Abstract
The continuous flow synthesis of a range of organic solutions of N,N-dialkyl-N-chloramines is described using either a bespoke meso-scale tubular reactor with static mixers or a continuous stirred tank reactor. Both reactors promote the efficient mixing of a biphasic solution of N,N-dialkylamine in organic solvent, and aqueous sodium hypochlorite to achieve near quantitative conversions, in 72–100% in situ yields, and useful productivities of around 0.05 mol/h with residence times from 3 to 20 minutes. Initial calorimetric studies have been carried out to inform on reaction exotherms, rates and safe operation. Amines which partition mainly in the organic phase require longer reaction times, provided by the CSTR, to compensate for low mass transfer rates in the biphasic system. The green metrics of the reaction have been assessed and compared to existing procedures and have shown the continuous process is improved over previous procedures. The organic solutions of N,N-dialkyl-N-chloramines produced continuously will enable their use in tandem flow reactions with a range of nucleophilic substrates.
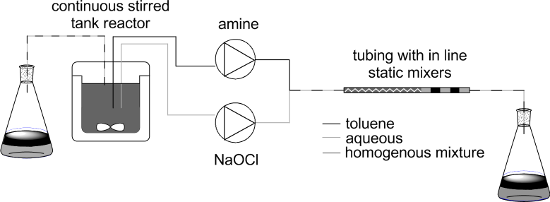
Graphical Abstract
Introduction
N-Chloramines provide a versatile and reactive class of reagents for use in electrophilic amination and other reactions. N-Chloro-N,N-dialkylamines have been shown to offer a broad range of products from reactions with i) unsaturated C–C bonds to give amines [1-3] and heterocycles [1,4]; ii) Grignard and organozinc reagents to give amines [1]; iii) aldehydes to give amides [5-7]; iv) base to give imines [8]; v) alkyl and aryl C–H bonds in the presence of acid and visible light to form heterocycles [9,10]. Furthermore they have also been used for chlorination of aromatics in the presence of acid [11], and as reagents in the syntheses of natural products [12,13].
Despite their versatility as reagents, the exothermicity and instability that occurs in both their formation and reaction, has reduced the interest in their use for reasons of process safety. Isolation of many N-chloramines is unadvisable with reports of unpredictable and rapid decomposition [11,14-16].
N-Chloramines are prepared conventionally by reaction of the amine precursor with an electrophilic chlorine source [14]. Despite its atom efficiency use of chlorine gas is undesirable due to its toxicity and strong oxidising properties, making it highly hazardous and operationally difficult to use. In addition, the hydrochloric acid byproduct requires additional processing to neutralise and separate it. N-Chlorosuccinimide is used frequently in the laboratory because it is a relatively stable solid that is easily weighed and added to reactions [3,17], however, it has a low atom economy, poor economics, and requires separation of the succinimide byproduct [18]. The tert-butyl hypochlorite (t-BuOCl) reagent is also used regularly, but is similarly expensive, wasteful and hazardous [14,19-21]. On the other hand sodium hypochlorite (NaOCl) is an inexpensive byproduct of chlorine manufacture, and offers a safer, greener source of electrophilic chlorine for producing N-chloramines [11,14,22]. Reaction times can, however, be long [14,23,24], and yields low [25]. Zhong et al. have reported a process using NaOCl to generate t-BuOCl in situ for chloramine formation, applying the methodology to a broad range of substrates in high yields [20].
Published literature on chloramine formation is limited to batch procedures, however, the use of continuous processes could offer significant advantages. Use of continuous flow reactors to achieve steady-state allows precise control over the reaction parameters to improve safety, selectivity, productivity and consistency. Reduced reactor volumes compared to conventional batch reactors limit the quantity of reacting material at any one time; particularly important for the preparation of hazardous or unstable products. Furthermore smaller volumes with higher surface areas enable faster heat removal and better temperature control to limit unwanted side reactions. Further kinetic control is achieved with reactant concentrations, feed rates, mixing regime and residence time resulting in higher product yields.
Herein we report the use of different types of bespoke continuous reactors for preparing different N-chloro-N,N-dialkylamines. The use of these materials in forming a range of different nitrogen containing products via a cascade/tandem procedure will be reported elsewhere. This atom efficient procedure, with sodium hydroxide and water the only byproducts, should have better green metrics than other methods. Using biphasic reaction conditions with the amine dissolved in a water immiscible, organic solvent such as toluene enabled facile separation of the organic soluble product from the water soluble NaOH (Scheme 1). Such separation of biphasic mixtures is known in continuous systems, for example membrane-based separators [26,27].
![[1860-5397-11-262-i1]](/bjoc/content/inline/1860-5397-11-262-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Two-phase reaction of N,N-dialkylamine and sodium hypochlorite.
Scheme 1: Two-phase reaction of N,N-dialkylamine and sodium hypochlorite.
Mixing is a key parameter for reactions in multiphasic systems and characterisation of material flows within different continuous reactors is widely studied by both modelling and experiment [28,29]. The rate of reaction of NaOCl and amines at high pH is very fast with a second order rate constant, kobs of 1.52 × 105 L·mol−1·min−1 reported for dimethylamine [30]. In this case it is the rate of mass transfer of the reagents, partitioned between the two liquid phases that limits the rate of product formation, rather than the chemical rate of reaction between the two species. Continuous liquid biphasic reactions are usually poorly mixed within micro-scale reactors, as low fluid velocities and frictional wall effects cause laminar flow and phase separation into alternating organic–aqueous segmented flow; whilst meso-scale reactors are much better suited, and provide more accurate and reliable information for scale-up. Interaction of the reactants occurs only at the phase interface, so efficient mixing increases the surface area and promotes product formation. Within batch or continuous stirred tank reactors an increase in mixing intensity is achieved with optimised impellor design and speed, alongside a reactor designed to disrupt the flow and increase turbulence. Within meso-scale tubular reactors the addition of in-line aids such as split and recombination streams [31,32], or static mixers [32,33], can enhance mixing and mass transfer between phases. Phase-transfer catalysts (PTCs) can also be used to promote reactions across phase boundaries [31], their use would, however, incur additional financial costs for the reaction, and also the need for purification and removal of the catalyst from the reaction solution.
