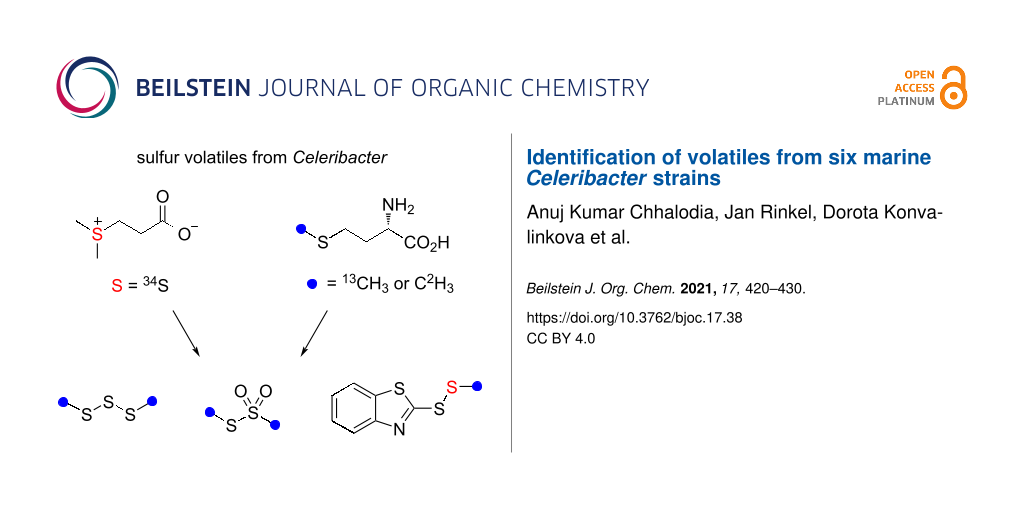
Abstract
The volatiles emitted from six marine Rhodobacteraceae species of the genus Celeribacter were investigated by GC–MS. Besides several known compounds including dimethyl trisulfide and S-methyl methanethiosulfonate, the sulfur-containing compounds ethyl (E)-3-(methylsulfanyl)acrylate and 2-(methyldisulfanyl)benzothiazole were identified and their structures were verified by synthesis. Feeding experiments with [methyl-2H3]methionine, [methyl-13C]methionine and [34S]-3-(dimethylsulfonio)propanoate (DMSP) resulted in the high incorporation into dimethyl trisulfide and S-methyl methanethiosulfonate, and revealed the origin of the methylsulfanyl group of 2-(methyldisulfanyl)benzothiazole from methionine or DMSP, while the biosynthetic origin of the benzothiazol-2-ylsulfanyl portion could not be traced. The heterocyclic moiety of this compound is likely of anthropogenic origin, because 2-mercaptobenzothiazole is used in the sulfur vulcanization of rubber. Also in none of the feeding experiments incorporation into ethyl (E)-3-(methylsulfanyl)acrylate could be observed, questioning its bacterial origin. Our results demonstrate that the Celeribacter strains are capable of methionine and DMSP degradation to widespread sulfur volatiles, but the analysis of trace compounds in natural samples must be taken with care.

Graphical Abstract
Introduction
Bacteria from the roseobacter group belong to the most abundant microbial species in marine ecosystems [1,2]. They are present from polar to tropical regions, in marine sediments, in estuarine and open ocean environments in different pelagic zones ranging from surface waters to depths of >2,000 m [3,4]. Some species are associated with other marine organisms, e.g., Thalassococcus halodurans DSM 26915T has been isolated from the marine sponge Halichondria panicea [5], and Phaeobacter gallaeciensis DSM 26640T is an isolate from the scallop Pecten maximus [6]. Important interactions are also observed between bacteria from the roseobacter group and various types of marine algae, e.g., the first described organisms Roseobacter litoralis DSM 6996T and R. denitrificans DSM 7001T were obtained from seaweed [7], while Dinoroseobacter shibae DSM 16493T and Marinovum algicola DSM 10251T are both isolates from the dinoflagellate Prorocentrum lima [8,9]. Especially in algal blooms bacteria of the roseobacter group are highly abundant [10], and here they belong to the main players involved in the enzymatic degradation of the algal sulfur metabolite 3-(dimethylsulfonio)propanoate (DMSP, Scheme 1) [11]. Its catabolism leads either through the demethylation pathway by action of the enzymes DmdABCD to methanethiol (MeSH, Scheme 1A) [12] or through lysis by DddD [13] or hydrolytic cleavage by one of the known DMSP lyases (DddW [14], DddP [15], DddQ [16], DddL [17], DddY [18] or DddK [19]) to dimethyl sulfide (DMS, Scheme 1B).
