Department of Drug Delivery, Helmholtz Centre for Infection Research and Helmholtz Institute for Pharmaceutical Research Saarland, Campus E8.1, 66123 Saarbruecken, Germany
Biopharmaceutics and Pharmaceutical Technology, Saarland University, Campus A 4.1, 66123 Saarbruecken, Germany
Department of Drug Delivery, Helmholtz Centre for Infection Research and Helmholtz Institute for Pharmaceutical Research Saarland, Campus E8.1, 66123 Saarbruecken, Germany
Biopharmaceutics and Pharmaceutical Technology, Saarland University, Campus A 4.1, 66123 Saarbruecken, Germany
1Institut Galien (UMR CNRS 8612) Faculté de Pharmacie, Université Paris-Sud, 5, rue Jean-Baptiste Clément, 92296 Châtenay-Malabry, France
2Department of Drug Delivery, Helmholtz Centre for Infection Research and Helmholtz Institute for Pharmaceutical Research Saarland, Campus E8.1, 66123 Saarbruecken, Germany
3Biopharmaceutics and Pharmaceutical Technology, Saarland University, Campus A 4.1, 66123 Saarbruecken, Germany
4BIOCIS (UMR CNRS 8076) Faculté de Pharmacie, Université Paris-Sud, 5, rue Jean-Baptiste Clément, 92296 Châtenay-Malabry, France
Corresponding author email
Associate Editor: S. C. Zimmerman Beilstein J. Org. Chem.2016,12, 1127–1135.https://doi.org/10.3762/bjoc.12.109 Received 25 Jan 2016,
Accepted 19 May 2016,
Published 06 Jun 2016
The synthesis of ω-di-(trideuteromethyl)-trisnorsqualenic acid has been achieved from natural squalene. The synthesis features the use of a Shapiro reaction of acetone-d6 trisylhydrazone as a key step to implement the terminal isopropylidene-d6 moiety. The obtained squalenic acid-d6 has been coupled to gemcitabine to provide the deuterated analogue of squalenoyl gemcitabine, a powerful anticancer agent endowed with self-assembling properties. The Raman spectra of both deuterated and non-deuterated squalenoyl gemcitabine nanoparticles displayed significant Raman scattering signals. They revealed no differences except from the deuterium peak patterns in the silent spectral region of cells. This paves the way for label-free intracellular trafficking studies of squalenoyl nanomedicines.
Application of nanotechnology to medicine holds promises to profoundly impact healthcare especially to treat severe diseases such as cancer, intracellular infections, neurodegenerative diseases, etc. Indeed, the nanometric size confers to drug delivery systems unique properties which improve the pharmacokinetics and the biodistribution of many active compounds, thus increasing specificity, therapeutic efficacy and reducing systemic exposure and toxicity [1,2]. In recent years, many drug delivery systems have been developed covering all aspects of medicine. Among them, lipid drug conjugates (LDC) were especially developed for the delivery of hydrophilic drugs by covalent coupling with lipid components [3,4]. In this context we recently found that the chemical conjugation of squalene, a natural and biocompatible triterpene, to a drug led to the formation of a prodrug that spontaneously self-assembled as nanoparticles in water. The advantage of this approach is a very high drug loading into the nanoparticles and the absence of burst release [5]. The proof of concept of this method has been done using gemcitabine (2), an anticancer chemotherapeutic drug used to treat various solid tumors [6]. Remarkably, the squalene conjugate of gemcitabine (GemSQ) self-assembled in aqueous media as nanoassemblies of around 100 nm mean particle size with a low polydispersity index. The nanosuspension exhibited impressively greater anticancer activity than free gemcitabine against different experimental tumor models [7-11] overcoming the main drawbacks of the parent drug such as its short biological half-life and its low intracellular diffusion [12,13]. Following these initial results, the squalenoylation method was extended to other nucleoside analogues such as antiretroviral agents, ddC, ddI and AZT [14] and to siRNA oligonucleotides [15]. More notably, squalenoylation of adenosine and the subsequent formation of NAs, allowed prolonged circulation of this nucleoside, providing neuroprotection in mice with induced focal cerebral ischemia and in rats undergoing spinal cord injury [16]. Interestingly, the “squalenisation platform” initially developed with highly hydrophilic therapeutics has been further extended to hydrophobic drugs such as beta-lactam antibiotics [17], paclitaxel [18], indolinone kinase inhibitors [19] or doxorubicin [20].