Results and Discussion
Calorimetry
In view of the potential dangers involved in making chloramines we began our studies with calorimetric analysis. The formation N-chloromorpholine was studied by feeding 1.1 M NaOCl (aq) into a 1 M morpholine (aq) at a rate of 1 g/min. The calorimeter jacket temperature was set at −15 °C, and power compensation used to maintain the reactor at 5 °C. 20 mL of the NaOCl solution were added to 20 mmol of morpholine over 20 min, and after subtracting the feed solution temperature (Q Dose) from compensatory power (Q Comp) the total was 2.76 W. For this calibrated calorimeter this equates to −1.47 kJ which gives a heat of reaction, ΔHr = −73.4 kJ·mol−1 for the formation of N-chloromorpholine (Figure 1). Calculation of the heat of reaction by bond forming/bond breaking calculation, according to mechanisms proposed in the literature, give a ΔHr of −72 kJ·mol−1 (Supporting Information File 1) [30].
![[1860-5397-11-262-1]](/bjoc/content/figures/1860-5397-11-262-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Calorimeter trace for the single phase reaction of morpholine (aq) and NaOCl (aq). Q Comp: compensatory power, Q Total: total power, Q Dose: power delivered by dosing of room temperature NaOCl (aq) to cooled reaction solution. Energy: heat energy.
Figure 1: Calorimeter trace for the single phase reaction of morpholine (aq) and NaOCl (aq). Q Comp: compensa...
Figure 1 shows the calorimetric trace for the formation of N-chloromorpholine. Following the addition of NaOCl (aq) there is a small delay in the reaction which may be mixing related. A rapid exotherm then occurs with 1.37 kJ energy released continuously over the 20 minute feed. When the NaOCl (aq) addition is stopped there is a further release of 0.09 kJ energy over 2 minutes indicating a small accumulation of heat. Analysis of the crude reaction product shows no side products, and no decomposition of the N-chloromorpholine product.
When the same reaction was carried out using a toluene–water biphasic system, the calorimeter trace was more complex with mass transfer effects making interpretation difficult (Supporting Information File 1).
The calorimetric analysis shows chloramine formation to be an energetic process with a significant associated exotherm illustrating the need for efficient temperature control during the reaction to ensure a safe process. In batch it would be necessary to cool the reaction with an ice bath or similar, however, the continuous reactors employed in this work, with increased surface area-to-volume ratio, allows effective heat dissipation under ambient conditions.
Continuous reactor
A nylon/PTFE tubular reactor was constructed that incorporates static mixers for enhanced mixing of the biphasic reaction solution, made of the acetal homopolymer, Delrin® that are solvent and oxidant resistant [34]. The set-up comprised one pump for the organic phase (amine/toluene) and one for the aqueous phase (NaOCl/water). The feeds were connected via a stainless steel T-piece to tubing (1/4 inch OD, 3/16 inch ID) containing static mixer tubes (1/8 inch ID, 3/16 inch OD) along the flow channel (Figure 2). The length of the reactor and number of static mixer inserts were adjusted to vary the residence time, thus maintaining sufficient flow rate to give effective mixing.
![[1860-5397-11-262-2]](/bjoc/content/figures/1860-5397-11-262-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Meso-scale static mixer set-up for continuous N-chloramine formation. (a) Pumps, (b) reagent solution reservoirs, (c) reactor tube containing static mixers, (d) collection vessel, (e) back pressure regulator (75 psi).
Figure 2: Meso-scale static mixer set-up for continuous N-chloramine formation. (a) Pumps, (b) reagent soluti...
Initial experiments using toluene and an aqueous dye were used to assess static mixer performance. Figure 3 shows the solutions being pumped simultaneously into a T-piece with equal flow rates. As the solution progresses into the tube containing the static mixers, it can be seen to be a well-mixed emulsion. Shortly after emanating from the mixed region, the biphase separates into a segmented flow before being collected into a flask.
![[1860-5397-11-262-3]](/bjoc/content/figures/1860-5397-11-262-3.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Effect of static mixers on biphasic solution.
Figure 3: Effect of static mixers on biphasic solution.
Initial reactions looked at the effect of increasing the number of static mixers within the reactor and also increasing residence time by decreasing the flow rate of reagents. The progress of the reaction was studied to determine the point at which steady state is achieved and to assess the consistency of the reaction over several reactor volumes (Figure 4). The reaction reaches steady state after 6 minutes (2 residence times) with fairly consistent conversion achieved thereafter. The N-chloromorpholine yield was measured by direct sampling of the toluene solution and measuring by quantitative 1H NMR.
![[1860-5397-11-262-4]](/bjoc/content/figures/1860-5397-11-262-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Progress of reaction for continuous formation of N-chloromorpholine. Morpholine (toluene) 0.9 M 1 mL/min, NaOCl (aq) 0.9 M, 1.1 mL/min, 6 mL total reactor volume, of which 0.8 mL is static mixers. Residence time = 2.9 min. Conversion refers to composition of organic phase of reaction solution.
Figure 4: Progress of reaction for continuous formation of N-chloromorpholine. Morpholine (toluene) 0.9 M 1 m...
Table 1 entry 1 shows the poor conversion observed using the T-piece alone, whilst higher conversions are seen with increased static mixed volume. Table 1 entry 2 shows higher chloramine formation with increased residence time (Tres) as a result of lower flow rates, with a maximum 89% conversion at a Tres of 20 min. Even at flow rates of 0.15 mL/min there appears to be intimate mixing of the aqueous–organic phases. Both intense mixing and long residence times are required because the N-benzylmethanamine partitions mainly in the organic phase with KD [organic]/[aqueous] = 28.8, causing the reaction to be limited by the mass transfer rate. For example mixing in the T-piece alone gives 11% conversion with N-benzylmethanamine, but 46% with 1,4-morpholine; also 65% conversion is seen with 0.8 min of intense mixing of N-benzylmethanamine with NaOCl, whilst 68% is seen with 1,4-morpholine mixed for half this time, because it partitions more favourably into the aqueous phase with KD = 0.01.
Table 1: Effect of reactor parameters on chloramine formation.a
| Entry | Product |
NaOCl
(equiv) |
Mixed volume
(mL) |
Total RV
(mL) |
Tres
(min) |
Amine conv.
(%)b |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-262-i2.svg?max-width=637&scale=1.0)
|
1.1 |
0c
0c 0.8 1.6 |
<0.1
6 6 6 |
<0.05
3 3 3 |
11
39 57 65 |
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-262-i3.svg?max-width=637&scale=1.0)
|
1.1 | 1.6 | 6 |
1.2d
3 6e 12f 20g |
41
65 69 78 89 |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-262-i4.svg?max-width=637&scale=1.0)
|
1.1 |
0c
0.8 |
<0.1
6 |
<0.05
3 |
46
68 |
aTres = residence time, RV = reactor volume. Reaction conditions: 1 M amine in toluene and 1.1 M NaOCl (aq) at equal flow rates of 1 mL/min, room temperature. bSteady state conversion by NMR vs internal standard. cT-piece only. dFlow rates = 2.5 mL/min. eFlow rates = 0.5 mL/min. fFlow rates = 0.25 mL/min. gFlow rates = 0.15 mL/min.
