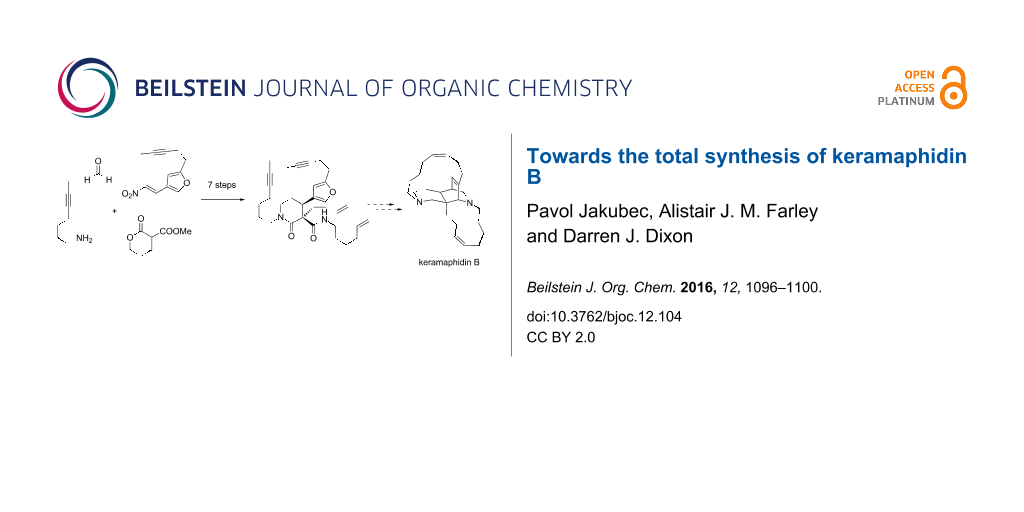
Abstract
The enantio- and diastereoselective Michael addition of a δ-valerolactone-derived pronucleophile to a substituted furanyl nitroolefin catalysed by a bifunctional cinchonine-derived thiourea has been used as the key stereocontrolling step in a new synthetic strategy to the heavily functionalised piperidine core of keramaphidin B.

Graphical Abstract
Introduction
Keramaphidin B (1) is a marine alkaloid first isolated by Kobayashi in 1994 from the Okinawan marine sponge Amphimedon sp and has been shown to be cytotoxic against KB human epidermoid carcinoma cells (IC50 0.28 μg/mL) and P388 murine leukemia cells (IC50 0.28 μg/mL) [1]. It is a member of the manzamine alkaloids and has an exquisite molecular structure comprising a 6,6,6,11,13 pentacycle possessing 4 stereogenic centres including one quaternary centre (Figure 1). In 1992, two years before its isolation, Baldwin and Whitehead, in their landmark paper entitled ‘On the Biosynthesis of Manzamines’ postulated that keramaphidin B was a common intermediate in the biosynthesis of the manzamine alkaloids [2]. Several years later, Baldwin and co-workers synthesised keramaphidin B following a biomimetic pathway via an intramolecular Diels–Alder reaction as the late stage key step. After extensive purification, the authors were able to isolate keramaphidin B in just 0.3% yield, but nevertheless they provided evidence for the biosynthesis [3]. A year later, Baldwin et al. completed an alternative synthesis by performing an intermolecular Diels–Alder reaction and a double late stage RCM reaction to close the two macrocyclic rings; albeit the last stage afforded 1 in 1% yield after separation of various oligomeric byproducts [4].
![[1860-5397-12-104-1]](/bjoc/content/figures/1860-5397-12-104-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Our group has had a long-standing research program dedicated towards the total syntheses of the manzamine alkaloids with a particular emphasis on the development of novel catalytic methods for simplifying their syntheses [5-10]. Owing to keramaphidin B’s attractive structure combined with its interesting biological profile and the lack of an efficient method for its synthesis, we selected 1 as a suitable synthetic target. Our plan was to design and implement a new synthetic route that integrated some of our newly developed bifunctional organocatalytic reactions, as well as cascade technologies, to rapidly and stereoselectively build up the core of this fascinating molecule. Herein we wish to report our preliminary synthetic efforts towards the stereoselective synthesis of the heavily functionalised piperidine core of keramaphidin B (1).