For the elucidation of the mechanisms involved in the efficacy of these promising nanomedicines, the precise knowledge regarding the cellular uptake, the intracellular localization and the determination of the subcellular interactions and trafficking is crucial. To fulfill this task, radioactive labeling or fluorescent probes have been thoroughly used. For example, the subcellular localization of the 3H-radiolabeled GemSQ conjugate has been evaluated by micro-autoradiography coupled to confocal imaging of fluorescently labeled cellular structures [21]. A dual radioactive labeling 3H,14C has been taken into profit to study the pharmacokinetics, the biodistribution and the metabolism of squalenoyl adenosine nanoparticles [22]. Nevertheless, the synthesis of labeled compounds is chemically challenging, expensive and submitted to drastic regulation rules. In addition, the use of fluorescent probes requires the covalent binding of large dye molecules (bodipy, cyanine, rhodamine etc, ...) to the drug conjugate, thus potentially modifying its physicochemical profile as well as the in vivo fate and the pharmacological activity. A simple encapsulation of an amphiphilic fluorochrome in LDC nanoparticles can be used as far as the colloidal stability of the nanocarrier is preserved, but cannot address the intracellular tracking of the loaded drug after carrier disassembling. Thus, specific tools such as fluorescence resonance energy transfer (FRET) and fluorescence quenching, have been developed to study the stability of nanoparticles [23].
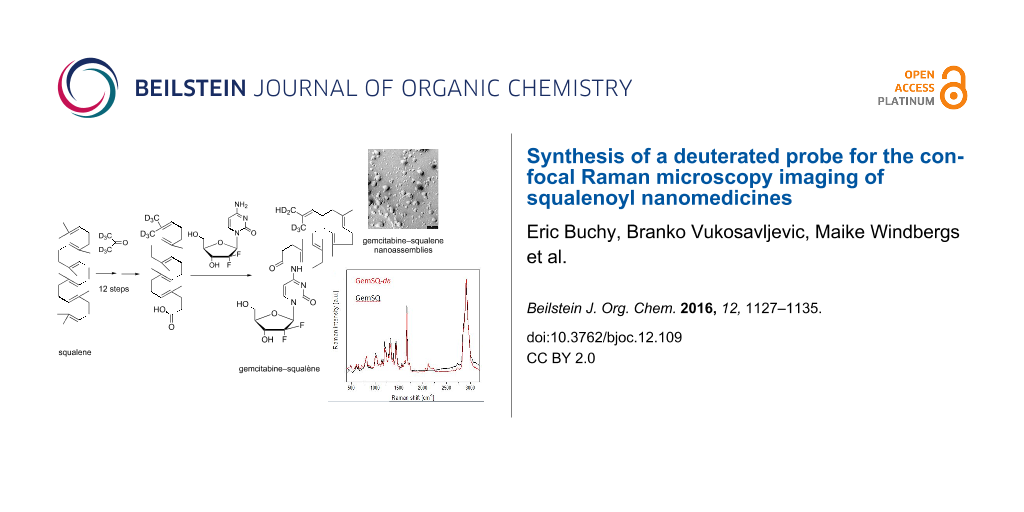
In this context, Raman spectroscopy is an interesting technique which is based on the detection of scattered laser light upon irradiating the sample. Nevertheless, because of the low intensity of the Raman scattering, efficient in vivo confocal Raman microspectroscopy of cells had to wait until laser technology and mathematical image processing have made enough progress [24,25]. In contrast to fluorescence spectroscopy, Raman spectroscopy is label-free, as its scattering effect is unique for a specific molecular structure. Raman spectra of cells usually consist of spectral contributions from proteins, lipids and polysaccharides. For the simultaneous detection of both the drug and the cell components with the aim to investigate how they interact, a significant spectral contrast is required. Unfortunately, it can be hardly achieved when dealing with low drug concentrations or biological-like structures such as peptide drugs or nucleoside analogues. To overcome this issue, deuterium can be introduced to a sample molecule, as it exhibits a significant Raman signal at around 2200 cm−1, which is in a so called “silent region” (1800–2800 cm−1) of most biological molecules. For example as early as 1976, specifically deuterated stearic acids have been used by Sunder et al. in Raman studies and Stiebing et al. studied the uptake of arachidonic acid in human macrophages [26,27]. Furthermore, deuterium does not change the physicochemical properties and thus does not perturb the structure of the described NAs. Consequently, deuterated squalenic acid is expected to be excellent as bioorthogonal Raman tag for squalene-based NAs. Since the Raman signal intensity is expected to increase with the number of deuterium atoms in the same chemical environment, we have undertaken an effort directed towards the synthesis of ω-di-(trideuteromethyl)trisnorsqualenic acid (SQCO2H-d6, 1) bearing six deuterium atoms on a non-labile position via isotopic exchange. We disclose herein the synthesis of this deuterated Raman probe from natural squalene, its coupling with gemcitabine and the Raman spectra of the deuterated GemSQ nanoassemblies, opening the way to perform intracellular imaging of squalenoyl nanomedicine (Figure 1).