Substrates
The formation of a range of N-chloro-N,N-dialkylamines was investigated. Some were found to react relatively slowly, partly for the mass transfer reasons discussed above, and possibly for electronic and steric reasons as well. The tube reactor did not conveniently allow sufficient residence times for full conversion of amine to chloramine to be achieved, and in such cases a continuous stirred tank reactor (CSTR), able to provide longer residence times, was used instead (Figure 5 and Figure 6).
![[1860-5397-11-262-5]](/bjoc/content/figures/1860-5397-11-262-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: CSTR set-up for N-chloramine formation. (a) Syringe pump, (b) collection vessels, (c) reactor (50 mL), (d) stirrer plate: stirring rate 1200 rpm.
Figure 5: CSTR set-up for N-chloramine formation. (a) Syringe pump, (b) collection vessels, (c) reactor (50 m...
![[1860-5397-11-262-6]](/bjoc/content/figures/1860-5397-11-262-6.jpg?scale=1.3&max-width=1024&background=FFFFFF)
Table 2 shows reaction parameters investigated for the reaction of different amines: residence time, reactor volume, number of static mixers and molar equivalents of NaOCl. Formation of N-chloromorpholine and N-chloropiperidine (Table 2, entries 1 and 2) proceeded with high yields under short reaction times using the static mixers. N-chloro-N-methyl-p-toluenesulfonamide and N-chloro-N-benzylmethanamine proceeded with good yields in both the tube reactor and CSTR, however, for the tube reactor 1.5 equiv NaOCl were required for complete reaction of N-methyl-p-toluenesulfonamide, and 20 min residence time was required for high yields of N-chloro-N-benzylmethylamine. Due to the low solubility of N-methyl-p-toluenesulfonamide in toluene the tube reactor was susceptible to blockage. This was overcome using EtOAc as the organic phase, and gave more consistent results. Formation of N-chlorodibutylamine proceeded slowly within the tube reactor and the longer residence times leant themselves to the CSTR. Table 2 entry 8 shows the need for 2 equiv NaOCl with 20 min residence time to give 35% conversion of the chloramine product with the tubular reactor. However, the conversion was improved markedly using the CSTR and enabled use of only 1.1 equiv of NaOCl to give quantitative formation of the product. The reaction of dibenzylamine with NaOCl proceeds slowly within the CSTR achieving only 40% conversion in 50 min, which may reflect its lower reactivity, since the partition coefficient is similar to that of dibutylamine. The productivities of N-chloramine formation are in the range of 0.04–0.06 mol/h.
Table 2: Formation of secondary chloramines.a
| Entry | Product | KD | Reactor type | Reactor vol. (mL)b |
Tres
(min) |
Amine conv.
(%)c |
Yield
(%)c |
|---|---|---|---|---|---|---|---|
| 1d |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-262-i5.svg?max-width=637&scale=1.0)
|
0.01 | Static mixers | 6 (0.8) | 4 | 100 | 84 |
| 2e |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-262-i6.svg?max-width=637&scale=1.0)
|
0.84 | Static mixers | 4 (1.6) | 4 | 100 | 94 |
| 3f |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-262-i7.svg?max-width=637&scale=1.0)
|
28.9 | Static mixers | 6 (1.6) | 3 | 97 | 100 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-262-i8.svg?max-width=637&scale=1.0)
|
28.9 | CSTR | 50 | 50 | 100 | 72–98g |
| 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-262-i9.svg?max-width=637&scale=1.0)
|
28.8 | Static mixers | 6 (1.6) | 20 | 89 | 87 |
| 6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-262-i10.svg?max-width=637&scale=1.0)
|
28.8 | CSTR | 50 | 25 | 100 | 100 |
| 7h |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-262-i11.svg?max-width=637&scale=1.0)
|
118 | Static mixers | 4 (1.6) | 20 | 35 | Not determined |
| 8 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-262-i12.svg?max-width=637&scale=1.0)
|
118 | CSTR | 50 | 50 | 100 | 100 |
| 9 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-262-i13.svg?max-width=637&scale=1.0)
|
114 | CSTR | 50 | 50 | 40h | Not determined |
aKD = [amine] in organic/[amine] in aqueous phase. Reaction conditions: Equal flow rates of amine and NaOCl solutions used. 1 M amine in toluene and 1.1 M NaOCl (aq) were used. Reactions conducted at room temperature. bStatic mixed volume in parentheses. cDetermined by NMR vs internal standard. dNaOCl (aq) 2.2 M, flow rate 0.5 mL/min, amine 1 M in toluene, flow rate 1 mL/min. e1.5 M NaOCl (aq). fEtOAc solvent instead of toluene used due to solubility of amine. gLow solubility of starting material in toluene caused problems with amine feed giving variation in yields. g2 M NaOCl (aq) used. h40% N-chlorodibenzylamine and 60% residual dibenzylamine observed by 1H NMR, no other products observed.
Green metrics
A key driver within the chemical industry is the need for greener, more sustainable processes. In order to assess the sustainability of the continuous process for chloramine formation described in this publication, the metrics of the reaction were assessed and compared with existing literature procedures [35]. The results are summarised in Table 3.
Table 3: Comparison of green metrics for different chloramine formation procedures.
| Entry | 1a | 2b | 3c | 4d | 5e |
|---|---|---|---|---|---|
| Amine |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-262-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-262-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-262-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-262-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-262-i18.svg?max-width=637&scale=1.0)
|
| Chlorine source | NaOCl | NaOCl/ t-BuOH | N-chloro succinimide | NaOCl | NaOCl |
| Reactor | Flow | Batch | Batch | Batch | Flow |
| Reaction solvent | toluene, H2O | TBME, H2O | Et2Of | H2O | toluene, H2O |
| Work-up solvents | – | H2O, brine | H2O | Et2O | – |
| Conversion | 100 | 100 | 100 | 100 | 100 |
| Yield | 94 | 100 | 90 | 88 | 84 |
| Reaction mass efficiency | 57.73 | 63.42 | 46.37 | 63.44 | 60.24 |
| Atom economy | 74.94 | 53.94 | 54.69 | 75.25 | 75.25 |
|
Mass intensity:
total |
18.17 | 37.5 | 80.3 | 12.39 | 15.09 |
| Mass intensity: reaction | 18.17 | 7.56 | 33.71 | 8.43 | 15.09 |
| Mass intensity: reaction chemicals | 1.73 | 2.03 | 2.16 | 1.58 | 1.66 |
| Mass intensity: reaction solvents | 16.43 | 5.54 | 31.55 | 6.85 | 13.43 |
| Mass intensity: work-up | – | 29.94 | 46.58 | 3.96 | – |
aSee Table 2, entry 3. bReference [20]. cReference [36]. dReference [11]. eSee Table 2, entry 1. fDCM has also been used for reactions of other amines with NCS [3].
