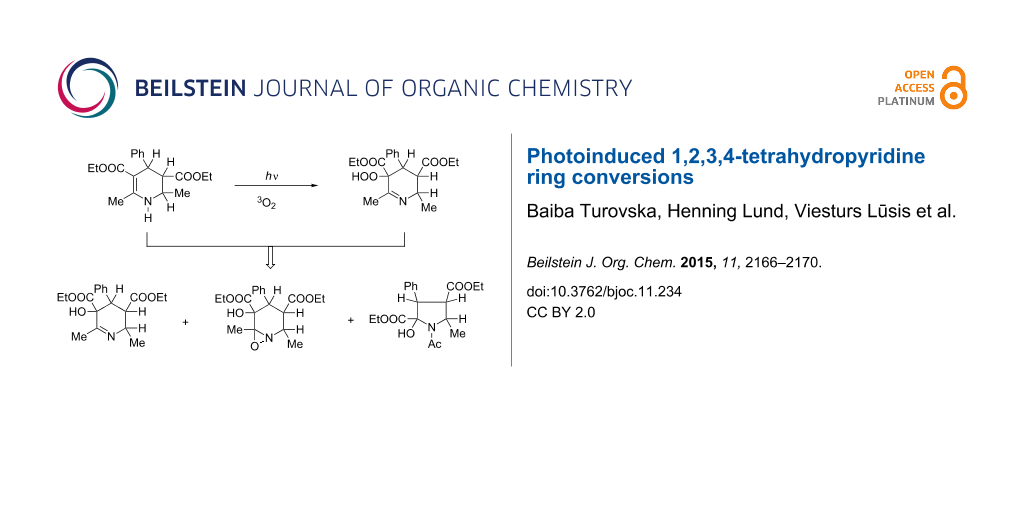
Abstract
Stable heterocyclic hydroperoxide can be easily prepared as a product of fast oxidation of a 1,2,3,4-tetrahydropyridine by 3O2 if the solution is exposed to sunlight. The driving force for the photoinduced electron transfer is calculated from electrochemical and spectroscopic data. The outcome of the reaction depends on the light intensity and the concentration of O2. In the solid state the heterocyclic hydroperoxide is stable; in solution it is involved in further reactions.

Graphical Abstract
Introduction
Increased attention has been paid to the chemistry of cyclic organic peroxides since it was found that naturally occurring representatives of this group possess biological activity, particular antimalarial [1,2]. Significantly less attention has been paid to organic aromatic or heterocyclic hydroperoxides, probably due to their low thermal stability and high reactivity.
Stable organic hydroperoxides were isolated in the early 1950s as products of autoxidation as well as catalytic oxygenation of indoles and tetrahydrocarbazoles [3-6]. In 1944 hydroperoxides were first obtained by H. Hock [7] and R. Udris [8-10] as the catalytic oxidation products of cumene. Heterocyclic hydroperoxides have been less represented, although some of these constitute the best choice for selective oxidations even in nature. When the peroxy functional group is placed near to electronegative groups, the oxidizing capability of hydroperoxides can be increased. This effect was particularly observed on heterocyclic systems.
Results and Discussion
Photosensitized aerobic oxidative aromatization [11-14] of Hantzsch 1,4-dihydropyridines has been extensively investigated, in the same time little attention has been paid to the corresponding reactions of tetrahydropyridines.
During the investigation of the electrochemical oxidation mechanism of tetrahydropyridine 1 [15], an extremely high sensitivity of the formed cation radicals towards traces of dioxygen was observed. In deaereated aprotic solvents 1 undergoes a reversible one-electron single-step oxidation [16] (+1.00 V in MeCN or +1.25 V in CH2Cl2) while the reversibility of the anodic process disappears immediately after the argon flow through/over the solution has been stopped (Figure 1).
![[1860-5397-11-234-1]](/bjoc/content/figures/1860-5397-11-234-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Electrochemical oxidation of 1 in deareated (blue) and O2 saturated (red) solutions of CH2Cl2/0.1 M TBAPF6, c = 5 × 10−4 M.
Figure 1: Electrochemical oxidation of 1 in deareated (blue) and O2 saturated (red) solutions of CH2Cl2/0.1 M...