Figure 1:
Structure of squalenic acid-d6, gemcitabine and GemSQ-d6 conjugate.
Figure 1:
Structure of squalenic acid-d6, gemcitabine and GemSQ-d6 conjugate.
Chemical synthesis of squalenic acid-d6 and GemSQ-d6 conjugate
In a first approach we decided to explore the selective Wittig mono-olefination of dialdehyde 5 readily accessible from the known 2,3;22,23-epoxysqualene (7) [28]. As depicted in Scheme 1, the synthetic sequence began with the treatment of squalene with two equivalents of NBS in a water/THF mixture. After separation of the bis-bromohydrin from the monoproduct, potassium carbonate treatment gave the expected diepoxide 7. Oxidative cleavage with periodic acid provided the corresponding dialdehyde 5 in 17% overall yield from squalene. The perdeuterated phosphonium salt 9 was obtained by simple condensation of commercially available 2-bromopropane-d7 (8) with triphenylphosphine [29]. To our surprise, condensation of dialdehyde 5 with one equivalent of the ylide 4 (9, n-BuLi, THF, −78 °C) did not afford any amount of the desired deuterated olefin but only polar material that could not be characterized. In an attempt to find more efficient reaction conditions, we investigated this reaction using the simple aldehyde 10 as a model compound, easily accessible from squalene according to the van Tamelen procedure [30]. The condensation of ylide 4 with 10 seemed to be an easy task, but many well-established procedures using various bases (n-BuLi, LiHMDS, NaH/DMSO, PhLi) [29,31,32] gave only intractable materials. We finally found that the treatment of 10 with the “instant ylide mixture” of Schlosser made by grinding a solid mixture of 9 and NaNH2[33] delivered the desired squalene-d6 (11) although in a low 18% yield. However, when applied to the dialdehyde 5 this procedure failed to give the expected Wittig adduct.
Scheme 1:
Retrosynthetic route to SQCO2H-d6 (1) and synthetic routes for the preparation of dialdehyde 5 and squalene-d6 11.
Scheme 1:
Retrosynthetic route to SQCO2H-d6 (1) and synthetic routes for the preparation of dialdehyde 5 and ...
Whatever the origin of this problem, we decided to explore a new strategy based on a more nucleophilic deuterated synthon. In this regard we targeted prop-1-en-2-yllithium-d5 (15) which is easily accessible through the Shapiro reaction of sulfonylhydrazone of acetone-d6[34,35]. Thus condensation of trisylhydrazine with acetone-d6 (99.8% D) gave the expected hydrazone 14 in 55% yield. To our delight, upon treatment with two equivalents of n-BuLi and warming to 0 °C, the trisylhydrazone 14 afforded the vinyllithium reagent 15 (along with N2 and the trisyl anion) which upon condensation with squalenaldehyde 10 furnished the desired allylic alcohol 16 in 59% yield. Reduction of the hydroxy group of 16 was straightforwardly achieved in 47% yield by treatment with a large excess of thionyl chloride followed by LiAlD4 reduction [36]. Having secured an efficient method to reinstall the isopropylidene end-group, we turned our attention to the application of this method to a two-end functionalized squalene derivative. However, when applied to dialdehyde 5 the Shapiro reaction led to a mixture of starting material (30%), allylic alcohol 18 (15%) and diol 17 (20%). This result could not by improved using reverse addition conditions (Scheme 2). Therefore, selective monoprotection of the dialdehyde 5 was next attempted. Unfortunately, treatment of the latter either with ethylene glycol or 2,2-dimethylpropandiol gave a mixture of di- and monoacetal whatever the conditions, as a new example of the lack of chemoselectivity of this long polyisoprenyl chain derivatives.