The yields of the continuous N-chloramine process were high, and comparable to literature batch procedures. The atom economy of our process was increased compared to the use of N-chlorosuccinimide [3], or NaOCl/t-BuOH, Table 3, entries 2 and 3 [20]. There are also improvements to the total mass intensity of the flow procedure, primarily by avoiding work-up and purification procedures. The mass efficiency remains low, but is comparable to literature procedures. Whilst increasing the reactant concentrations is possible, the safe removal of the heat of reaction must be considered, moreover the flow process is much more productive than batch. Nevertheless the N-chloramine solution that is generated should be used directly, so is ideally coupled with a second flow process, and the results of this will be reported elsewhere.
Conclusion
The facile synthesis of N,N-dialkyl-N-chloramines is described using either a tubular reactor with static mixers or a continuous stirred tank reactor; both are able to promote efficient mixing of the biphasic reaction solution. Those amines which partition mainly in the organic phase required longer reaction times to compensate for the reduced mass transfer between the organic and aqueous phases, and good yields with useful productivities were achieved for most. The green metrics of the reaction have been assessed and compared to existing procedures for chloramine formation, and have shown the continuous process is improved over previous procedures. Work to expand the scope of N-chloramines, and their subsequent use in flow reactions with alkenes to make amines, aldehydes to make amides, and base to make imines is ongoing.
Experimental
All reagents were used as received from suppliers without purification. Sodium hypochlorite solution 10–15% available chlorine was purchased from Sigma-Aldrich. The accurate NaOCl concentration was determined by titration (Supporting Information File 1) and diluted to the required concentration with distilled water. CDCl3 purchased from Sigma-Aldrich was used for NMR analysis. NMR spectra (1H and 13C) were obtained on either a Bruker Advance 500 MHz, 400 MHz or a Bruker DPX 300 MHz spectrometer. NMR spectra were referenced to either TMS or CHCl3. The partitioning of the amine reagents in the organic phase of a biphasic organic/aqueous solution was determined by GC analysis using an Agilent HP 6890 with FID (Supporting Information File 1). Calorimetry experiments were carried out using HEL AutoMATE parallel reactors with HEL WinISO 2225 and HEL IQ 1.2.16 software. For the tube reactor, Harvard syringe pumps (model 981074) or JASCO PU-980 HPLC pumps were connected via a syringe and 1.5 inch, 21 guage disposable needle to PTFE tubing with 1/16 inch OD. The 1/16 inch stainless steel T-piece and stainless steel 1/16 inch to 1/4 inch increasing connector were obtained from Swagelok. The reactor tube comprises of PTFE tubing 1/4 inch OD and in-line plastic static mixers supplied by Nordson EFD (3/16 inch OD, mixer element diameter 1/8 inch, 3.5 inch length). For the CSTR, the same Harvard syringe pumps were used along with PTFE tubing with 1/8 inch OD.
General procedure for N-chloramine formation using the tube reactor and static mixers
The required number of static mixers (3/16 inch OD, 1/8 inch mixing element diameter 3.5 inch length) was inserted into a length of 1/4 inch PTFE tubing to give the overall required reactor volume. The reactor was connected to 1/16 inch tubing via a 1/4–1/16 inch reducing adapter. The tubing was split with a T-piece to 2 pumps (either piston or syringe pumps). A 1 M solution of amine in toluene was prepared and fed via pump 1 at the required flow rate. A 1.1 M solution of aqueous NaOCl was fed via pump 2 at the required flow rate. The reaction solution was flowed through the reactor and collected in residence time fractions. The organic phase was separated from each fraction to avoid further interaction with the NaOCl solution. The organic phase was analysed by 1H NMR. For non-volatile products yields were obtained by removal of toluene from the organic phase of each residence time fraction and weighing the resulting product. For volatile products yields were obtained by 1H NMR analysis. 100 μL or the organic phase was weighed and analysed by 1H NMR. The ratio of toluene/starting material/product and mass of 100 μL of the solution was used to determine the N-chloramine concentration in the solution. From this the yield of N-chloramine in the full organic phase could be determined.
General procedure for N-chloramine formation using the CSTR
Through the lid of a 50 mL jacketed glass vessel containing a stirrer bar was inserted 2 lengths of 1/8 inch PTFE tubing connected to syringe pumps. A third length of tubing was inserted so that it ended flush with the base of the lid to act as an overflow tube out of the reactor into a collection vessel. The reactor was secured on a stirrer plate.
A 1 M solution of amine in toluene was prepared and fed via pump 1 at the required flow rate. A 1.1 M solution of aqueous NaOCl was fed via pump 2 at the required flow rate. The reaction solution was flowed into the reactor, and once full was eluted from the reactor via the overflow tube and collected. The solution was collected in residence time fractions. The organic phase was separated from each fraction to avoid further interaction with the NaOCl solution. The organic phase was analysed by 1H NMR. For non-volatile products yields were obtained by removal of toluene from the organic phase of each residence time fraction and weighing the resulting product. For volatile products, yields were obtained by 1H NMR analysis. 100 μL or the organic phase was weighed and analysed by 1H NMR. The ratio of toluene/starting material/product and mass of 100 μL of the solution was used to determine the N-chloramine concentration in the organic solution. From this the yield of N-chloramine in the full organic phase could be determined.
N-Chloro-1,4-morpholine
Prepared according to the general procedure using the tube reactor with 2 static mixers, a 6 mL reactor volume and a residence time of 4 min, 2.2 M NaOCl (aq) at 0.5 mL/min and 1 M morpholine in toluene at 1 mL/min. Due to the high volatility of N-chloromorpholine and its reported instability the product was not isolated and was instead obtained as a solution in toluene (0.84 M, 84%) by separation of the organic phase form the aqueous phase of the reaction solution. The yield of product was determined by NMR analysis as in the typical procedure for volatile products. The toluene solution was analysed by NMR and data matches that reported in the literature [17]. 1H NMR (CDCl3, 500 MHz) δ ppm 3.69 (br s, 4H, CH2OCH2), 3.12 (br s, 4H CH2NClCH2); 13C NMR (CDCl3, 125 MHz) δ ppm 67.72 (CH2OCH2), 63.03 (CH2NClCH2).