![[1860-5397-17-38-i1]](/bjoc/content/inline/1860-5397-17-38-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Sulfur metabolism in bacteria from the roseobacter group. A) DMSP demethylation by DmdABCD, B) DMSP hydrolysis by DddP and lysis by DddW, DddP, DddQ, DddL, DddY or DddK, and C) structures of DHPS and sulfur-containing secondary metabolites.
Scheme 1: Sulfur metabolism in bacteria from the roseobacter group. A) DMSP demethylation by DmdABCD, B) DMSP...
It has already been pointed out in the 1970s and 1980s that atmospheric DMS is important for the global sulfur cycle [20] and influences the climate on Earth, known as CLAW hypothesis according to the authors’ initials (Carlson, Lovelock, Andreae, Warren) [21], which underpins the relevance of this algal–bacterial interaction. Isotopic labeling experiments demonstrated that also in laboratory cultures roseobacter group bacteria efficiently degrade DMSP into sulfur volatiles [22,23], but also from other sulfur sources including 2,3-dihydroxypropane-1-sulfonic acid (DHPS, Scheme 1C) labeling was efficiently incorporated into sulfur volatiles [24,25]. Notably, DHPS is produced in large quantities by the marine diatom Thalassiosira pseudonana [26], and diatoms from this genus live in symbiotic relationship with bacteria of the roseobacter group [27]. Another interesting aspect of sulfur metabolism in marine bacteria from the roseobacter group is the production of the sulfur-containing antibiotic tropodithietic acid (TDA) in Phaeobacter piscinae DSM 103509T [28], a compound that is in equilibrium with its tautomer thiotropocin [29] that was first described from Pseudomonas sp. CB-104 [30]. Its biosynthesis depends on the clustered tda genes [31] and has been studied by feeding experiments with labeled precursors to the wildtype and gene knockout strains of P. inhibens DSM 17395T, demonstrating the formation of TDA from phenylalanine through phenylacetyl-CoA and the phenylacetyl-CoA catabolon [32,33]. These experiments also led to a suggestion for the mechanism for sulfur incorporation, but further research is required for a deep understanding of TDA biosynthesis. Besides its function as an antibiotic, TDA acts as a signaling molecule, similar to N-acylhomoserine lactones, at concentrations 100 times lower than required for a significant antibiotic activity [34]. The biosynthesis of tropone [35] and of the algicidal sulfur-containing roseobacticides [36] are most likely connected to the TDA pathway. Interestingly, in the interaction with marine algae P. inhibens can change its lifestyle from a symbiotic relationship during which the antibiotic TDA and growth stimulants are produced to a pathogenic interaction promoted by lignin degradation products in fading algal blooms that induce roseobacticide biosynthesis [36]. All these examples demonstrate the importance of sulfur metabolism for marine bacteria from the roseobacter group. Here we report on the volatiles emitted by six Celeribacter species with a special focus on sulfur volatiles. The results from feeding studies with labeled precursors demonstrate that the Celeribacter strains can form sulfur volatiles from methionine and DMSP, but also showed that some of the detected sulfur compounds are not or only partly of bacterial origin.
