357. doi:10.1021/ja3039536
Return to citation in text: [1] -
Guisado‐Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759–4762. doi:10.1002/anie.201001864
Return to citation in text: [1] [2] [3] -
Konstandaras, N.; Dunn, M. H.; Guerry, M. S.; Barnett, C. D.; Cole, M. L.; Harper, J. B. Org. Biomol. Chem. 2020, 18, 66–75. doi:10.1039/c9ob02258a
Return to citation in text: [1] [2] -
Bouffard, J.; Keitz, B. K.; Tonner, R.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R. H.; Bertrand, G. Organometallics 2011, 30, 2617–2627. doi:10.1021/om200272m
Return to citation in text: [1] [2] -
Yamaguchi, K.; Sonoda, O.; Minoura, Y. J. Polym. Sci., Part A-1: Polym. Chem. 1972, 10, 63–76. doi:10.1002/pol.1972.150100105
Return to citation in text: [1] -
Yavari, I.; Seyfi, S.; Hossaini, Z. Tetrahedron Lett. 2010, 51, 2193–2194. doi:10.1016/j.tetlet.2010.02.107
Return to citation in text: [1] -
Golabi, P.; Akbarzadeh, R.; Dehghani, H. J. Alloys Compd. 2015, 647, 539–547. doi:10.1016/j.jallcom.2015.06.135
Return to citation in text: [1] -
Mazars, F.; Hrubaru, M.; Tumanov, N.; Wouters, J.; Delaude, L. Eur. J. Org. Chem. 2021, 2025–2033. doi:10.1002/ejoc.202100274
Return to citation in text: [1] [2] [3] [4] -
Yuan, D.; Huynh, H. V. Organometallics 2012, 31, 405–412. doi:10.1021/om2010029
Return to citation in text: [1] -
Huynh, H. V. Chem. Rev. 2018, 118, 9457–9492. doi:10.1021/acs.chemrev.8b00067
Return to citation in text: [1] -
Allen, F. H.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. Typical interatomic distances: organic compounds. International Tables for Crystallography; International Union of Crystallography: Chester, UK, 2006; Vol. C, pp 790–811. doi:10.1107/97809553602060000621
Return to citation in text: [1] [2] -
Nakayama, J.; Kitahara, T.; Sugihara, Y.; Sakamoto, A.; Ishii, A. J. Am. Chem. Soc. 2000, 122, 9120–9126. doi:10.1021/ja001213r
Return to citation in text: [1] -
Schmidt, A. Adv. Heterocycl. Chem. 2003, 85, 67–171. doi:10.1016/s0065-2725(03)85002-x
Return to citation in text: [1] -
Ramsden, C. A., Ed. Heterocyclic Mesomeric Betaines and Mesoionic Compounds; Advances in Heterocyclic Chemistry, Vol. 137; Academic Press: Cambridge, MA, USA, 2022. doi:10.1016/s0065-2725(22)00027-7
Return to citation in text: [1] -
Hans, M.; Lorkowski, J.; Demonceau, A.; Delaude, L. Beilstein J. Org. Chem. 2015, 11, 2318–2325. doi:10.3762/bjoc.11.252
Return to citation in text: [1]
| 39. | Sereda, O.; Blanrue, A.; Wilhelm, R. Chem. Commun. 2009, 1040–1042. doi:10.1039/b817991c |
| 41. | Dagmara Konieczna, D.; Blanrue, A.; Wilhelm, R. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 596–604. doi:10.5560/znb.2014-4014 |
| 42. | Aydogan Gokturk, P.; Donmez, S. E.; Ulgut, B.; Türkmen, Y. E.; Suzer, S. New J. Chem. 2017, 41, 10299–10304. doi:10.1039/c7nj01996c |
| 58. | Siemeling, U.; Memczak, H.; Bruhn, C.; Vogel, F.; Träger, F.; Baio, J. E.; Weidner, T. Dalton Trans. 2012, 41, 2986–2994. doi:10.1039/c2dt11976e |
| 63. | Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s |
| 64. | Bock, V. D.; Hiemstra, H.; van Maarseveen, J. H. Eur. J. Org. Chem. 2006, 51–68. doi:10.1002/ejoc.200500483 |
| 65. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 24. | Crowley, J. D.; Lee, A.-L.; Kilpin, K. J. Aust. J. Chem. 2011, 64, 1118–1132. doi:10.1071/ch11185 |