Scheme 2:
Implementation of the deuterated isopropylidene end-group by the Shapiro reaction of trisylhydrazone of acetone-d6.
Scheme 2:
Implementation of the deuterated isopropylidene end-group by the Shapiro reaction of trisylhydrazon...
The differentiation of both ends of squalene was thus performed starting from trisnorsqualenaldehyde 10. Protection of 10 as 2,2-dimethyl-1,3-dioxane derivative gave 19 in 96% yield which was further elaborated into aldehyde 20 in 16% overall yield according to the three-step van Tamelen sequence (i. NBS, THF, H2O; ii. K2CO3, MeOH; iii. H3IO6, Et2O) [30]. Interestingly enough, the 1,3-dioxane group survived the strongly acidic conditions of the oxidative cleavage. We next turned to the elaboration of the isopropylidene-d6 moiety. In the event, the Shapiro reaction using trisylhydrazide 14 delivered the expected allylic alcohol 21 in 70% yield. The latter afforded the deuterated ketal 22 in 52% yield, upon sequential treatment with thionyl chloride and LiAlD4 as described above. With the success of the implementation of the terminal deuterated isopropylidene group we turned our attention to the deprotection of the ketal and the oxidation of the aldehyde group. This seemingly trivial task, turned out to be unexpectedly challenging. All conditions tried (HCl 3 N, THF, 20 °C; HCl 3 N, THF, reflux; HCl 6 N, dioxane, 20 °C; HCO2H, reflux; FeCl3·6H2O/SiO2[37], Jones reagent) either let the starting material unchanged or induced a complete decomposition. Even treatment with 1,2-ethanedithiol in the presence of BF3·OEt2 failed to give the corresponding thioketal [38]. These results clearly showed that another protecting group must be used, avoiding the use of an acidic catalyst that triggered the cyclization cascade of the polyisoprenyl chain (Scheme 3).
Scheme 3:
Attempted synthesis of 1 via the protection of the aldehyde 10 as a 5,5-dimethyl-1,3-dioxane.
Scheme 3:
Attempted synthesis of 1 via the protection of the aldehyde 10 as a 5,5-dimethyl-1,3-dioxane.
Despite this setback, the synthetic route seemed suitable to produce the desired material. Thus, the synthetic pathway described above was reimplemented starting from tert-butyldiphenylsiloxysqualene 23 readily obtained in 85% yield from squalenaldehyde 10 by NaBH4 reduction followed by protection with tert-butyldiphenylsilyl chloride. Functionalisation of the opposite extremity of the polyisoprenoid chain using the van Tamelen sequence (i. 1 equiv NBS, THF, H2O; ii. K2CO3, MeOH; iii. H3IO6, Et2O) afforded the aldehyde 26 in 16% overall yield. Uneventfully, the Shapiro reaction with trisylhydrazone 14 produced the allylic alcohol 27. Thionyl chloride treatment followed by LiAlD4 reduction delivered directly the alcohol 28 in 41% yield through concomitant reduction of the intermediate allylic chloride and cleavage of the silyl protecting group. The reductive cleavage of tert-butyldiphenylsilyl ethers by LiAlH4 has been previously noticed [39]. Jones oxidation straightforwardly completed the synthesis of SQCO2H-d61. Mass spectral analysis confirmed the presence of the six deuterium atoms (m/z = 405.3631 for [C27H37D6O2−]) along a small amount (~5%) of SQCO2H-d5. GemSQ-d63 was next synthetized using activation with ethyl chloroformate as previously reported [7]. However, to optimize the process a large excess of gemcitabine was used in the reaction to increase the yield to 72% in respect of the more valuable SQCO2H-d6 (Scheme 4).
Scheme 4:
Synthesis of squalenic acid-d61 and conjugation to gemcitabine.
Scheme 4:
Synthesis of squalenic acid-d61 and conjugation to gemcitabine.
Nanoparticle formulation of the GemSQ-d6 conjugate and Raman spectroscopy
The GemSQ and GemSQ-d6 nanoassembly suspensions (2 mg·mL−1) were prepared in a single step by nanoprecipitation of an ethanolic solution (2–4 mg·mL−1) in milli-Q water [7]. After spontaneous formation of the NAs the organic solvent was evaporated under vacuum (Figure 2A). Single Raman spectra of the raw substances as well as of the particles were recorded (Figure 2B). As depicted in Figure 2C and Figure 2D, the Raman spectra of the deuterated and non-deuterated compounds revealed no differences except the deuterium peaks in the silent region.