N-Chloropiperidine
Prepared according to the general procedure using the tube reactor with 4 static mixers, a 4 mL reactor volume and a residence time of 4 min, 1.5 M NaOCl (aq) at 0.5 mL/min and 1 M piperidine in toluene at 0.5 mL/min. Due to the high volatility of N-chloropiperidine the product was not isolated and was instead obtained as a solution in toluene (0.94 M, 94%) by separation of the organic phase from the aqueous phase of the reaction solution. The yield of product was determined by NMR analysis as in the typical procedure for volatile products. The toluene solution was analysed by NMR and data matches that reported in the literature [17]. 1H NMR (CDCl3, 300 MHz) δ ppm 3.49–2.63 (br m, 4H, CH2NClCH2), 1.84 (quin, J = 5.8 Hz, 4H, 2 x CH2CH2CH2), 1.75–1.32 (br m, 2H, NCH2CH2CH2); 13C NMR (CDCl3, 125 ppm) δ ppm 64.00 (2 × CH2NCl), 27.68 (2 × CH2CH2NCl), 23.07 (CH2(CH2)2NCl).
N-Benzyl-N-chloromethanamine
Prepared according to the general procedures. 1) Tube reactor with 4 static mixers, a 6 mL reactor volume and a residence time of 3 min, 1.1 M NaOCl (aq) at 0.15 mL/min and 1 M N-benzylmethylamine in toluene at 0.15 mL/min. 2) CSTR with a reactor volume of 50 mL, and residence time of 25 min. 1.1 M NaOCl (aq) at 1 mL/min and 1 M N-benzylmethylamine in toluene at 1 mL/min were used. Due to the volatility of N-benzyl-N-chloromethanamine the product was not isolated and was instead obtained as a solution in toluene (1 M, quantitative yield) by separation of the organic phase from the aqueous phase of the reaction solution. The yield of product was determined by NMR analysis as in the typical procedure or volatile products. The toluene solution was analysed by NMR and data matches that reported in the literature [17]. 1H NMR (CDCl3, 300 MHz) δ ppm 7.32–7.28 (m, 5H, CHAr), 3.99 (s, 2H, CH2), 2.88 (s, 3H, CH3); 13C NMR (CDCl3, 125 MHz) δ ppm 139.37 (CAr), 128.58 (2 × CHAr), 128.49 (2 × CHAr), 127.32 (CHAr), 55.85 (CH2), 35.68 (CH3).
N-Chloro-N-methyl-p-toluenesulfonamide
Prepared according to the general procedures. 1) Tube reactor with 4 static mixers, a 6 mL reactor volume and a residence time of 3 min, 1.5 M NaOCl (aq) at 1 mL/min and 1 M N-benzylmethylamine in EtOAc at 1 mL/min. 2) CSTR with a reactor volume of 50 mL, and residence time of 25 min. 1.1 M NaOCl (aq) at 1 mL/min and 1 M N-benzylmethylamine in toluene at 1 mL/min were used. The product was isolated for each reactor volume by separation of the organic phase for each reactor volume of solution and removal of the solvent by rotary evaporation to give a white solid (1 reactor volume gives 666 mg, 3.0 mmol, quantitative yield). NMR data matches that reported in the literature [37]. 1H NMR (CDCl3, 500 MHz) δ ppm 7.82 (d, J = 8.4 Hz, 2H, CHAr), 7.42 (d, J = 8.4 Hz, 2H, CHAr), 3.09 (s, 3H, NCH3), 2.48 (s, 3H, Ar-CH3); 13C NMR (CDCl3, 125 MHz) δ ppm 145.75 (CAr), 129.80 (2CHAr), 129.78 (2 CHAr), 128.28 (CAr), 45.51 (NCH3), 21.70 (ArCH3).
N-Chloro-N,N-dibutylamine
Prepared according to the general procedures. 1) Tube reactor with 4 static mixers, a 4 mL reactor volume and a residence time of 20 min, 2 M NaOCl (aq) at 0.1 mL/min and 1 M dibutylamine in toluene at 0.1 mL/min. 2) CSTR with a reactor volume of 50 mL, and residence time of 50 min. 1.1 M NaOCl (aq) at 0.5 mL/min and 1 M dibutylamine in toluene 0.5 mL/min were used. The product was isolated for each reactor volume by separation of the organic phase for each reactor volume of solution and removal of the solvent by rotary evaporation to give a colourless oil (1 reactor volume gives 8.17 g, 50 mmol, quantitative yield). NMR data matches that reported in the literature [17]. 1H NMR (CDCl3, 300 MHz) δ ppm 3.07–3.02 (m, 4H, 2 × NClCH2), 1.85–1.74 (m, 4H, 2 × NCH2CH2CH2), 1.54–1.45 (m, 4H, 2 × CH2CH3), 1.11–1.05 (t, J = 7.4 Hz, 6H, 2 × CH3); 13C NMR (CDCl3, 125 MHz) δ ppm 64.04 (2 × CH2NCl), 30.01 (2 × CH2CH2NCl), 20.03 (2 × CH2(CH2)2NCl), 13.90 (2 × CH3).
N-Chloro-N,N-dibenzylamine
Prepared according to the general procedure using the CSTR with a reactor volume of 50 mL, and residence time of 50 min, 1.1 M NaOCl (aq) at 0.5 mL/min and 1 M dibenzylamine in toluene 0.5 mL/min were used. The product was not isolated and was instead obtained as a solution in toluene (0.4 M, 40% conversion) by separation of the organic phase from the aqueous phase of the reaction solution and removal of the solvent by rotary evaporation. The conversion to product was determined by NMR analysis of dried product (orange oil) and data obtained matches that reported in the literature [38]. 1H NMR (CDCl3, 300 MHz) 40% conversion to chloramine δ ppm 7.56–7.44 (m, 20H, CHAr in amine and chloramine), 4.28 (s, 4H, 2 × NClCH2 in chloramine), 3.80 (s, 4H 2 × NHCH2 in amine); 13C NMR (CDCl3, 125 MHz) δ ppm chloramine: 137.17 (2 × CAr), 129.18 (4 × CHAr), 128.52 (4 × CHAr), 127.95 (2 × CHAr), 67.24 (2 × CH2NCl); amine: 136.26 (2 × CAr), 128.47 (4 × CHAr), 128.39 (4 × CHAr), 127.19 (2 × CHAr), 52.94 (2 × CH2NH).