Results and Discussion
Headspace analysis
The volatiles released by six marine Celeribacter type strains, including C. marinus DSM 100036T, C. neptunius DSM 26471T, C. manganoxidans DSM 27541T, C. baekdonensis DSM 27375T, C. halophilus DSM 26270T and C. indicus DSM 27257T, were collected through a closed-loop stripping apparatus (CLSA) on charcoal [37]. After extraction with dichloromethane the obtained extracts were analyzed by GC–MS (Figure 1). The compounds were identified by the comparison of the recorded EI mass spectra to library spectra and of retention indices [38] to tabulated literature data (Table 1), or by a direct comparison to authentic standards. The structures of the identified compounds are shown in Figure 2.
![[1860-5397-17-38-1]](/bjoc/content/figures/1860-5397-17-38-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Total ion chromatograms of headspace extracts from A) C. marinus DSM 100036T, B) C. neptunius DSM 26471T, C) C. manganoxidans DSM 27541T, D) C. baekdonensis DSM 27375T, E) C. halophilus DSM 26270T, and F) C. indicus DSM 27257T. Peaks arising from known contaminants are indicated by asterisks.
Figure 1: Total ion chromatograms of headspace extracts from A) C. marinus DSM 100036T, B) C. neptunius DSM 2...
Table 1: Volatiles from Celeribacter spp.
| Compounda | Ib | I(lit.)b | Id.c | Occurrenced | |||||
| 3-hydroxypentan-2-one (4) | 812 | 815 [39] | ri, ms | C | |||||
| hexanal (7) | 813 | 806 [39] | ri, ms | B | |||||
| 2-hydroxypentan-3-one (5) | 818 | 818 [40] | ri, ms | C | |||||
| methylpyrazine (1) | 831 | 826 [41] | ri, ms | A | B | C | D | E | F |
| furfural (24) | 841 | 841 [42] | ri, ms | B | C | ||||
| furan-2-ylmethanol (23) | 861 | 863 [43] | ri, ms | A | B | C | D | E | F |
| cyclohexanol (26) | 888 | 886 [44] | ri, ms | A | |||||
| 2-hydroxyhexan-3-one (6) | 899 | 900 [40] | ri, ms | A | F | ||||
| heptanal (8) | 906 | 901 [45] | ri, ms | C | |||||
| 2,5-dimethylpyrazine (2) | 912 | 908 [45] | ri, ms | A | B | C | D | E | F |
| 2-acetylfuran (25) | 913 | 909 [45] | ri, ms | C | D | ||||
| pentan-4-olide (14) | 953 | 956 [46] | ri, ms | A | D | F | |||
| 3-methylbutan-4-olide (21) | 957 | 958 [47] | ri, ms | A | F | ||||
| 6-methylheptan-2-one (35) | 959 | 962 [48] | ri, ms | D | |||||
| benzaldehyde (28) | 961 | 952 [45] | ri, ms | A | B | C | D | E | |
| dimethyl trisulfide (37) | 970 | 968 [49] | ri, ms | A | B | C | D | E | F |
| 6-methylhept-5-en-2-one (34) | 988 | 981 [45] | ri, ms | A | B | C | D | E | F |
| trimethylpyrazine (3) | 1000 | 1000 [45] | ri, ms | A | B | C | D | E | F |
| 2-acetylthiazole (39) | 1017 | 1014 [45] | ri, ms | A | B | C | D | ||