Figure 2:
A) Sketch depicting the procedure of preparing the NAs. B) Single Raman spectra of GemSQ-d6 NAs, GemSQ NAs, gemcitabine and squalenic acid, respectively. C) Single Raman spectra of deuterated GemSQ-d6 NAs (red) and GemSQ NAs (black). D) Close-up showing the difference between deuterated and non-deuterated compounds.
Figure 2:
A) Sketch depicting the procedure of preparing the NAs. B) Single Raman spectra of GemSQ-d6 NAs, Ge...
A synthesis of squalenic acid-d6 was developed through the Shapiro reaction of the sulfonylhydrazone of acetone-d6 with an ω-silyloxysqualene aldehyde derivative followed by a regioselective reduction of the obtained allylic alcohol. This material was obtained from natural squalene in 0.6% yield over 12 steps. The synthesized deuterated squalenic acid was coupled to gemcitabine to provide the corresponding deuterated squalenoyl conjugate. Raman spectra of the nanoassemblies made of this conjugate were recorded, showing significant Raman peaks in the silent region of the cells thus making this material a potential Raman probe. The use of this material as Raman probe in the study of the intracellular trafficking of GemSQ-based NAs is currently in progress and will be reported in due course.
Experimental
(4E,8E,12E,16E)-21-(2H3)methyl-4,8,13,17-tetramethyl(22,22,22-2H3)docosa-4,8,12,16,20-pentaenoic acid (1): An ice-cooled solution of alcohol 28 (55 mg, 0.14 mmol) in acetone (2 mL) was treated with a few drops of Jones reagent (CrO3/H2SO4 6 M) until the mixture took a persistent dark red color. After complete disappearance of the starting material a few drops of isopropanol were added. The mixture was taken into brine (10 mL) and extracted with Et2O (4 × 15 mL), dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel eluting with petroleum ether/Et2O 80:20 to give trisnorsqualenic acid-d6 (1) as a colorless oil (31.5 mg, 55%). 1H NMR (CDCl3, 300 MHz) δ 5.19–5.07 (m, 5H, =CH), 2.45 (t, J = 7.6 Hz, 2H, CH2CO2H), 2.30 (t, J = 7.6 Hz, 2H, CH2CH2CO2H), 2.15–1.95 (m, 16H, =HCCH2CH2C(CH3)), 1.62 (s, 3H, (CH3)C=), 1.60 (br s, 9H, (CH3)C=); 13C NMR (CDCl3, 75 MHz) δ 179.2 (C, CO2H), 135.3 (C, CH2(CH3)C=), 135.1 (C, CH2(CH3)C=), 135.0 (C, CH2(CH3)C=), 133.0 (C, CH2(CH3)C=), 131.2 (C, (CD3)2C=), 125.5 (CH, HC=), 124.6 (2CH, HC=), 124.4 (2CH, HC=), 39.9 (2CH2), 39.7 (CH2), 34.4(CH2, CH2CH2CO2H), 33.0 (CH2, CH2CO2H), 28.4 (2CH2), 26.9 (CH2), 26.8 (2CH2), 16.1 (3CH3), 16.0 (CH3); IR (film, cm−1) ν: 3500–2600 (broad), 2962, 2916, 2856, 2222, 2188, 1709, 1666, 1450, 1411, 1302, 1299, 1259, 1211, 1096, 1049, 955, 846, 736, 704; HRMS–ESI−: calcd for C27H37D6O2: 405.3645; found: 405.3631.