Results and Discussion
Our overall synthetic strategy is presented in Figure 2. We envisaged that a late stage alkyne RCM reaction of 2 and cis-selective hydrogenation of the internal alkyne would present an efficient method for the synthesis of the 13-membered ring.
![[1860-5397-12-104-2]](/bjoc/content/figures/1860-5397-12-104-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis of keramaphidin B.
Figure 2: Retrosynthetic analysis of keramaphidin B.
The synthesis of the 11-membered ring could be achieved by a Z-selective alkene RCM reaction [5] to afford spirocyclic bislactam 4 from metathesis precursor 5. Bisalkene 5 could in turn be synthesised by an aminolysis/oxidation/olefination sequence of the terminal alcohol 6, following traceless nitro group removal.
5-Nitropiperidin-2-one 6 in turn could be accessed by a nitro-Mannich lactamisation cascade reaction between Michael adduct 7, formaldehyde and a suitable primary amine, following our well-established protocol [6-12]. The key quaternary stereocentre of keramaphidin B, we envisaged, would be installed through an enantio- and diastereoselective organocatalytic Michael addition [13-15] between pronucleophile 8 and the known substituted furanyl nitroolefin 9 under the control of a cinchona-derived bifunctional Brønsted base/H-bond donor organocatalyst developed in our group and others [16-19].
Bifunctional organocatalysed Michael addition studies
In our previous total syntheses of nakadomarin A [5,7,20] and manzamine A [10] the stereochemical configuration of the quaternary carbon was established by a diastereoselective Michael addition between a chiral, single enantiomer, cyclic β-amido ester and a nitroolefin, and, in the case of nakadomarin A the reaction could be rendered catalytic using a bifunctional cinchonine-derived urea catalyst. We reasoned that a similar catalytic approach could be used to fix the absolute stereochemical configuration in the Michael addition reaction between δ-valerolactone pronucleophile 8 and nitro-olefin 9, although a degree of uncertainty as to the relative stereochemical outcome of the catalyst-controlled Michael reaction remained present. Accordingly, we chose to probe reactivity and establish relative stereocontrol using a close model system comprising pronucleophile 8 and furanyl nitroolefin 11 (Scheme 1).
![[1860-5397-12-104-i1]](/bjoc/content/inline/1860-5397-12-104-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Enantio- and diastereoselective bifunctional thiourea 12 organocatalysed Michael addition. (a) CO(OMe)2, LHMDS, THF, −78 °C to rt, 83%; (b) 20 mol % 12, toluene, −20 °C, 24 h, 95:5 dr (13:14), 90:10 er for 13, 99% yield; (c) butylamine, aq formaldehyde, MeOH, reflux, 1 h, 63%.
Scheme 1: Enantio- and diastereoselective bifunctional thiourea 12 organocatalysed Michael addition. (a) CO(O...
The δ-valerolactone pronucleophile 8 was synthesised by the enolate acylation of δ-valerolactone (10) with dimethyl carbonate, using LHMDS as the base, in 83% yield. The furanyl nitroolefin 11 was readily synthesised on multigram scale via a Henry condensation, according to literature procedures [20]. We were delighted to observe that the Michael addition reaction using our previously reported cinchonine-derived bifunctional thiourea catalyst 12 afforded the addition product 13 in high yield with good levels of diastereo- and enantioselectivity (95:5 dr, 90:10 er for the major diastereomer 13). The relative stereochemical configuration of the minor diastereomeric product 14 was determined by single X-ray crystallographic analysis of rac-14 (see Supporting Information File 1) and revealed that the major diastereomer 13 indeed possessed the necessary stereochemical configuration [21] for accessing keramaphidin B, assuming the chemoselectivity of the nitro-Mannich lactamisation favoured attack at the more reactive δ-lactone carbonyl instead of that of the methyl ester.
Pleasingly, this was indeed realised at the next stage; performing a nitro-Mannich lactamisation cascade on 13 with formaldehyde and butylamine in methanol afforded lactam 15 in 63% yield, possessing the hydroxypropyl chain attached to the quaternary stereocentre, poised for further functionalisation.