| 25. | Donnelly, K. F.; Petronilho, A.; Albrecht, M. Chem. Commun. 2013, 49, 1145–1159. doi:10.1039/c2cc37881g |
| 26. | Marichev, K. O.; Patil, S. A.; Bugarin, A. Tetrahedron 2018, 74, 2523–2546. doi:10.1016/j.tet.2018.04.013 |
| 27. | Vivancos, Á.; Segarra, C.; Albrecht, M. Chem. Rev. 2018, 118, 9493–9586. doi:10.1021/acs.chemrev.8b00148 |
| 28. | Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2018, 51, 3236–3244. doi:10.1021/acs.accounts.8b00480 |
| 75. | Mazars, F.; Hrubaru, M.; Tumanov, N.; Wouters, J.; Delaude, L. Eur. J. Org. Chem. 2021, 2025–2033. doi:10.1002/ejoc.202100274 |
| 25. | Donnelly, K. F.; Petronilho, A.; Albrecht, M. Chem. Commun. 2013, 49, 1145–1159. doi:10.1039/c2cc37881g |
| 69. | Guisado‐Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759–4762. doi:10.1002/anie.201001864 |
| 70. | Konstandaras, N.; Dunn, M. H.; Guerry, M. S.; Barnett, C. D.; Cole, M. L.; Harper, J. B. Org. Biomol. Chem. 2020, 18, 66–75. doi:10.1039/c9ob02258a |
| 71. | Bouffard, J.; Keitz, B. K.; Tonner, R.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R. H.; Bertrand, G. Organometallics 2011, 30, 2617–2627. doi:10.1021/om200272m |
| 71. | Bouffard, J.; Keitz, B. K.; Tonner, R.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R. H.; Bertrand, G. Organometallics 2011, 30, 2617–2627. doi:10.1021/om200272m |
| 72. | Yamaguchi, K.; Sonoda, O.; Minoura, Y. J. Polym. Sci., Part A-1: Polym. Chem. 1972, 10, 63–76. doi:10.1002/pol.1972.150100105 |
| 73. | Yavari, I.; Seyfi, S.; Hossaini, Z. Tetrahedron Lett. 2010, 51, 2193–2194. doi:10.1016/j.tetlet.2010.02.107 |
| 74. | Golabi, P.; Akbarzadeh, R.; Dehghani, H. J. Alloys Compd. 2015, 647, 539–547. doi:10.1016/j.jallcom.2015.06.135 |
| 69. | Guisado‐Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759–4762. doi:10.1002/anie.201001864 |
| 25. | Donnelly, K. F.; Petronilho, A.; Albrecht, M. Chem. Commun. 2013, 49, 1145–1159. doi:10.1039/c2cc37881g |
| 69. | Guisado‐Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759–4762. doi:10.1002/anie.201001864 |
| 70. | Konstandaras, N.; Dunn, M. H.; Guerry, M. S.; Barnett, C. D.; Cole, M. L.; Harper, J. B. Org. Biomol. Chem. 2020, 18, 66–75. doi:10.1039/c9ob02258a |
| 66. | Barral, K.; Moorhouse, A. D.; Moses, J. E. Org. Lett. 2007, 9, 1809–1811. doi:10.1021/ol070527h |
| 67. | Lorkowski, J.; Żak, P.; Kubicki, M.; Pietraszuk, C.; Jędrzkiewicz, D.; Ejfler, J. New J. Chem. 2018, 42, 10134–10141. doi:10.1039/c8nj00981c |
| 68. | Brown, D. G.; Sanguantrakun, N.; Schulze, B.; Schubert, U. S.; Berlinguette, C. P. J. Am. Chem. Soc. 2012, 134, 12354–12357. doi:10.1021/ja3039536 |
| 40. | Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110 |
| 75. | Mazars, F.; Hrubaru, M.; Tumanov, N.; Wouters, J.; Delaude, L. Eur. J. Org. Chem. 2021, 2025–2033. doi:10.1002/ejoc.202100274 |
| 75. | Mazars, F.; Hrubaru, M.; Tumanov, N.; Wouters, J.; Delaude, L. Eur. J. Org. Chem. 2021, 2025–2033. doi:10.1002/ejoc.202100274 |
| 40. | Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110 |
| 75. | Mazars, F.; Hrubaru, M.; Tumanov, N.; Wouters, J.; Delaude, L. Eur. J. Org. Chem. 2021, 2025–2033. doi:10.1002/ejoc.202100274 |