(4E,8E,12E,16E)-N-{1-[(2R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-oxo-1,2-dihydropyrimidin-4-yl}-21-(2H3)methyl-4,8,13,17-tetramethyl(22,22,22-2H3)docosa-4,8,12,16,20-pentaenamide (3): To a solution cooled at −5 °C of trisnorsqualenic acid-d6 (1) (31.5 mg, 0.077 mmol) in dry THF (0.6 mL) was sequentially added triethylamine (60 mg, 0.23 mmol) and ethyl chloroformate (10 mg, 0.093 mmol). The reaction mixture was stirred for 30 min at this temperature and a solution of gemcitabine base (60.6 mg, 0.23 mmol) in DMF (2 mL) was added. The reaction mixture was stirred for 4 days at 20 °C and concentrated under reduced pressure. The residue was directly chromatographed over silica gel eluting with cyclohexane/AcOEt 4:1 followed by neat AcOEt to provide GemSQ-d6 (3) as a colorless oil (36.0 mg, 72%). 1H NMR (CDCl3, 400 MHz) δ 9.15 (br s, 1H, NHCO), 8.10 (d, J = 7.5 Hz, 1H, H-6), 7.47 (d, J = 7.5 Hz, 1H, H-5), 6.18 (t, J = 7.4 Hz, 1H, H-1’), 5.20–5.06 (m, 5H, =CH), 4.55–4.41 (m, 1H, H-3’), 4.15–3.95 (m, 3H, H-4’, H-5’, OH), 3.91 (d, 1H, J = 10.8 Hz, H-5’), 2.55 (2H, t, J = 7.6 Hz, CH2CON), 2.32 (2H, t, J = 7.4 Hz, CH2CH2CON), 2.10–1.91 (m, 16H, =CCH2CH2C(CH3)), 1.60 (3H, s, =C(CH3)), 1.59 (s, 6H, =C(CH3)), 1.58 (3H, s, =C(CH3)); 13C NMR (CDCl3, 75 MHz) δ 173.6 (C, CONH), 163.1 (C, C-4), 155.8 (C, C-2), 145.6 (CH, C-6), 135.3 (C, (CH3)C= CH2(CH3)C=), 135.0 (2C, CH2(CH3)C=), 132.8 (C, CH2(CH3)C=), 131.2 (C, (CD3)2C=), 126.0 (CH, HC=), 124.5 (2CH, HC=), 124.4 (2 HC=), 122.5 (CF2, t, J = 258 Hz, C-2’), 97.8 (CH, C-5), 81.8 (CH, C-4’), 69.3 (CH, m, C-3’), 60.0 (CH2, C-5’), 39.9 (2CH2), 39.7 (CH2), 36.7 (CH2, NHCOCH2CH2), 34.5 (CH2, NHCOCH2CH2), 29.8 (CH2), 28.4 (2CH2), 27.0 (CH2), 26.9 (2CH2), 26.8 (2CH2), 16.2 (2CH3), 16.1 (CH3), 16.0 (CH3); IR (film, cm−1) ν: 3500–3000 (broad), 2979, 2932, 2872, 2852, 2222, 2191, 1724, 1683, 1660, 1618, 1561, 1494, 1433, 1397, 1383, 1365, 1337, 1320, 1312, 1272, 1206, 1194, 1134, 1086, 1069, 1051, 915, 893, 813, 787, 738; HRMS–ESI−: calcd for C36H46D6N3O5F2: 650.4257; found: 650.4230.
Preparation of nanoassemblies from GemSQ and GemSQ-d6: In a similar manner to the procedure published [19] the prodrugs based nanoparticle suspensions (2 mg·mL−1) were prepared in a single step by dropwise addition of an ethanol solution (4 mg·mL−1) in milli-Q water (1 mL) under vigorous stirring (500 rpm). Formation of NAs occurred immediately. After being stirred for 2 min, the nanoparticle suspension was then transferred into a weighted round bottom flask and ethanol was evaporated using a Rotavapor with a preheated water bath (35 °C) setting the vacuum to about 15–50 mbar for about 5 min. Then, the flask was dipped into a water bath (water temperature 37 °C) for about 3–5 minutes. Evaporation was continued till the weight of the contents decreased to 0.8–0.9 g. Then, the volume of the suspension in the flask was made-up to 1.0 g using either 5% dextrose solution or milli-Q water. The colloidal dispersions were stored at 4 °C.
Raman microscopy measurements. Confocal Raman microscopy measurements were performed with a WITec alpha 300R+ (WITec GmbH, Ulm, Germany). The excitation source was a diode laser with a wavelength of 532 nm adjusted to a power of 40 mW before the objective. A confocal pinhole of 50 μm rejected signals from out-of-focus regions. An objective with 50× magnification (N.A. 0.8, Epiplan Neofluar, Zeiss, Germany) was applied for acquiring single Raman spectra of the pure compounds with an integration time of 2 s and 10 accumulations. All spectra were background subtracted and normalized to the most intense peak.
Supporting Information
Supporting Information File 1:
Experimental procedures and 1H and 13C NMR spectral data for compounds 1, 3, 11, 25, 27, 28.
The research leading to these results has received funding from the European Research Council under the European Community's Seventh Framework Program FP7/2007-2013 Grant Agreement no.249835. The financial supports of the Ministère de la Recherche et de la Technologie (fellowship to E.B.) is gratefully acknowledged.