Moreover, it was found that tetrahydropyridine 1 (Figure 2) reacts with dioxygen if the solution is exposed to intense sunlight. In order to study this reaction, the solution of 1 (0.5 g, 1.51 mmol) was left under an irradiation of intense sunlight and continuously purged with dioxygen (~9.1 mmol) [17,18] in chloroform (25 mL). Crystals suitable for X-ray analysis were obtained after evaporation of the chloroform by a stream of dioxygen; elemental analysis, 1H, 13C NMR spectra and X-ray analysis confirmed the structure of hydroperoxide 2 (Figure 2) as the only product of the reaction. Hydroperoxide 2 is a colourless crystalline compound which is stable in the solid state but unstable in solution.
![[1860-5397-11-234-2]](/bjoc/content/figures/1860-5397-11-234-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The X-ray structures of compounds 1 and 2.
Figure 2: The X-ray structures of compounds 1 and 2.
The oxidation of tetrahydropyridine 1 is photoinduced; it does not take place in the dark and it depends on the intensity of the light. No reaction could be initiated by light in a deareated solution.
The reaction of dioxygen (3O2) having a triplet ground state with tetrahydropyridine 1 having a singlet ground state is spin forbidden. On the other hand, the electron transfer from the organic compound to 3O2 resulting in the formation of a radical cation of the organic donor and the radical anion of O2 (O2−•) is spin permitted [19], however, it is not thermodynamically favourable.
The driving force for the photoinduced electron transfer can be calculated from electrochemical and spectroscopic measurements made in the same solvent according to Weller’s approximation [20]
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-234-i4.svg?max-width=637&scale=1.18182)
where ![[Graphic 2]](/bjoc/content/inline/1860-5397-11-234-i5.svg?max-width=637&scale=1.18182) is the difference between the first oxidation potential of the donor and the first reduction potential of the acceptor;
is the difference between the first oxidation potential of the donor and the first reduction potential of the acceptor;
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-234-i6.svg?max-width=637&scale=1.18182)
In CH2Cl2 the reduction potential of O2 is −1.18 V and E0 of 1 is +1.14 V.
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-234-i7.svg?max-width=637&scale=1.18182)
The reaction becomes thermodynamically favourable if the organic molecule is excited to a singlet state. The oxidation potential of excited molecules is shifted to negative values (ΔE = ~2 V) compared to their ground state [21-23]. For example the oxidation potential of 9,10-dihydro-10-methylacridine is +0.80 V [23], which is shifted to −3.10 V in the singlet excited state [23].
The recorded UV–vis spectrum of 1 (Figure 3) has only one absorption band at λmax = 282 nm in the range of 250–900 nm which corresponds to the spin allowed S→S* transition (ε = 19000).
![[1860-5397-11-234-3]](/bjoc/content/figures/1860-5397-11-234-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Decrease of the UV absorption band of compound 1 under irradiation (254 nm) in air-saturated CHCl3, c = 5 × 10−5 M.
Figure 3: Decrease of the UV absorption band of compound 1 under irradiation (254 nm) in air-saturated CHCl3, ...
If a series of UV spectra is recorded after subsequent periods of 1 min irradiation of the sample with an external UV lamp (254 nm, 8 W), the absorption band decreases and no new band appears in the range of 250–900 nm (Figure 3). If the irradiation of the sample is stopped for 1 min, the reaction halts, and the recorded UV spectrum exactly repeats the previous one.
It might be suggested that the initiation step, excitation of 1 at 254 nm, leading to the formation of the singlet state of 1*, is followed by single-electron transfer from 1* to 3O2 generating 1+• and O2− • in solution. Such reactions between strong nucleophiles and strong electrophiles, especially the annihilation reactions between ion radicals, have not been studied extensively, probably due to the difficulties of generating anion and cation radicals simultaneously [24,25].
The reactivity of superoxide is widely studied by chemists and biochemists. It has been shown electrochemically that O2− • is a product of the reversible one-electron reduction of dioxygen in aprotic solvents [26,27]. Superoxide O2−• is a highly reactive molecule [26,28] and it acts as a strong Brönsted base removing a proton from substrates to an extent equivalent to that of the conjugate base with a pKa value of approximately 23 in water [29,30].