Having established that the cinchonine-derived bifunctional Brønsted base/thiourea organocatalyst 12 was effective for installing two stereocentres including the quaternary carbon in a model system, we next performed the reaction using the substituted furanyl nitroolefin 9 (Scheme 2).
![[1860-5397-12-104-i2]](/bjoc/content/inline/1860-5397-12-104-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of bis alkene 5. (a) 12 (20 mol %), toluene, −20 °C, 36 h, 95:5 dr, 92% yield; (b) aq HCHO, MeOH, reflux, 70 °C, 1 h, >95:5 dr, 91:9 er, 56% yield; (c) AIBN, Bu3SnH, toluene, reflux, 0.33 h, 71% yield; (d) Ti(OiPr)4, toluene, 50 °C, 2 h, 84% yield; (e) 19 (neat), 130 °C, 24 h, 67% yield; (f) (COCl)2, DMSO, NEt3, CH2Cl2, −78 °C to rt, 0.5 h, 88% yield; (g) Cp2Ti(CH3)2, THF/toluene, reflux, 0.33 h, 42% yield.
Scheme 2: Synthesis of bis alkene 5. (a) 12 (20 mol %), toluene, −20 °C, 36 h, 95:5 dr, 92% yield; (b) aq HCH...
The organocatalysed Michael addition of 8 with substituted furanyl nitroolefin 9 [20] under the control of bifunctional organocatalyst 12 proceeded efficiently and, although the reaction time was slightly increased relative to the model system, identical levels of diastereo- and enantiocontrol were observed in the formation of 7 (92% yield). Treatment of the major diastereomeric product 7 with hept-5-yn-1-amine (16) and formaldehyde in boiling methanol afforded the lactam 6 in 56% yield as a single diastereomer in 91:9 er. Having played a key role in the Michael addition reaction and the nitro-Mannich lactamisation cascade, at this stage the nitro functionality had fully served its purpose. Accordingly, traceless reductive cleavage of the nitro group [22] using tributyltin hydride and AIBN was carried out to afford piperidin-2-one 17 in 71% yield. Lactonisation under Lewis acidic conditions afforded spirocyclic malonamate 18 possessing the correct relative stereochemistry for keramaphidin B, in 84% yield. Aminolysis under neat conditions with hex-5-en-1-amine (19) gave the primary alcohol 20 (67% yield), which was subsequently subjected to a Swern oxidation to yield the aldehyde 21 in 88% yield. Finally, treatment of the aldehyde with Petasis reagent afforded the target bisalkene RCM precursor 5 in a satisfactory 42% yield.
Conclusion
In conclusion we have utilized a bifunctional cinchona-derived thiourea organocatalyst 12 for governing the key Michael addition towards the synthesis of keramaphidin B. The catalyst imparted high levels of enantio- and diastereocontrol at the newly formed contiguous tertiary and quaternary stereocentres. The stereochemical integrity of the newly formed stereogenic centres was not compromised during a subsequent three-step nitro-Mannich lactamisation cascade, aminolysis and lactonisation sequence. Further manipulation of the pendant functional groups allowed the synthesis of target compound 5 bearing two alkynes and two terminal alkenes for successive RCM reactions to construct the 11- and 13-membered rings of keramaphidin B. RCM precursor 5 – possessing all of the necessary masked functionality already installed about a piperidin-2-one framework – represents an advanced intermediate for the potential future synthesis of keramaphidin B, and our work towards this goal will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures, analytical data, copies of NMR spectra and single X-ray crystal diffraction data of 14. | ||
| Format: PDF | Size: 2.2 MB | Download |
Acknowledgements
This work was supported by the EPSRC (Leadership Fellowship to D.J.D., Post-Doctoral Fellowship to P.J. and Doctoral Training Grant [EP/M50659X/1] to A.J.M.F.). We also thank Matt Rattley, Jinchao Yang for X-ray structure determination and the Oxford Chemical Crystallography Service for use of the instrumentation. X-ray crystallographic data have been deposited in the Cambridge Crystallographic Data Centre database (http://www.ccdc.cam.ac.uk/) under accession code 1481419.