References
Koo, O. M.; Rubinstein, I.; Onyuksel, H. Nanomed.: Nanotechnol. Biol. Med.2005,1, 193–212. doi:10.1016/j.nano.2005.06.004
Return to citation in text:
[1]
Farokhzad, O. C.; Langer, R. ACS Nano2009,3, 16–20. doi:10.1021/nn900002m
Return to citation in text:
[1]
Müller, R. H.; Mehnert, W.; Lucks, J. S.; Schwarz, C.; Mühlen, Z.; Weyhers, H.; Freitas, C.; Rühl, D. Eur. J. Pharm. Biopharm.1995,41, 62–69.
Return to citation in text:
[1]
Olbrich, C.; Gessner, A.; Kayser, O.; Müller, R. H. J. Drug Targeting2002,10, 387–396. doi:10.1080/1061186021000001832
Return to citation in text:
[1]
Desmaële, D.; Gref, R.; Couvreur, P. J. Controlled Release2012,161, 609–618. doi:10.1016/j.jconrel.2011.07.038
Return to citation in text:
[1]
Burris, H. A., III; Moore, M. J.; Andersen, J.; Green, M. R.; Rothenberg, M. L.; Modiano, M. R.; Cripps, M. C.; Portenoy, R. K.; Storniolo, A. M.; Tarassoff, P.; Nelson, R.; Dorr, F. A.; Stephens, C. D.; Von Hoff, D. D. J. Clin. Oncol.1997,15, 2403–2413.
Return to citation in text:
[1]
Reddy, L. H.; Renoir, J.-M.; Marsaud, V.; Lepetre-Mouelhi, S.; Desmaële, D.; Couvreur, P. Mol. Pharmaceutics2009,6, 1526–1535. doi:10.1021/mp900099e
Return to citation in text:
[1]
Reddy, L. H.; Ferreira, H.; Dubernet, C.; Mouelhi, S. L.; Desmaële, D.; Rousseau, B.; Couvreur, P. Anti-Cancer Drugs2008,19, 999–1006. doi:10.1097/CAD.0b013e3283126585
Return to citation in text:
[1]
Reddy, L. H.; Marque, P.-E.; Dubernet, C.; Lepêtre Mouelhi, S.; Desmaële, D.; Couvreur, P. J. Pharmacol. Exp. Ther.2008,325, 484–490. doi:10.1124/jpet.107.133751
Return to citation in text:
[1]
Réjiba, S.; Reddy, L. H.; Bigand, C.; Parmentier, C.; Couvreur, P.; Hajri, A. Nanomedicine2011,7, 841–849. doi:10.1016/j.nano.2011.02.012
Return to citation in text:
[1]
Gourdeau, H.; Clarke, M. L.; Ouellet, F.; Mowles, D.; Selner, M.; Richard, A.; Lee, N.; Mackey, J. R.; Young, J. D.; Jolivet, J.; Lafrenière, R. G.; Cass, C. E. Cancer Res.2001,61, 7217–7224.