| 78. | Allen, F. H.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. Typical interatomic distances: organic compounds. International Tables for Crystallography; International Union of Crystallography: Chester, UK, 2006; Vol. C, pp 790–811. doi:10.1107/97809553602060000621 |
| 40. | Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110 |
| 79. | Nakayama, J.; Kitahara, T.; Sugihara, Y.; Sakamoto, A.; Ishii, A. J. Am. Chem. Soc. 2000, 122, 9120–9126. doi:10.1021/ja001213r |
| 80. | Schmidt, A. Adv. Heterocycl. Chem. 2003, 85, 67–171. doi:10.1016/s0065-2725(03)85002-x |
| 81. | Ramsden, C. A., Ed. Heterocyclic Mesomeric Betaines and Mesoionic Compounds; Advances in Heterocyclic Chemistry, Vol. 137; Academic Press: Cambridge, MA, USA, 2022. doi:10.1016/s0065-2725(22)00027-7 |
| 40. | Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110 |
| 78. | Allen, F. H.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. Typical interatomic distances: organic compounds. International Tables for Crystallography; International Union of Crystallography: Chester, UK, 2006; Vol. C, pp 790–811. doi:10.1107/97809553602060000621 |
| 40. | Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110 |
| 22. | Teng, Q.; Huynh, H. V. Dalton Trans. 2017, 46, 614–627. doi:10.1039/c6dt04222h |
| 76. | Yuan, D.; Huynh, H. V. Organometallics 2012, 31, 405–412. doi:10.1021/om2010029 |
| 77. | Huynh, H. V. Chem. Rev. 2018, 118, 9457–9492. doi:10.1021/acs.chemrev.8b00067 |
| 82. | Hans, M.; Lorkowski, J.; Demonceau, A.; Delaude, L. Beilstein J. Org. Chem. 2015, 11, 2318–2325. doi:10.3762/bjoc.11.252 |
| 1. | Arduengo, A. J., III; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361–363. doi:10.1021/ja00001a054 |
| 7. | Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165 |
| 24. | Crowley, J. D.; Lee, A.-L.; Kilpin, K. J. Aust. J. Chem. 2011, 64, 1118–1132. doi:10.1071/ch11185 |
| 25. | Donnelly, K. F.; Petronilho, A.; Albrecht, M. Chem. Commun. 2013, 49, 1145–1159. doi:10.1039/c2cc37881g |
| 26. | Marichev, K. O.; Patil, S. A.; Bugarin, A. Tetrahedron 2018, 74, 2523–2546. doi:10.1016/j.tet.2018.04.013 |
| 27. | Vivancos, Á.; Segarra, C.; Albrecht, M. Chem. Rev. 2018, 118, 9493–9586. doi:10.1021/acs.chemrev.8b00148 |
| 28. | Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2018, 51, 3236–3244. doi:10.1021/acs.accounts.8b00480 |
| 6. | Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z |
| 3. | Nolan, S. P., Ed. N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527671229 |
| 4. | Díez-González, S., Ed. N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, 2nd ed.; RSC Catalysis Series, Vol. 27; Royal Society of Chemistry: Cambridge, UK, 2017. doi:10.1039/9781782626817 |
| 5. | Nolan, S. P.; Cazin, C. S. J., Eds. N-Heterocyclic Carbenes in Catalytic Organic Synthesis; Science of Synthesis; Thieme: Stuttgart, Germany, 2017. doi:10.1055/b-004-132254 |
| 22. | Teng, Q.; Huynh, H. V. Dalton Trans. 2017, 46, 614–627. doi:10.1039/c6dt04222h |
| 2. | Bertrand, G., Ed. Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents; Marcel Dekker: New York, NY, USA, 2002. |
| 23. | Mathew, P.; Neels, A.; Albrecht, M. J. Am. Chem. Soc. 2008, 130, 13534–13535. doi:10.1021/ja805781s |
| 17. | Gründemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J. W.; Crabtree, R. H. Chem. Commun. 2001, 2274–2275. doi:10.1039/b107881j |
| 18. | Gründemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J. W.; Crabtree, R. H. J. Am. Chem. Soc. 2002, 124, 10473–10481. doi:10.1021/ja026735g |