Supporting Information
| Supporting Information File 1: Details of the titration method for determination of NaOCl strength, determination of amine partition coefficients, GC analytical conditions and calorimetry. | ||
| Format: PDF | Size: 390.6 KB | Download |
Acknowledgements
The research for this work has received funding from the Innovative Medicines Initiative joint undertaking project Chem21 under grant agreement no. 115360, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution.
References
-
Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076–1091. doi:10.1021/cs400138b
Return to citation in text: [1] [2] [3] -
Göttlich, R. Synthesis 2000, 1561–1564. doi:10.1055/s-2000-7605
Return to citation in text: [1] -
Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848–1854. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V
Return to citation in text: [1] [2] [3] [4] -
Liu, X.-Y.; Gao, P.; Shen, Y.-W.; Liang, Y.-M. Adv. Synth. Catal. 2011, 353, 3157–3160. doi:10.1002/adsc.201100382
Return to citation in text: [1] -
Porcheddu, A.; De Luca, L. Adv. Synth. Catal. 2012, 354, 2949–2953. doi:10.1002/adsc.201200659
Return to citation in text: [1] -
Vanjari, R.; Guntreddi, T.; Singh, K. N. Green Chem. 2014, 16, 351–356. doi:10.1039/C3GC41548A
Return to citation in text: [1] -
Vanjari, R.; Guntreddi, T.; Singh, K. N. Org. Lett. 2013, 15, 4908–4911. doi:10.1021/ol4023886
Return to citation in text: [1] -
Bartsch, R. A.; Cho, B. R. J. Am. Chem. Soc. 1979, 101, 3587–3591. doi:10.1021/ja00507a025
Return to citation in text: [1] -
Anderson, P. S.; Lundell, G. F.; Cias, J. L.; Robinson, F. M. Tetrahedron Lett. 1971, 12, 2787–2790. doi:10.1016/S0040-4039(01)96980-1
Return to citation in text: [1] -
Qin, Q.; Yu, S. Org. Lett. 2015, 17, 1894–1897. doi:10.1021/acs.orglett.5b00582
Return to citation in text: [1] -
Lindsay Smith, J. R.; McKeer, L. C.; Taylor, J. M. Org. Synth. 1989, 67, 222–228. doi:10.15227/orgsyn.067.0222
Return to citation in text: [1] [2] [3] [4] -
Cossy, J.; Tresnard, L.; Gomez Pardo, D. Tetrahedron Lett. 1999, 40, 1125–1128. doi:10.1016/S0040-4039(98)02585-4
Return to citation in text: [1] -
Leal, R. A.; Beaudry, D. R.; Alzghari, S. K.; Sarpong, R. Org. Lett. 2012, 14, 5350–5353. doi:10.1021/ol302535r
Return to citation in text: [1] -
Enders, D.; Schaumann, E., Eds. Science of Science - Houben-Weyl Methods of Molecular Transformations: Compounds with one saturated carbon-heteroatom bond; Thieme: Stuuttgart, Germany, 2008; Vol. 40b, pp 901–919.
Return to citation in text: [1] [2] [3] [4] [5] -
Urben, P. G.; Pitt, M. J., Eds. Bretherick's Handbook of Reactive Chemical Hazards; Academic Press: Oxford, UK, 2006; Vol. 2, pp 167–168.
Return to citation in text: [1] -
Kovacic, P.; Lowery, M. K.; Field, K. W. Chem. Rev. 1970, 70, 639–665. doi:10.1021/cr60268a002
Return to citation in text: [1] -
Barker, T. J.; Jarvo, E. R. J. Am. Chem. Soc. 2009, 131, 15598–15599. doi:10.1021/ja907038b
Return to citation in text: [1] [2] [3] [4] [5] -
Paquette, L. A., Ed. Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons: New York, 1995; Vol. 2.
Return to citation in text: [1] -
Zimmer, H.; Audrieth, L. F. J. Am. Chem. Soc. 1954, 76, 3856–3857. doi:10.1021/ja01643a082
Return to citation in text: [1] -
Zhong, Y.-L.; Zhou, H.; Gauthier, D. R.; Lee, J.; Askin, D.; Dolling, U. H.; Volante, R. P. Tetrahedron Lett. 2005, 46, 1099–1101. doi:10.1016/j.tetlet.2004.12.088
Return to citation in text: [1] [2] [3] [4] -
Paquette, L. A., Ed. Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons: New York, 1995; Vol. 2, pp 889–891.
Return to citation in text: [1] -
Zhu, R.; Xu, Z.; Ding, W.; Liu, S.; Shi, X.; Lu, X. Chin. J. Chem. 2014, 32, 1039–1048. doi:10.1002/cjoc.201400471
Return to citation in text: [1] -
Padegimas, S. J.; Kovacic, P. J. Org. Chem. 1972, 37, 2672–2676. doi:10.1021/jo00982a008
Return to citation in text: [1] -
Pedder, D. J.; Fales, H. M.; Jaouni, T.; Blum, M.; MacConnell, J.; Crewe, R. M. Tetrahedron 1976, 32, 2275–2279. doi:10.1016/0040-4020(76)88001-5
Return to citation in text: [1] -
Lee, S. J.; Terrazas, M. S.; Pippel, D. J.; Beak, P. J. Am. Chem. Soc. 2003, 125, 7307–7312. doi:10.1021/ja0300463
Return to citation in text: [1] -
Adamo, A.; Heider, P. L.; Weeranoppanant, N.; Jensen, K. F. Ind. Eng. Chem. Res. 2013, 52, 10802–10808. doi:10.1021/ie401180t
Return to citation in text: [1] -
Hamlin, T. A.; Lazarus, G. M. L.; Kelly, C. B.; Leadbeater, N. E. Org. Process Res. Dev. 2014, 18, 1253–1258. doi:10.1021/op500190j
Return to citation in text: [1] -
Nagy, K. D.; Shen, B.; Jamison, T. F.; Jensen, K. F. Org. Process Res. Dev. 2012, 16, 976–981. doi:10.1021/op200349f
Return to citation in text: [1] -
Schwolow, S.; Hollmann, J.; Schenkel, B.; Röder, T. Org. Process Res. Dev. 2012, 16, 1513–1522. doi:10.1021/op300107z
Return to citation in text: [1] -
Weil, I.; Morris, J. C. J. Am. Chem. Soc. 1949, 71, 1664–1671. doi:10.1021/ja01173a033
Return to citation in text: [1] [2] -
Zhang, Y.; Born, S. C.; Jensen, K. F. Org. Process Res. Dev. 2014, 18, 1476–1481. doi:10.1021/op500158h
Return to citation in text: [1] [2] -
Darvas, F.; Dorman, G.; Hessel, V. Flow Chemistry Fundamentals; Walter De Gruyter and Co KG: Berlin, 2014.