| benzyl alcohol (27) | 1033 | 1026 [45] | ri, ms | C | |||||
| 4-methylhex-5-en-4-olide (22) | 1039 | 1034 [45] | ri, ms | B | C | ||||
| salicylaldehyde (29) | 1042 | 1039 [45] | ri, ms | B | D | ||||
| hexan-4-olide (15) | 1052 | 1056 [50] | ri, ms | A | |||||
| S-methyl methanethiosulfonate (38) | 1061 | 1068 [51] | ri, ms | A | B | C | D | F | |
| acetophenone (30) | 1065 | 1059 [45] | ri, ms | A | B | C | |||
| nonanal (9) | 1103 | 1100 [45] | ri, ms | A | B | C | F | ||
| 2-phenylethanol (32) | 1111 | 1106 [45] | ri, ms | B | C | ||||
| phenylacetone (33) | 1127 | 1124 [52] | ri, ms | A | |||||
| ethyl (E)-3-(methylsulfanyl)acrylate (42) | 1177 | 1144 [53] | ms | A | C | ||||
| decanal (10) | 1203 | 1201 [45] | ri, ms | A | B | C | F | ||
| benzothiazole (40) | 1221 | 1222 [54] | ri, ms | A | B | C | D | E | F |
| octan-4-olide (16) | 1252 | 1250 [45] | ri, ms | A | |||||
| o-aminoacetophenone (31) | 1292 | 1296 [55] | ri, ms | A | |||||
| undecanal (11) | 1298 | 1305 [45] | ri, ms | B | C | ||||
| nonan-4-olide (17) | 1354 | 1358 [45] | ri, ms | A | C | ||||
| dodecanal (12) | 1400 | 1408 [45] | ri, ms | A | B | C | |||
| geranylacetone (36) | 1445 | 1453 [45] | ri, ms | A | B | C | |||
| decan-4-olide (18) | 1461 | 1465 [45] | ri, ms | A | C | ||||
| undecan-4-olide (19) | 1568 | 1569 [45] | ri, ms | C | |||||
| tetradecanal (13) | 1605 | 1611 [45] | ri, ms | B | C | ||||
| dodecan-4-olide (20) | 1673 | 1676 [45] | ri, ms | A | C | ||||
| 2-(methyldisulfanyl)benzothiazole (41) | 1860 | std | A | B | D | ||||
aIdentified by GC–MS, known typical contaminants such as plasticizers are not included and all listed compounds were not detected in blank runs with medium plates (except traces of benzaldehyde); bretention index on a HP5-MS GC column and comparison to literature data from the same or a similar type of GC column; cidentification based on ri: matching retention index (difference between measured retention index and literature data ≤10 points), ms: mass spectrum matching to a database spectrum, std: direct comparison to an authentic standard; doccurrence in A: C. marinus DSM 100036T, B: C. neptunius DSM 26471T, C: C. manganoxidans DSM 27541T, D: C. baekdonensis DSM 27375T, E: C. halophilus DSM 26270T, and F: C. indicus DSM 27257T.
![[1860-5397-17-38-2]](/bjoc/content/figures/1860-5397-17-38-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of the identified volatile compounds in the headspace extracts from six Celeribacter type strains.
Figure 2: Structures of the identified volatile compounds in the headspace extracts from six Celeribacter typ...