A number of weakly acidic organic compounds are deprotonated efficiently in the presence of superoxide including Hantzsch 1,4-dihydropyridines [31,32]. Cation radicals have increased acidity comparing to the parent molecules from which they are derived by oxidation [33-35]. Consequently, their deprotonation proceeds more efficiently. Photooxygenation of 1 can be described as shown in Scheme 1.
![[1860-5397-11-234-i1]](/bjoc/content/inline/1860-5397-11-234-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photoinduced reaction of 1 in O2 saturated CHCl3 under irradiation by intensive sunlight.
Scheme 1: Photoinduced reaction of 1 in O2 saturated CHCl3 under irradiation by intensive sunlight.
In an air saturated CHCl3 solution, the concentration of dioxygen is about 2 mmol/L [17,18] and if it becomes comparable with the concentration of 1, the reaction takes a different course (Scheme 2).
![[1860-5397-11-234-i2]](/bjoc/content/inline/1860-5397-11-234-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Heterocycle transformations of 1 in air saturated CHCl3 solutions.
Scheme 2: Heterocycle transformations of 1 in air saturated CHCl3 solutions.
It has been suggested that peracids attack the carbon–carbon double bond in enamines to give the corresponding epoxides, however, in most cases they have not been isolated [36].
The hydroperoxide 2 reacts in the same way. Fission of the epoxide ring may be induced by the base itself producing 3. Attack of a second molecule of hydroperoxide 2 on the imine group of 3 oxidizes it to oxaziridine intermediate 4 (Figure 4), which then undergoes a slow nucleophilic ring fission followed by cyclization (Scheme 3) to give 5 (Figure 4).
![[1860-5397-11-234-i3]](/bjoc/content/inline/1860-5397-11-234-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of conversion of oxaziridine 4 to 5.
Scheme 3: Proposed mechanism of conversion of oxaziridine 4 to 5.
![[1860-5397-11-234-4]](/bjoc/content/figures/1860-5397-11-234-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The X-ray structures of compounds 4 and 5.
Figure 4: The X-ray structures of compounds 4 and 5.
The reaction depicted in Scheme 2 proceeds as long as the hydroperoxide 2 is present in the solution, and the crude product contains the mixture of 3, 4 and 5. The same product mixture was obtained if the solution of 1 is treated with concentrated H2O2 or Fenton reagent.
The reaction starts even if the irradiation wavelengths only partially cover the absorption band of 1. It explains also the observed fact that during the summer as well as under bright laboratory light the photooxygenation of tetrahydropyridine 1 in the presence of dioxygen proceeds spontaneously in CHCl3, CH2Cl2 or CH3CN solutions. Although much of the light emitted by the sun in UV below 300 nm is absorbed by ordinary glass, the transmitted intensity of the sunlight in summer at 300 nm is sufficient to initiate the photooxygenation of 1.
Direct reactions of dioxygen with organic substrates in the absence of a catalyst are usually slow, unless the substrate is a particularly good reducing agent. Excited state species are easier to oxidize than the corresponding ground state species.
The reaction between photochemically generated radical cations and radical anions by electron transfer from photoexcited electron donor to electron acceptors is often reversible [37] thus reproducing the reactant pair without the formation of a chemical bond. As the dioxygen has a large reorganization energy for electron transfer [38,39], the reverse reaction may be retarded, promoting to the subsequent heterocycle conversions.
Conclusion
Several synthetic routes have been explored towards chiral hydroperoxides as they have been utilized successfully in a variety of asymmetric oxidations. We have found a sunlight activated reaction of 1,2,3,4-tetrahydropiridine with dioxygen (3O2) producing stable heterocyclic hydroperoxide in excellent yield. The reaction has significant advantages as it uses cost-free reagents: light and dioxygen. The same reaction demonstrated the oxidizing capacity of the hydroperoxide when its concentration is less or comparable with the tetrahydropyridine leading to another versatile intermediate in organic synthesis – oxaziridine – as the inherent strain of the ring and the relatively weak N–O bond makes it unusually reactive.
Supporting Information
Synthesis and characterization of all products, copies of 1H and 13C NMR spectra of newly synthesized products.