Return to citation in text: [1] [2] -
Van Waes, F. E. A.; Seghers, S.; Dermaut, W.; Cappuyns, B.; Stevens, C. V. J. Flow Chem. 2014, 4, 118–124. doi:10.1556/JFC-D-14-00006
Return to citation in text: [1] -
In-line static mixers obtained from Nordsen EFD. 3/16 inch OD, 1/8 inch ID, 839 cm length.
Return to citation in text: [1] -
McElroy, C. R.; Constantinou, A.; Jones, L. C.; Summerton, L.; Clark, J. H. Green Chem. 2015, 17, 3111–3121. doi:10.1039/C5GC00340G
Return to citation in text: [1] -
Grandl, J.; Sakr, E.; Kotzyba-Hibert, F.; Krieger, F.; Bertrand, S.; Bertrand, D.; Vogel, H.; Goeldner, M.; Hovius, R. Angew. Chem., Int. Ed. 2007, 46, 3505–3508. doi:10.1002/anie.200604807
Return to citation in text: [1] -
Pastoriza, C.; Antelo, J. M.; Crugeiras, J. J. Phys. Org. Chem. 2013, 26, 551–559. doi:10.1002/poc.3127
Return to citation in text: [1] -
Pandiancherri, S.; Lupton, D. W. Tetrahedron Lett. 2011, 52, 671–674. doi:10.1016/j.tetlet.2010.11.142
Return to citation in text: [1]
| 36. | Grandl, J.; Sakr, E.; Kotzyba-Hibert, F.; Krieger, F.; Bertrand, S.; Bertrand, D.; Vogel, H.; Goeldner, M.; Hovius, R. Angew. Chem., Int. Ed. 2007, 46, 3505–3508. doi:10.1002/anie.200604807 |
| 11. | Lindsay Smith, J. R.; McKeer, L. C.; Taylor, J. M. Org. Synth. 1989, 67, 222–228. doi:10.15227/orgsyn.067.0222 |
| 3. | Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848–1854. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V |
| 1. | Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076–1091. doi:10.1021/cs400138b |
| 2. | Göttlich, R. Synthesis 2000, 1561–1564. doi:10.1055/s-2000-7605 |
| 3. | Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848–1854. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V |
| 8. | Bartsch, R. A.; Cho, B. R. J. Am. Chem. Soc. 1979, 101, 3587–3591. doi:10.1021/ja00507a025 |
| 14. | Enders, D.; Schaumann, E., Eds. Science of Science - Houben-Weyl Methods of Molecular Transformations: Compounds with one saturated carbon-heteroatom bond; Thieme: Stuuttgart, Germany, 2008; Vol. 40b, pp 901–919. |
| 23. | Padegimas, S. J.; Kovacic, P. J. Org. Chem. 1972, 37, 2672–2676. doi:10.1021/jo00982a008 |
| 24. | Pedder, D. J.; Fales, H. M.; Jaouni, T.; Blum, M.; MacConnell, J.; Crewe, R. M. Tetrahedron 1976, 32, 2275–2279. doi:10.1016/0040-4020(76)88001-5 |
| 17. | Barker, T. J.; Jarvo, E. R. J. Am. Chem. Soc. 2009, 131, 15598–15599. doi:10.1021/ja907038b |
| 5. | Porcheddu, A.; De Luca, L. Adv. Synth. Catal. 2012, 354, 2949–2953. doi:10.1002/adsc.201200659 |
| 6. | Vanjari, R.; Guntreddi, T.; Singh, K. N. Green Chem. 2014, 16, 351–356. doi:10.1039/C3GC41548A |
| 7. | Vanjari, R.; Guntreddi, T.; Singh, K. N. Org. Lett. 2013, 15, 4908–4911. doi:10.1021/ol4023886 |
| 25. | Lee, S. J.; Terrazas, M. S.; Pippel, D. J.; Beak, P. J. Am. Chem. Soc. 2003, 125, 7307–7312. doi:10.1021/ja0300463 |
| 38. | Pandiancherri, S.; Lupton, D. W. Tetrahedron Lett. 2011, 52, 671–674. doi:10.1016/j.tetlet.2010.11.142 |
| 1. | Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076–1091. doi:10.1021/cs400138b |
| 14. | Enders, D.; Schaumann, E., Eds. Science of Science - Houben-Weyl Methods of Molecular Transformations: Compounds with one saturated carbon-heteroatom bond; Thieme: Stuuttgart, Germany, 2008; Vol. 40b, pp 901–919. |
| 19. | Zimmer, H.; Audrieth, L. F. J. Am. Chem. Soc. 1954, 76, 3856–3857. doi:10.1021/ja01643a082 |
| 20. | Zhong, Y.-L.; Zhou, H.; Gauthier, D. R.; Lee, J.; Askin, D.; Dolling, U. H.; Volante, R. P. Tetrahedron Lett. 2005, 46, 1099–1101. doi:10.1016/j.tetlet.2004.12.088 |
| 21. | Paquette, L. A., Ed. Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons: New York, 1995; Vol. 2, pp 889–891. |
| 17. | Barker, T. J.; Jarvo, E. R. J. Am. Chem. Soc. 2009, 131, 15598–15599. doi:10.1021/ja907038b |
| 1. | Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076–1091. doi:10.1021/cs400138b |
| 4. | Liu, X.-Y.; Gao, P.; Shen, Y.-W.; Liang, Y.-M. Adv. Synth. Catal. 2011, 353, 3157–3160. doi:10.1002/adsc.201100382 |
| 11. | Lindsay Smith, J. R.; McKeer, L. C.; Taylor, J. M. Org. Synth. 1989, 67, 222–228. doi:10.15227/orgsyn.067.0222 |
| 14. | Enders, D.; Schaumann, E., Eds. Science of Science - Houben-Weyl Methods of Molecular Transformations: Compounds with one saturated carbon-heteroatom bond; Thieme: Stuuttgart, Germany, 2008; Vol. 40b, pp 901–919. |
| 22. | Zhu, R.; Xu, Z.; Ding, W.; Liu, S.; Shi, X.; Lu, X. Chin. J. Chem. 2014, 32, 1039–1048. doi:10.1002/cjoc.201400471 |