While the headspace extracts from C. marinus, C. neptunius and C. manganoxidans were particularly rich, the extracts from C. baekdonensis, C. halophilus and C. indicus contained fewer compounds. Most of the observed volatiles are well known [56,57] and were thus readily identified from their mass spectra and retention indices. Pyrazines including methylpyrazine (1), 2,5-dimethylpyrazine (2) and trimethylpyrazine (3) were present in the extracts from all six strains. Notably, also several α-hydroxyketones that have been described as biosynthetic precursors to pyrazines [40], represented by 3-hydroxypentan-2-one (4), 2-hydroxypentan-3-one (5) and 2-hydroxyhexan-3-one (6), were observed in some of the investigated strains. A series of aldehydes ranging from hexanal (7) to tetradecanal (13) was found in strain specific patterns, with all identified compounds present in the bouquet from C. manganoxidans. A similar series of γ-lactones spanning from pentan-4-olide (14) to dodecan-4-olide (20), in addition to 3-methylbutan-4-olide (21) and 4-methylhex-5-en-4-olide (22), was detected in strain-specific patterns, with almost all of these compounds present in C. marinus; only C. halophilus did not emit lactones. Furans included furan-2-ylmethanol (23), furfural (24), and 2-acetylfuran (25). Cyclohexanol (26) was observed only once in C. marinus, and aromatic compounds included benzyl alcohol (27), benzaldehyde (28) and salicylaldehyde (29), acetophenone (30) and o-aminoacetophenone (31), 2-phenylethanol (32), and phenylacetone (33). 6-Methylhept-5-en-2-one (34) was detected in all strains, while its saturated analog 6-methylheptan-2-one (35) was only emitted by C. baekdonensis and geranylacetone (36) only by the three productive species C. marinus, C. neptunius, and C. manganoxidans. Compounds 34 and 36 have been described as non-enzymatic degradation products arising from the side chain in menaquinones [58]. Sulfur-containing compounds included dimethyl trisulfide (37), released by all six species, S-methyl methanethiosulfonate (38), 2-acetylthiazole (39), and benzothiazole (40), the latter also in the extracts from all six strains. In addition, the extracts from the three species C. marinus, C. neptunius and C. baekdonensis contained an additional volatile (41) whose mass spectrum (Figure 3A) was not included in our libraries. Furthermore, ethyl 3-(methylsulfanyl)acrylate (42) was found in C. marinus and C. manganoxidans, but the measured retention index (I = 1177) did not allow to distinguish between the E and the Z isomer for which retention indices of I = 1144 (E) and I = 1158 (Z) were reported [53]. Therefore, for an unambiguous structural assignment for compounds 41 and 42 the synthesis of reference compounds was required.
![[1860-5397-17-38-3]](/bjoc/content/figures/1860-5397-17-38-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: EI mass spectra of A) unlabeled 2-(methyldisulfanyl)benzothiazole (41) and of labeled 41 after feeding of B) (methyl-2H3)methionine, C) (methyl-13C)methionine and D) (34S)DMSP.
Figure 3: EI mass spectra of A) unlabeled 2-(methyldisulfanyl)benzothiazole (41) and of labeled 41 after feed...
Synthesis of reference compounds
The mass spectrum of the component 41 showed strong similarities to the library mass spectrum of 2-mercaptobenzothiazole that has a molecular weight of 167 Da. The isotope pattern of the molecular ion at m/z = 213 indicated the presence of three sulfur atoms. The strong base peak at m/z = 167 in the mass spectrum of 41 suggested a benzothiazol-2-ylsulfanyl moiety, while the mass difference to the molecular ion pointed to the connection to a methylsulfanyl group. Taken together, this analysis resulted in the structural proposal of 2-(methyldisulfanyl)benzothiazole for 41. For the structural verification a synthesis was performed by a BF3·OEt2-catalyzed reaction of bis(benzothiazol-2-yl)disulfane with dimethyl disulfide, giving access to 41 with a yield of 64% (Scheme 2). The synthetic compound 41 showed an identical mass spectrum and retention index compared to the volatile in the Celeribacter extracts. The Z and E stereoisomers of 42 were obtained by the Michael addition of NaSMe to ethyl propiolate (45), yielding a mixture of stereoisomers inseparable by silica gel column chromatography (92%). The major stereoisomer was found to be (Z)-42 (dr 94:6), whose preferred formation may be a result of a chalcogen–chalcogen interaction between the sulfur and an ester oxygen. This phenomenon was first described in supramolecular structures by Gleiter [59] and later also used to explain the outcome of organocatalytic reactions [60]. The pure stereoisomers of 42 were isolated by preparative HPLC, for which the best separation was achieved using a YMC ChiralART Cellulose-SC column. This yielded 70% of (Z)-42 and 6% of (E)-42, and their analysis by GC–MS showed retention indices of I = 1177 for (E)-42 and I = 1200 for (Z)-42, revealing that the compound in the headspace extracts of C. marinus DSM 100036T and C. manganoxidans DSM 27541T was identical to (E)-42.