References
-
Kobayashi, J.; Tsuda, M.; Kawasaki, N.; Matsumoto, K.; Adachi, T. Tetrahedron Lett. 1994, 35, 4383–4386. doi:10.1016/S0040-4039(00)73362-4
Return to citation in text: [1] -
Baldwin, J. E.; Whitehead, R. C. Tetrahedron Lett. 1992, 33, 2059–2062. doi:10.1016/0040-4039(92)88141-Q
Return to citation in text: [1] -
Baldwin, J. E.; Claridge, T. D. W.; Culshaw, A. J.; Heupel, F. A.; Lee, V.; Spring, D. R.; Whitehead, R. C.; Boughtflower, R. J.; Mutton, I. M.; Upton, R. J. Angew. Chem., Int. Ed. 1998, 37, 2661–2663. doi:10.1002/(SICI)1521-3773(19981016)37:19<2661::AID-ANIE2661>3.0.CO;2-D
Return to citation in text: [1] -
Baldwin, J. E.; Claridge, T. D. W.; Culshaw, A. J.; Heupel, F. A.; Lee, V.; Spring, D. R.; Whitehead, R. C. Chem. – Eur. J. 1999, 5, 3154–3161. doi:10.1002/(SICI)1521-3765(19991105)5:11<3154::AID-CHEM3154>3.0.CO;2-7
Return to citation in text: [1] -
Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563
Return to citation in text: [1] [2] [3] -
Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w
Return to citation in text: [1] [2] -
Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s
Return to citation in text: [1] [2] [3] -
Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2009, 11, 4512–4515. doi:10.1021/ol901640v
Return to citation in text: [1] [2] -
Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2011, 13, 6406–6409. doi:10.1021/ol202710g
Return to citation in text: [1] [2] -
Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D. J. J. Am. Chem. Soc. 2012, 134, 17482–17485. doi:10.1021/ja308826x
Return to citation in text: [1] [2] [3] -
Jakubec, P.; Cockfield, D. M.; Helliwell, M.; Raftery, J.; Dixon, D. J. Beilstein J. Org. Chem. 2012, 8, 567–578. doi:10.3762/bjoc.8.64
Return to citation in text: [1] -
Clark, P. G. K.; Vieira, L. C. C.; Tallant, C.; Fedorov, O.; Singleton, D. C.; Rogers, C. M.; Monteiro, O. P.; Bennett, J. M.; Baronio, R.; Müller, S.; Daniels, D. L.; Méndez, J.; Knapp, S.; Brennan, P. E.; Dixon, D. J. Angew. Chem., Int. Ed. 2015, 54, 6217–6221. doi:10.1002/anie.201501394
Return to citation in text: [1] -
Marqués-López, E.; Herrera, R. P.; Christmann, M. Nat. Prod. Rep. 2010, 27, 1138–1167. doi:10.1039/b924964h
Return to citation in text: [1] -
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653
Return to citation in text: [1] -
Chen, D. Y.-K.; Ma, D. Beilstein J. Org. Chem. 2013, 9, 1192–1193. doi:10.3762/bjoc.9.134
Return to citation in text: [1] -
Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j
Return to citation in text: [1] -
Li, B.-J.; Jiang, L.; Liu, M.; Chen, Y.-C.; Ding, L.-S.; Wu, Y. Synlett 2005, 603–606. doi:10.1055/s-2005-863710
Return to citation in text: [1] -
McCooey, S. H.; Connon, S. J. Angew. Chem., Int. Ed. 2005, 44, 6367–6370. doi:10.1002/anie.200501721
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s
Return to citation in text: [1] -
Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h
Return to citation in text: [1] [2] [3] -
The absolute stereochemical configuration of the tertiary stereocentre was assigned by analogy; see references [7,11,16,20].