| 37. | Pastoriza, C.; Antelo, J. M.; Crugeiras, J. J. Phys. Org. Chem. 2013, 26, 551–559. doi:10.1002/poc.3127 |
| 11. | Lindsay Smith, J. R.; McKeer, L. C.; Taylor, J. M. Org. Synth. 1989, 67, 222–228. doi:10.15227/orgsyn.067.0222 |
| 14. | Enders, D.; Schaumann, E., Eds. Science of Science - Houben-Weyl Methods of Molecular Transformations: Compounds with one saturated carbon-heteroatom bond; Thieme: Stuuttgart, Germany, 2008; Vol. 40b, pp 901–919. |
| 15. | Urben, P. G.; Pitt, M. J., Eds. Bretherick's Handbook of Reactive Chemical Hazards; Academic Press: Oxford, UK, 2006; Vol. 2, pp 167–168. |
| 16. | Kovacic, P.; Lowery, M. K.; Field, K. W. Chem. Rev. 1970, 70, 639–665. doi:10.1021/cr60268a002 |
| 3. | Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848–1854. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V |
| 17. | Barker, T. J.; Jarvo, E. R. J. Am. Chem. Soc. 2009, 131, 15598–15599. doi:10.1021/ja907038b |
| 17. | Barker, T. J.; Jarvo, E. R. J. Am. Chem. Soc. 2009, 131, 15598–15599. doi:10.1021/ja907038b |
| 12. | Cossy, J.; Tresnard, L.; Gomez Pardo, D. Tetrahedron Lett. 1999, 40, 1125–1128. doi:10.1016/S0040-4039(98)02585-4 |
| 13. | Leal, R. A.; Beaudry, D. R.; Alzghari, S. K.; Sarpong, R. Org. Lett. 2012, 14, 5350–5353. doi:10.1021/ol302535r |
| 18. | Paquette, L. A., Ed. Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons: New York, 1995; Vol. 2. |
| 17. | Barker, T. J.; Jarvo, E. R. J. Am. Chem. Soc. 2009, 131, 15598–15599. doi:10.1021/ja907038b |
| 11. | Lindsay Smith, J. R.; McKeer, L. C.; Taylor, J. M. Org. Synth. 1989, 67, 222–228. doi:10.15227/orgsyn.067.0222 |
| 3. | Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848–1854. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V |
| 9. | Anderson, P. S.; Lundell, G. F.; Cias, J. L.; Robinson, F. M. Tetrahedron Lett. 1971, 12, 2787–2790. doi:10.1016/S0040-4039(01)96980-1 |
| 10. | Qin, Q.; Yu, S. Org. Lett. 2015, 17, 1894–1897. doi:10.1021/acs.orglett.5b00582 |
| 14. | Enders, D.; Schaumann, E., Eds. Science of Science - Houben-Weyl Methods of Molecular Transformations: Compounds with one saturated carbon-heteroatom bond; Thieme: Stuuttgart, Germany, 2008; Vol. 40b, pp 901–919. |
| 20. | Zhong, Y.-L.; Zhou, H.; Gauthier, D. R.; Lee, J.; Askin, D.; Dolling, U. H.; Volante, R. P. Tetrahedron Lett. 2005, 46, 1099–1101. doi:10.1016/j.tetlet.2004.12.088 |
| 28. | Nagy, K. D.; Shen, B.; Jamison, T. F.; Jensen, K. F. Org. Process Res. Dev. 2012, 16, 976–981. doi:10.1021/op200349f |
| 29. | Schwolow, S.; Hollmann, J.; Schenkel, B.; Röder, T. Org. Process Res. Dev. 2012, 16, 1513–1522. doi:10.1021/op300107z |
| 20. | Zhong, Y.-L.; Zhou, H.; Gauthier, D. R.; Lee, J.; Askin, D.; Dolling, U. H.; Volante, R. P. Tetrahedron Lett. 2005, 46, 1099–1101. doi:10.1016/j.tetlet.2004.12.088 |
| 26. | Adamo, A.; Heider, P. L.; Weeranoppanant, N.; Jensen, K. F. Ind. Eng. Chem. Res. 2013, 52, 10802–10808. doi:10.1021/ie401180t |
| 27. | Hamlin, T. A.; Lazarus, G. M. L.; Kelly, C. B.; Leadbeater, N. E. Org. Process Res. Dev. 2014, 18, 1253–1258. doi:10.1021/op500190j |
| 35. | McElroy, C. R.; Constantinou, A.; Jones, L. C.; Summerton, L.; Clark, J. H. Green Chem. 2015, 17, 3111–3121. doi:10.1039/C5GC00340G |
| 20. | Zhong, Y.-L.; Zhou, H.; Gauthier, D. R.; Lee, J.; Askin, D.; Dolling, U. H.; Volante, R. P. Tetrahedron Lett. 2005, 46, 1099–1101. doi:10.1016/j.tetlet.2004.12.088 |
| 30. | Weil, I.; Morris, J. C. J. Am. Chem. Soc. 1949, 71, 1664–1671. doi:10.1021/ja01173a033 |
| 34. | In-line static mixers obtained from Nordsen EFD. 3/16 inch OD, 1/8 inch ID, 839 cm length. |
| 32. | Darvas, F.; Dorman, G.; Hessel, V. Flow Chemistry Fundamentals; Walter De Gruyter and Co KG: Berlin, 2014. |
| 33. | Van Waes, F. E. A.; Seghers, S.; Dermaut, W.; Cappuyns, B.; Stevens, C. V. J. Flow Chem. 2014, 4, 118–124. doi:10.1556/JFC-D-14-00006 |
| 31. | Zhang, Y.; Born, S. C.; Jensen, K. F. Org. Process Res. Dev. 2014, 18, 1476–1481. doi:10.1021/op500158h |
| 30. | Weil, I.; Morris, J. C. J. Am. Chem. Soc. 1949, 71, 1664–1671. doi:10.1021/ja01173a033 |
| 31. | Zhang, Y.; Born, S. C.; Jensen, K. F. Org. Process Res. Dev. 2014, 18, 1476–1481. doi:10.1021/op500158h |
| 32. | Darvas, F.; Dorman, G.; Hessel, V. Flow Chemistry Fundamentals; Walter De Gruyter and Co KG: Berlin, 2014. |
© 2015 Blacker and Jolley; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)