![[1860-5397-17-38-i2]](/bjoc/content/inline/1860-5397-17-38-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of sulfur-containing compounds detected in the Celeribacter headspace extracts. A) Synthesis of 2-(methyldisulfanyl)benzothiazole (41) and B) synthesis of ethyl (Z)- and (E)-3-(methylsulfanyl)acrylate (42).
Scheme 2: Synthesis of sulfur-containing compounds detected in the Celeribacter headspace extracts. A) Synthe...
Feeding experiments with isotopically labeled precursors
The biosynthesis of sulfur volatiles in C. marinus was investigated in a series of feeding experiments with isotopically labeled precursors. Feeding of (methyl-2H3)methionine resulted in the efficient incorporation of labeling into 37 (79% incorporation rate, Figure S1B in Supporting Information File 1), 38 (78%, Figure S1F in Supporting Information File 1) and the S-methyl group of 41 (84%), as indicated by a shift of the molecular ion from m/z = 213 to 216 (Figure 3B, deuterated compounds can be separated from their non-deuterated analogs by gas chromatography [61]). The base peak appears at m/z = 168, demonstrating its formation with participation of one deuterium from the S-methyl group. Analogous results were obtained by feeding of (methyl-13C)methionine, showing incorporation into 37 (74%, Figure S1C in Supporting Information File 1), 38 (71%, Figure S1G in Supporting Information), and the MeS group of 41 (71%, Figure 3C; the signal at m/z = 213 represents unlabeled 41 that, in contrast to a deuterated compound, cannot be separated from 13C-labeled 41 by gas chromatography). Furthermore, feeding of [34S]DMSP gave an incorporation into the MeS groups of 37 (50%, Figure S1D), into both sulfur atoms of 38 (47%, Figure S1H in Supporting Information File 1), but only into one sulfur atom of 41 (46%), as indicated by the molecular ion at m/z = 215, while no signals at m/z = 217 and 219 were visible that would account for the incorporation of labeling into two or three of the sulfur atoms in 41 (Figure 3D; also here the signal at m/z = 213 represents inseparable unlabeled 41). In this experiment, the base peak did not change which allowed the localization of labeling specifically in the MeS group of 41.
The fact that no incorporation was observed for the other two sulfur atoms of 41 prompted us to further investigate the biosynthetic origin of the benzothiazol-2-ylsulfanyl portion of 41 to establish its natural origin. Several feeding experiments with central primary metabolites including (13C6)glucose, (13C5)ribose and (indole-2H5)tryptophan were performed, but none of these experiments resulted in a detectable incorporation of labeling. Conclusively, a non-biological origin of this part of the molecule seems likely, which may also explain why the detection of 41 in Celeribacter was not always reproducible. Notably, 2-mercaptobenzothiazole is used in the sulfur vulcanization of rubber and could react spontaneously with MeSH of bacterial origin in the presence of oxygen to form 41, giving a reasonable explanation for its formation.
Also none of the feeding experiments with the various labeled precursors resulted in an incorporation of labeling into the sulfur volatiles 39, 40, and 42, which also questioned their natural origin. This finding is rather surprising for 42, especially regarding the feeding experiment with (34S)DMSP, because its formation would be explainable by a DMSP degradation through the demethylation pathway, for which all relevant enzymes are encoded in the six Celeribacter strains (only a DmdA homolog is missing in C. indicus, Table S1 in Supporting Information File 1), and e.g., transesterification of the DmdC product with EtOH (Scheme 1A). Compound 42 is not a widespread sulfur volatile, but has been reported before from pineapples [53], pears [62], passion fruits [63], and apples [64].