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Gesmundo, N. J.; Nicewicz, D. A. Beilstein J. Org. Chem. 2014, 10, 1272–1281. doi:10.3762/bjoc.10.128
Return to citation in text: [1] -
Terent'ev, A. O.; Borisov, D. A.; Vil’, V. A.; Dembitsky, V. M. Beilstein J. Org. Chem. 2014, 10, 34–114. doi:10.3762/bjoc.10.6
Return to citation in text: [1] -
Witkop, B.; Patrick, J. B. J. Am. Chem. Soc. 1951, 73, 2188–2195. doi:10.1021/ja01149a079
Return to citation in text: [1] -
Witkop, B.; Patrick, J. B. J. Am. Chem. Soc. 1951, 73, 2196–2200. doi:10.1021/ja01149a080
Return to citation in text: [1] -
Witkop, B.; Patrick, J. B.; Rosenblum, M. J. Am. Chem. Soc. 1951, 73, 2641–2647. doi:10.1021/ja01150a065
Return to citation in text: [1] -
Mentel, M.; Breinbauer, R. Curr. Org. Chem. 2007, 11, 159–176. doi:10.2174/138527207779316426
Return to citation in text: [1] -
Hock, H.; Lang, S. Ber. Dtsch. Chem. Ges. B 1944, 77, 257–264. doi:10.1002/cber.19440770321
Return to citation in text: [1] -
Stradiņš, J. P. Iz istorii estestvoznanija i techniki Pribaltiki; Zinatne: Riga, Latvia, 1968.
Return to citation in text: [1] -
Udris, R. J.; Sergeyev, P. G.; Kruzhalov, B. D. Author’s certificate of USSR No. 106666, January 7, 1947.
Return to citation in text: [1] -
Udris, R. J.; Kruzhalov, B. D.; Sergeyev, P. G. Author’s certificate of USSR No. 106712, January 7, 1947.
Return to citation in text: [1] -
Memarian, H. R.; Abdoli-Senejani, M.; Tangestaninejad, S. J. Iran. Chem. Soc. 2006, 3, 285–292. doi:10.1007/BF03247221
Return to citation in text: [1] -
Han, B.; Liu, Z.; Liu, Q.; Yang, L.; Liu, Z.-L.; Yu, W. Tetrahedron 2006, 62, 2492–2496. doi:10.1016/j.tet.2005.12.056
Return to citation in text: [1] -
Pizarro, N.; Günther, G.; Núñez-Vergara, L. J. J. Photochem. Photobiol., A 2007, 189, 23–29. doi:10.1016/j.jphotochem.2007.01.003
Return to citation in text: [1] -
Memarian, H. R.; Abdoli-Senejani, M. Ultrason. Sonochem. 2008, 15, 110–114. doi:10.1016/j.ultsonch.2006.06.008
Return to citation in text: [1] -
Rosentreter, U. Synthesis 1985, 210–212. doi:10.1055/s-1985-31160
Return to citation in text: [1] -
Turovska, B.; Goba, I.; Lielpetere, A.; Turovskis, I.; Lusis, V.; Muceniece, D.; Stradiņš, J. Chem. Heterocycl. Compd. 2014, 49, 1640–1652. doi:10.1007/s10593-014-1415-5
Return to citation in text: [1] -
Hara, M.; Samori, S.; Xichen, C.; Fujitsuka, M.; Majima, T. J. Org. Chem. 2005, 70, 4370–4374. doi:10.1021/jo050317n
Return to citation in text: [1] [2] -
Hanaki, H.; Fukatsu, Y.; Harada, M.; Sawaki, Y. Tetrahedron Lett. 2004, 45, 5791–5793. doi:10.1016/j.tetlet.2004.06.022