Return to citation in text: [1] -
Ono, N.; Kaji, A. Synthesis 1986, 693–704. doi:10.1055/s-1986-31754
Return to citation in text: [1]
| 1. | Kobayashi, J.; Tsuda, M.; Kawasaki, N.; Matsumoto, K.; Adachi, T. Tetrahedron Lett. 1994, 35, 4383–4386. doi:10.1016/S0040-4039(00)73362-4 |
| 5. | Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563 |
| 6. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 7. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s |
| 8. | Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2009, 11, 4512–4515. doi:10.1021/ol901640v |
| 9. | Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2011, 13, 6406–6409. doi:10.1021/ol202710g |
| 10. | Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D. J. J. Am. Chem. Soc. 2012, 134, 17482–17485. doi:10.1021/ja308826x |
| 4. | Baldwin, J. E.; Claridge, T. D. W.; Culshaw, A. J.; Heupel, F. A.; Lee, V.; Spring, D. R.; Whitehead, R. C. Chem. – Eur. J. 1999, 5, 3154–3161. doi:10.1002/(SICI)1521-3765(19991105)5:11<3154::AID-CHEM3154>3.0.CO;2-7 |
| 7. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s |
| 11. | Jakubec, P.; Cockfield, D. M.; Helliwell, M.; Raftery, J.; Dixon, D. J. Beilstein J. Org. Chem. 2012, 8, 567–578. doi:10.3762/bjoc.8.64 |
| 16. | Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j |
| 20. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h |
| 3. | Baldwin, J. E.; Claridge, T. D. W.; Culshaw, A. J.; Heupel, F. A.; Lee, V.; Spring, D. R.; Whitehead, R. C.; Boughtflower, R. J.; Mutton, I. M.; Upton, R. J. Angew. Chem., Int. Ed. 1998, 37, 2661–2663. doi:10.1002/(SICI)1521-3773(19981016)37:19<2661::AID-ANIE2661>3.0.CO;2-D |
| 21. | The absolute stereochemical configuration of the tertiary stereocentre was assigned by analogy; see references [7,11,16,20]. |
| 2. | Baldwin, J. E.; Whitehead, R. C. Tetrahedron Lett. 1992, 33, 2059–2062. doi:10.1016/0040-4039(92)88141-Q |
| 20. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h |
| 16. | Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j |
| 17. | Li, B.-J.; Jiang, L.; Liu, M.; Chen, Y.-C.; Ding, L.-S.; Wu, Y. Synlett 2005, 603–606. doi:10.1055/s-2005-863710 |
| 18. | McCooey, S. H.; Connon, S. J. Angew. Chem., Int. Ed. 2005, 44, 6367–6370. doi:10.1002/anie.200501721 |
| 19. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 10. | Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D. J. J. Am. Chem. Soc. 2012, 134, 17482–17485. doi:10.1021/ja308826x |
| 13. | Marqués-López, E.; Herrera, R. P.; Christmann, M. Nat. Prod. Rep. 2010, 27, 1138–1167. doi:10.1039/b924964h |
| 14. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 15. | Chen, D. Y.-K.; Ma, D. Beilstein J. Org. Chem. 2013, 9, 1192–1193. doi:10.3762/bjoc.9.134 |
| 20. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h |
| 6. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 7. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s |
| 8. | Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2009, 11, 4512–4515. doi:10.1021/ol901640v |
| 9. | Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2011, 13, 6406–6409. doi:10.1021/ol202710g |
| 10. | Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D. J. J. Am. Chem. Soc. 2012, 134, 17482–17485. doi:10.1021/ja308826x |
| 11. | Jakubec, P.; Cockfield, D. M.; Helliwell, M.; Raftery, J.; Dixon, D. J. Beilstein J. Org. Chem. 2012, 8, 567–578. doi:10.3762/bjoc.8.64 |
| 12. | Clark, P. G. K.; Vieira, L. C. C.; Tallant, C.; Fedorov, O.; Singleton, D. C.; Rogers, C. M.; Monteiro, O. P.; Bennett, J. M.; Baronio, R.; Müller, S.; Daniels, D. L.; Méndez, J.; Knapp, S.; Brennan, P. E.; Dixon, D. J. Angew. Chem., Int. Ed. 2015, 54, 6217–6221. doi:10.1002/anie.201501394 |
| 5. | Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563 |
| 5. | Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563 |
| 7. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s |
| 20. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h |
© 2016 Jakubec et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)