| 20. | Han, Y.; Huynh, H. V. Dalton Trans. 2011, 40, 2141–2147. doi:10.1039/c0dt01037e |
| 13. | Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/c1cc16460k |
| 14. | César, V.; Tourneux, J.-C.; Vujkovic, N.; Brousses, R.; Lugan, N.; Lavigne, G. Chem. Commun. 2012, 48, 2349–2351. doi:10.1039/c2cc17870b |
| 15. | Mummel, S.; Lederle, F.; Hübner, E. G.; Namyslo, J. C.; Nieger, M.; Schmidt, A. Angew. Chem., Int. Ed. 2021, 60, 18882–18887. doi:10.1002/anie.202107495 |
| 16. | Mummel, S.; Lederle, F.; Hübner, E. G.; Namyslo, J. C.; Nieger, M.; Schmidt, A. Eur. J. Org. Chem. 2023, 26, e202300216. doi:10.1002/ejoc.202300216 |
| 21. | Iglesias, M.; Albrecht, M. Dalton Trans. 2010, 39, 5213–5215. doi:10.1039/c0dt00027b |
| 9. | Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2015, 48, 256–266. doi:10.1021/ar5003494 |
| 10. | Melaimi, M.; Jazzar, R.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2017, 56, 10046–10068. doi:10.1002/anie.201702148 |
| 11. | Morvan, J.; Mauduit, M.; Bertrand, G.; Jazzar, R. ACS Catal. 2021, 11, 1714–1748. doi:10.1021/acscatal.0c05508 |
| 12. | Singh, R. K.; Khan, T. K.; Misra, S.; Singh, A. K. J. Organomet. Chem. 2021, 956, 122133. doi:10.1016/j.jorganchem.2021.122133 |
| 8. | Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. doi:10.1002/anie.200501841 |
| 7. | Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165 |
| 19. | Crabtree, R. H. Coord. Chem. Rev. 2013, 257, 755–766. doi:10.1016/j.ccr.2012.09.006 |
| 46. | Borer, L. L.; Kong, J.; Sinn, E. Inorg. Chim. Acta 1986, 122, 145–148. doi:10.1016/s0020-1693(00)81631-x |
| 47. | Delaude, L.; Sauvage, X.; Demonceau, A.; Wouters, J. Organometallics 2009, 28, 4056–4064. doi:10.1021/om9002363 |
| 48. | Naeem, S.; Thompson, A. L.; Delaude, L.; Wilton‐Ely, J. D. E. T. Chem. – Eur. J. 2010, 16, 10971–10974. doi:10.1002/chem.201001235 |
| 49. | Naeem, S.; Thompson, A. L.; White, A. J. P.; Delaude, L.; Wilton-Ely, J. D. E. T. Dalton Trans. 2011, 40, 3737–3747. doi:10.1039/c1dt10048c |
| 50. | Champion, M. J. D.; Solanki, R.; Delaude, L.; White, A. J. P.; Wilton-Ely, J. D. E. T. Dalton Trans. 2012, 41, 12386–12394. doi:10.1039/c2dt31413d |
| 51. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2016, 45, 18346–18355. doi:10.1039/c6dt03428d |
| 52. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 1779–1788. doi:10.1039/c6dt04780g |
| 53. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 9036–9048. doi:10.1039/c7dt01889d |
| 54. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Polyhedron 2021, 197, 115055. doi:10.1016/j.poly.2021.115055 |
| 55. | Zain Aldin, M.; Zaragoza, G.; Deschamps, W.; Tomani, J.-C. D.; Souopgui, J.; Delaude, L. Inorg. Chem. 2021, 60, 16769–16781. doi:10.1021/acs.inorgchem.1c02648 |
| 30. | Sheldrick, W. S.; Schönberg, A.; Singer, E.; Eckert, P. Chem. Ber. 1980, 113, 3605–3609. doi:10.1002/cber.19801131118 |
| 31. | Krasuski, W.; Nikolaus, D.; Regitz, M. Liebigs Ann. Chem. 1982, 1451–1465. doi:10.1002/jlac.198219820805 |
| 32. | Kuhn, N.; Bohnen, H.; Henkel, G. Z. Naturforsch., B: J. Chem. Sci. 1994, 49, 1473–1480. doi:10.1515/znb-1994-1105 |
| 33. | Küçükbay, H.; Çetinkaya, E.; Durmaz, R. Arzneim. Forsch. 1995, 45, 1331–1334. |