Return to citation in text: [1] [2] -
Fukuzumi, S.; Okamoto, K.; Tokuda, Y.; Gros, C. P.; Guilard, R. J. Am. Chem. Soc. 2004, 126, 17059–17066. doi:10.1021/ja046422g
Return to citation in text: [1] -
Rehm, D.; Weller, A. Isr. J. Chem. 1970, 8, 259–271. doi:10.1002/ijch.197000029
Return to citation in text: [1] -
Qin, C.; Clark, A. E. Chem. Phys. Lett. 2007, 438, 26–30. doi:10.1016/j.cplett.2007.02.063
Return to citation in text: [1] -
Rubio, N.; Jiménez-Banzo, A.; Torres, T.; Nonell, S. J. Photochem. Photobiol., A 2007, 185, 214–219. doi:10.1016/j.jphotochem.2006.06.007
Return to citation in text: [1] -
Fukuzumi, S.; Ishikawa, M.; Tanaka, T. J. Chem. Soc., Perkin Trans. 2 1989, 1037–1045. doi:10.1039/P29890001037
Return to citation in text: [1] [2] [3] -
Amatore, C.; Brown, A. R. J. Am. Chem. Soc. 1996, 118, 1482–1486. doi:10.1021/ja952600h
Return to citation in text: [1] -
Mayeda, E. A.; Bard, A. J. J. Am. Chem. Soc. 1973, 95, 6223–6226. doi:10.1021/ja00800a012
Return to citation in text: [1] -
Sawyer, D. T.; Chiericato, G., Jr.; Angelis, C. T.; Nanni, E. J., Jr.; Tsuchiya, T. Anal. Chem. 1982, 54, 1720–1724. doi:10.1021/ac00248a014
Return to citation in text: [1] [2] -
Maricle, D. L.; Hodgson, W. G. Anal. Chem. 1965, 37, 1562–1565. doi:10.1021/ac60231a027
Return to citation in text: [1] -
Meskina, M. Y.; Baider, L. M.; Skibida, I. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1990, 39, 646–650. doi:10.1007/BF00960318
Return to citation in text: [1] -
Frimer, A. A.; Farkash-Solomon, T.; Aljadeff, G. J. Org. Chem. 1986, 51, 2093–2098. doi:10.1021/jo00361a030
Return to citation in text: [1] -
Sawyer, D. T.; Gibian, M. J. Tetrahedron 1979, 35, 1471–1481. doi:10.1016/0040-4020(79)80032-0
Return to citation in text: [1] -
Ortiz, M. E.; Núñez-Vergara, L. J.; Camargo, C.; Squella, J. A. Pharm. Res. 2004, 21, 428–435. doi:10.1023/B:PHAM.0000019295.32103.e4
Return to citation in text: [1] -
Ortiz, M. E.; Núñez-Vergara, L. J.; Squella, J. A. J. Electroanal. Chem. 2002, 519, 46–52. doi:10.1016/S0022-0728(01)00720-3
Return to citation in text: [1] -
Anne, A.; Fraoua, S.; Hapiot, P.; Moiroux, J.; Saveant, J.-M. J. Am. Chem. Soc. 1995, 117, 7412–7421. doi:10.1021/ja00133a014
Return to citation in text: [1] -
Hapiot, P.; Moiroux, J.; Saveant, J. M. J. Am. Chem. Soc. 1990, 112, 1337–1343. doi:10.1021/ja00160a007
Return to citation in text: [1] -
Anne, A.; Moiroux, J. Can. J. Chem. 1995, 73, 531–538. doi:10.1139/v95-068
Return to citation in text: [1] -
Pitacco, G.; Valentin, E. In The Chemistry of Enamines; Rappoport, Z., Ed.; John Wiley & Sons, Ltd.: NY, USA, 1994.