Return to citation in text:
[1]
Raouane, M.; Desmaele, D.; Gilbert-Sirieix, M.; Gueutin, C.; Zouhiri, F.; Bourgaux, C.; Lepeltier, E.; Gref, R.; Ben Salah, R.; Clayman, G.; Massaad-Massade, L.; Couvreur, P. J. Med. Chem.2011,54, 4067–4076. doi:10.1021/jm2000272
Return to citation in text:
[1]
Gaudin, A.; Yemisci, M.; Eroglu, H.; Lepêtre-Mouelhi, S.; Turgoglu, O. F.; Dönmez-Demir, B.; Caban, S.; Fevzi Sargon, M.; Garcia-Argote, S.; Pieters, G.; Loreau, O.; Rousseau, B.; Tagit, O.; Hildebrandt, N.; Le Dantec, Y.; Mougin, J.; Valetti, S.; Chacun, H.; Nicolas, V.; Desmaële, D.; Andrieux, K.; Capan, Y.; Dalkara, T.; Couvreur, P. Nat. Nanotechnol.2014,9, 1054–1062. doi:10.1038/nnano.2014.274
Return to citation in text:
[1]
Sémiramoth, N.; Di Meo, C.; Zouhiri, F.; Saïd-Hassane, F.; Valetti, S.; Gorges, R.; Nicolas, V.; Poupaert, J. H.; Chollet-Martin, S.; Desmaële, D.; Gref, R.; Couvreur, P. ACS Nano2012,6, 3820–3831. doi:10.1021/nn204928v
Return to citation in text:
[1]
Caron, J.; Maksimenko, A.; Wack, S.; Lepeltier, E.; Bourgaux, C.; Morvan, E.; Leblanc, K.; Couvreur, P.; Desmaële, D. Adv. Healthcare Mater.2013,2, 172–185. doi:10.1002/adhm.201200099
Return to citation in text:
[1]
Buchy, E.; Valetti, S.; Mura, S.; Mougin, J.; Troufflard, C.; Couvreur, P.; Desmaële, D. Eur. J. Org. Chem.2015, 202–212. doi:10.1002/ejoc.201403088
Return to citation in text:
[1]
[2]
Maksimenko, A.; Dosio, F.; Mougin, J.; Ferrero, A.; Wack, S.; Reddy, L. H.; Weyn, A.-A.; Lepeltier, E.; Bourgaux, C.; Stella, B.; Cattel, L.; Couvreur, P. Proc. Natl. Acad. Sci. U. S. A.2014,111, E217–E226. doi:10.1073/pnas.1313459110
Return to citation in text:
[1]
Bildstein, L.; Marsaud, V.; Chacun, H.; Lepêtre-Mouelhi, S.; Desmaële, D.; Couvreur, P.; Dubernet, C. Soft Matter2010,6, 5570–5580. doi:10.1039/C0SM00342E
Return to citation in text:
[1]
Gaudin, A.; Lepetre-Mouelhi, S.; Mougin, J.; Parrod, M.; Pieters, G.; Garcia-Argote, S.; Loreau, O.; Goncalves, J.; Chacun, H.; Courbebaisse, Y.; Clayette, P.; Desmaële, D.; Rousseau, B.; Andrieux, K.; Couvreur, P. J. Controlled Release2015,212, 50–58. doi:10.1016/j.jconrel.2015.06.016
Return to citation in text:
[1]
Li, Y.; Budamagunta, M. S.; Luo, J.; Xiao, W.; Voss, J. C.; Lam, K. S. ACS Nano2012,6, 9485–9495. doi:10.1021/nn302317j
Return to citation in text:
[1]
Antonio, K. A.; Schultz, Z. D. Anal. Chem.2014,86, 30–46. doi:10.1021/ac403640f
Return to citation in text:
[1]
Kann, B.; Offerhaus, H. L.; Windbergs, M.; Otto, C. Adv. Drug Delivery Rev.2015,89, 71–90. doi:10.1016/j.addr.2015.02.006
Return to citation in text:
[1]
Mendelsohn, R.; Sunder, S.; Berstein, H. J. Biochim. Biophys. Acta1976,443, 613–617.
Return to citation in text:
[1]
Stiebing, C.; Matthäus, C.; Krafft, C.; Keller, A.-A.; Weber, K.; Lorkowski, S.; Popp, J. Anal. Bioanal. Chem.2014,406, 7037–7046. doi:10.1007/s00216-014-7927-0
Return to citation in text:
[1]
Burris, H. A., III; Moore, M. J.; Andersen, J.; Green, M. R.; Rothenberg, M. L.; Modiano, M. R.; Cripps, M. C.; Portenoy, R. K.; Storniolo, A. M.; Tarassoff, P.; Nelson, R.; Dorr, F. A.; Stephens, C. D.; Von Hoff, D. D. J. Clin. Oncol.1997,15, 2403–2413.
Gourdeau, H.; Clarke, M. L.; Ouellet, F.; Mowles, D.; Selner, M.; Richard, A.; Lee, N.; Mackey, J. R.; Young, J. D.; Jolivet, J.; Lafrenière, R. G.; Cass, C. E. Cancer Res.2001,61, 7217–7224.


![[1860-5397-12-109-1]](/bjoc/content/figures/1860-5397-12-109-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-109-i1]](/bjoc/content/inline/1860-5397-12-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-109-i2]](/bjoc/content/inline/1860-5397-12-109-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-109-i3]](/bjoc/content/inline/1860-5397-12-109-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-109-i4]](/bjoc/content/inline/1860-5397-12-109-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-109-2]](/bjoc/content/figures/1860-5397-12-109-2.png?scale=2.0&max-width=1024&background=FFFFFF)