| 34. | Enders, D.; Breuer, K.; Runsink, J.; Teles, J. H. Liebigs Ann. 1996, 2019–2028. doi:10.1002/jlac.199619961212 |
| 35. | Kuhn, N.; Niquet, E.; Steimann, M.; Walker, I. Z. Naturforsch., B: J. Chem. Sci. 1999, 54, 1181–1187. doi:10.1515/znb-1999-0915 |
| 36. | Küçükbay, H.; Durmaz, R.; Orhan, E.; Günal, S. Farmaco 2003, 58, 431–437. doi:10.1016/s0014-827x(03)00068-5 |
| 37. | Akkurt, M.; Öztürk, S.; Küçükbay, H.; Orhan, E.; Büyükgüngör, O. Acta Crystallogr., Sect. E: Struct. Rep. Online 2004, 60, o219–o221. doi:10.1107/s160053680400073x |
| 38. | Nyce, G. W.; Csihony, S.; Waymouth, R. M.; Hedrick, J. L. Chem. – Eur. J. 2004, 10, 4073–4079. doi:10.1002/chem.200400196 |
| 39. | Sereda, O.; Blanrue, A.; Wilhelm, R. Chem. Commun. 2009, 1040–1042. doi:10.1039/b817991c |
| 40. | Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, 1882–1891. doi:10.1002/ejic.200801110 |
| 41. | Dagmara Konieczna, D.; Blanrue, A.; Wilhelm, R. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 596–604. doi:10.5560/znb.2014-4014 |
| 42. | Aydogan Gokturk, P.; Donmez, S. E.; Ulgut, B.; Türkmen, Y. E.; Suzer, S. New J. Chem. 2017, 41, 10299–10304. doi:10.1039/c7nj01996c |
| 43. | Yılmaz, Ü.; Küçükbay, H. J. Turk. Chem. Soc., Sect. A 2018, 5, 1037–1042. doi:10.18596/jotcsa.447056 |
| 44. | Fujihara, T.; Sugaya, T.; Nagasawa, A.; Nakayama, J. Acta Crystallogr., Sect. E: Struct. Rep. Online 2004, 60, m282–m284. doi:10.1107/s160053680400217x |
| 45. | Naeem, S.; Delaude, L.; White, A. J. P.; Wilton-Ely, J. D. E. T. Inorg. Chem. 2010, 49, 1784–1793. doi:10.1021/ic9021504 |
| 62. | Kuchenbeiser, G.; Soleilhavoup, M.; Donnadieu, B.; Bertrand, G. Chem. – Asian J. 2009, 4, 1745–1750. doi:10.1002/asia.200900338 |
| 8. | Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. doi:10.1002/anie.200501841 |
| 45. | Naeem, S.; Delaude, L.; White, A. J. P.; Wilton-Ely, J. D. E. T. Inorg. Chem. 2010, 49, 1784–1793. doi:10.1021/ic9021504 |
| 59. | Cabeza, J. A.; García‐Álvarez, P.; Guadalupe Hernández‐Cruz, M. Eur. J. Inorg. Chem. 2012, 2928–2932. doi:10.1002/ejic.201200245 |
| 60. | Shi, Y.-C.; Shi, Y. Inorg. Chim. Acta 2015, 434, 92–96. doi:10.1016/j.ica.2015.04.024 |
| 61. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 13002–13009. doi:10.1039/c7dt03202a |
| 57. | Neuba, A.; Ortmeyer, J.; Konieczna, D. D.; Weigel, G.; Flörke, U.; Henkel, G.; Wilhelm, R. RSC Adv. 2015, 5, 9217–9220. doi:10.1039/c4ra09033k |
| 58. | Siemeling, U.; Memczak, H.; Bruhn, C.; Vogel, F.; Träger, F.; Baio, J. E.; Weidner, T. Dalton Trans. 2012, 41, 2986–2994. doi:10.1039/c2dt11976e |
| 45. | Naeem, S.; Delaude, L.; White, A. J. P.; Wilton-Ely, J. D. E. T. Inorg. Chem. 2010, 49, 1784–1793. doi:10.1021/ic9021504 |
| 51. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2016, 45, 18346–18355. doi:10.1039/c6dt03428d |
| 52. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 1779–1788. doi:10.1039/c6dt04780g |
| 51. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2016, 45, 18346–18355. doi:10.1039/c6dt03428d |
| 52. | Beltrán, T. F.; Zaragoza, G.; Delaude, L. Dalton Trans. 2017, 46, 1779–1788. doi:10.1039/c6dt04780g |
| 56. | Ortmeyer, J.; Flörke, U.; Henkel, G.; Wilhelm, R.; Neuba, A. Eur. J. Inorg. Chem. 2017, 3191–3197. doi:10.1002/ejic.201700328 |
© 2023 Touj et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.