Return to citation in text: [1] -
Ohkubo, K.; Nanjo, T.; Fukuzumi, S. Bull. Chem. Soc. Jpn. 2006, 79, 1489–1500. doi:10.1246/bcsj.79.1489
Return to citation in text: [1] -
Lind, J.; Shen, X.; Merenyi, G.; Jonsson, B. O. J. Am. Chem. Soc. 1989, 111, 7654–7655. doi:10.1021/ja00201a078
Return to citation in text: [1] -
Eberson, L.; Gonzalez-Luque, R.; Lorentzon, J.; Merchan, M.; Roos, B. O. J. Am. Chem. Soc. 1993, 115, 2898–2902. doi:10.1021/ja00060a043
Return to citation in text: [1]
| 33. | Anne, A.; Fraoua, S.; Hapiot, P.; Moiroux, J.; Saveant, J.-M. J. Am. Chem. Soc. 1995, 117, 7412–7421. doi:10.1021/ja00133a014 |
| 34. | Hapiot, P.; Moiroux, J.; Saveant, J. M. J. Am. Chem. Soc. 1990, 112, 1337–1343. doi:10.1021/ja00160a007 |
| 35. | Anne, A.; Moiroux, J. Can. J. Chem. 1995, 73, 531–538. doi:10.1139/v95-068 |
| 29. | Frimer, A. A.; Farkash-Solomon, T.; Aljadeff, G. J. Org. Chem. 1986, 51, 2093–2098. doi:10.1021/jo00361a030 |
| 30. | Sawyer, D. T.; Gibian, M. J. Tetrahedron 1979, 35, 1471–1481. doi:10.1016/0040-4020(79)80032-0 |
| 31. | Ortiz, M. E.; Núñez-Vergara, L. J.; Camargo, C.; Squella, J. A. Pharm. Res. 2004, 21, 428–435. doi:10.1023/B:PHAM.0000019295.32103.e4 |
| 32. | Ortiz, M. E.; Núñez-Vergara, L. J.; Squella, J. A. J. Electroanal. Chem. 2002, 519, 46–52. doi:10.1016/S0022-0728(01)00720-3 |
| 1. | Gesmundo, N. J.; Nicewicz, D. A. Beilstein J. Org. Chem. 2014, 10, 1272–1281. doi:10.3762/bjoc.10.128 |
| 2. | Terent'ev, A. O.; Borisov, D. A.; Vil’, V. A.; Dembitsky, V. M. Beilstein J. Org. Chem. 2014, 10, 34–114. doi:10.3762/bjoc.10.6 |
| 11. | Memarian, H. R.; Abdoli-Senejani, M.; Tangestaninejad, S. J. Iran. Chem. Soc. 2006, 3, 285–292. doi:10.1007/BF03247221 |
| 12. | Han, B.; Liu, Z.; Liu, Q.; Yang, L.; Liu, Z.-L.; Yu, W. Tetrahedron 2006, 62, 2492–2496. doi:10.1016/j.tet.2005.12.056 |
| 13. | Pizarro, N.; Günther, G.; Núñez-Vergara, L. J. J. Photochem. Photobiol., A 2007, 189, 23–29. doi:10.1016/j.jphotochem.2007.01.003 |
| 14. | Memarian, H. R.; Abdoli-Senejani, M. Ultrason. Sonochem. 2008, 15, 110–114. doi:10.1016/j.ultsonch.2006.06.008 |
| 26. | Sawyer, D. T.; Chiericato, G., Jr.; Angelis, C. T.; Nanni, E. J., Jr.; Tsuchiya, T. Anal. Chem. 1982, 54, 1720–1724. doi:10.1021/ac00248a014 |
| 27. | Maricle, D. L.; Hodgson, W. G. Anal. Chem. 1965, 37, 1562–1565. doi:10.1021/ac60231a027 |
| 8. | Stradiņš, J. P. Iz istorii estestvoznanija i techniki Pribaltiki; Zinatne: Riga, Latvia, 1968. |
| 9. | Udris, R. J.; Sergeyev, P. G.; Kruzhalov, B. D. Author’s certificate of USSR No. 106666, January 7, 1947. |
| 10. | Udris, R. J.; Kruzhalov, B. D.; Sergeyev, P. G. Author’s certificate of USSR No. 106712, January 7, 1947. |
| 26. | Sawyer, D. T.; Chiericato, G., Jr.; Angelis, C. T.; Nanni, E. J., Jr.; Tsuchiya, T. Anal. Chem. 1982, 54, 1720–1724. doi:10.1021/ac00248a014 |
| 28. | Meskina, M. Y.; Baider, L. M.; Skibida, I. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1990, 39, 646–650. doi:10.1007/BF00960318 |
| 7. | Hock, H.; Lang, S. Ber. Dtsch. Chem. Ges. B 1944, 77, 257–264. doi:10.1002/cber.19440770321 |
| 23. | Fukuzumi, S.; Ishikawa, M.; Tanaka, T. J. Chem. Soc., Perkin Trans. 2 1989, 1037–1045. doi:10.1039/P29890001037 |
| 3. | Witkop, B.; Patrick, J. B. J. Am. Chem. Soc. 1951, 73, 2188–2195. doi:10.1021/ja01149a079 |
| 4. | Witkop, B.; Patrick, J. B. J. Am. Chem. Soc. 1951, 73, 2196–2200. doi:10.1021/ja01149a080 |
| 5. | Witkop, B.; Patrick, J. B.; Rosenblum, M. J. Am. Chem. Soc. 1951, 73, 2641–2647. doi:10.1021/ja01150a065 |
| 6. | Mentel, M.; Breinbauer, R. Curr. Org. Chem. 2007, 11, 159–176. doi:10.2174/138527207779316426 |
| 24. | Amatore, C.; Brown, A. R. J. Am. Chem. Soc. 1996, 118, 1482–1486. doi:10.1021/ja952600h |
| 25. | Mayeda, E. A.; Bard, A. J. J. Am. Chem. Soc. 1973, 95, 6223–6226. doi:10.1021/ja00800a012 |
| 19. | Fukuzumi, S.; Okamoto, K.; Tokuda, Y.; Gros, C. P.; Guilard, R. J. Am. Chem. Soc. 2004, 126, 17059–17066. doi:10.1021/ja046422g |
| 21. | Qin, C.; Clark, A. E. Chem. Phys. Lett. 2007, 438, 26–30. doi:10.1016/j.cplett.2007.02.063 |
| 22. | Rubio, N.; Jiménez-Banzo, A.; Torres, T.; Nonell, S. J. Photochem. Photobiol., A 2007, 185, 214–219. doi:10.1016/j.jphotochem.2006.06.007 |
| 23. | Fukuzumi, S.; Ishikawa, M.; Tanaka, T. J. Chem. Soc., Perkin Trans. 2 1989, 1037–1045. doi:10.1039/P29890001037 |
| 37. | Ohkubo, K.; Nanjo, T.; Fukuzumi, S. Bull. Chem. Soc. Jpn. 2006, 79, 1489–1500. doi:10.1246/bcsj.79.1489 |
| 17. | Hara, M.; Samori, S.; Xichen, C.; Fujitsuka, M.; Majima, T. J. Org. Chem. 2005, 70, 4370–4374. doi:10.1021/jo050317n |
| 18. | Hanaki, H.; Fukatsu, Y.; Harada, M.; Sawaki, Y. Tetrahedron Lett. 2004, 45, 5791–5793. doi:10.1016/j.tetlet.2004.06.022 |
| 23. | Fukuzumi, S.; Ishikawa, M.; Tanaka, T. J. Chem. Soc., Perkin Trans. 2 1989, 1037–1045. doi:10.1039/P29890001037 |
| 38. | Lind, J.; Shen, X.; Merenyi, G.; Jonsson, B. O. J. Am. Chem. Soc. 1989, 111, 7654–7655. doi:10.1021/ja00201a078 |
| 39. | Eberson, L.; Gonzalez-Luque, R.; Lorentzon, J.; Merchan, M.; Roos, B. O. J. Am. Chem. Soc. 1993, 115, 2898–2902. doi:10.1021/ja00060a043 |
| 16. | Turovska, B.; Goba, I.; Lielpetere, A.; Turovskis, I.; Lusis, V.; Muceniece, D.; Stradiņš, J. Chem. Heterocycl. Compd. 2014, 49, 1640–1652. doi:10.1007/s10593-014-1415-5 |
| 17. | Hara, M.; Samori, S.; Xichen, C.; Fujitsuka, M.; Majima, T. J. Org. Chem. 2005, 70, 4370–4374. doi:10.1021/jo050317n |
| 18. | Hanaki, H.; Fukatsu, Y.; Harada, M.; Sawaki, Y. Tetrahedron Lett. 2004, 45, 5791–5793. doi:10.1016/j.tetlet.2004.06.022 |
| 20. | Rehm, D.; Weller, A. Isr. J. Chem. 1970, 8, 259–271. doi:10.1002/ijch.197000029 |
| 36. | Pitacco, G.; Valentin, E. In The Chemistry of Enamines; Rappoport, Z., Ed.; John Wiley & Sons, Ltd.: NY, USA, 1994. |
© 2015 Turovska et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)