Conclusion
Six marine Celeribacter strains were investigated for their volatiles, leading to the identification of 42 compounds from different classes, including several sulfur volatiles. However, feeding experiments with isotopically labeled precursors suggested that only the widespread compounds dimethyl trisulfide (37) and S-methyl methanethiosulfonate (38) are of natural origin, while no labeling from any of the fed precursors was incorporated into 2-acetylthiazole (39), benzothiazole (40), and ethyl (E)-3-(methylsulfanyl)acrylate (42), thus questioning their natural source from Celeribacter. These results demonstrate that the six Celeribacter strains are able to degrade methionine and DMSP with formation of MeSH as a source for the likely non-enzymatic oxidation in the presence of air to 37 and 38, opening possibilities for future studies on methionine and DMSP degrading enzymes and pathways in Celeribacter. Our study also shows that the results from trace compound analyses must be taken with care and contaminations from other sources must always be taken into consideration. For the unusual compound 2-(methyldisulfanyl)benzothiazole (41) the incorporation of labeling was observed only into the MeS group, while the benzothiazol-2-ylsulfanyl portion is likely of anthropogenic origin from the rubber vulcanization agent 2-mercaptobenzothiazole that reacts with MeSH from the bacterial metabolism.
Experimental
Strains, culture conditions, and feeding experiments
All six Celeribacter type strains were cultivated at 28 °C on marine broth agar plates. In case of feeding experiments, the isotopically labeled compound (1 mM) was added to the agar medium before inoculation.
Collection of volatiles
The volatiles emitted by Celeribacter spp. agar plate cultures were collected on charcoal filters (Chromtech, Idstein, Germany, precision charcoal filters charged with 5 mg of charcoal) by use of a closed-loop stripping apparatus as developed by Grob and Zürcher [37]. After a collection time of 24 h the charcoal was extracted with CH2Cl2 (50 μL) and the extract was analyzed by GC–MS.
GC–MS
GC–MS analyses were carried out through a 7890B GC – 5977A MD system (Agilent, Santa Clara, CA, USA). The GC was equipped with a HP5-MS fused silica capillary column (30 m, 0.25 mm i.d., 0.50 μm film) and operated with the settings 1) inlet pressure: 77.1 kPa, He flow: 23.3 mL min−1, 2) injection volume: 2 μL, 3) splitless injection, 4) temperature program: 5 min isothermic at 50 °C, then increasing with 5 °C min−1 to 320 °C, and 5) He carrier gas flow: 1.2 mL min−1. The parameters of the MS were 1) transfer line temperature: 250 °C, 2) ion source temperature: 230 °C, 3) quadrupole temperature: 150 °C, and 4) electron energy: 70 eV. Retention indices were calculated from retention times in comparison to those of a homologous series of n-alkanes (C7–C40).
General synthetic and analytical methods
Reactions were carried out in oven-dried flasks under Ar atmosphere and using distilled and dried solvents. Chemicals were obtained from Sigma-Aldrich (St. Louis, USA). Column chromatography was performed on silica gel (0.04–0.06 nm) purchased from Acros Organics (Geel, Belgium) with distilled solvents. NMR spectroscopy was performed on a Bruker (Billerica, USA) Avance III HD Ascend (500 MHz) spectrometer. Solvent peaks were used for referencing (1H NMR: CDCl3 residual proton signal δ = 7.26 ppm, 13C NMR: CDCl3 δ = 77.16 ppm) [65]. Multiplicities are indicated by s (singlet) and d (doublet), coupling constants J are given in Hz. IR spectra were recorded on a Bruker α spectrometer equipped with a diamond-ATR probe, and qualitative signal intensities are reported by w (weak), m (medium), and s (strong). HPLC purification of compound 42 was performed on an Azura HPLC system (Knauer, Berlin, Germany) equipped with a UV–vis detector MWL 2.1L (deuterium lamp, 190–700 nm) and a YMC ChiralART Cellulose-SC column (5 μm; 250 × 20 mm) with a guard column of the same type (30 × 20 mm). The elution was performed with hexane/propanol 60:40 (isocratic) at a flow rate of 10 mL min−1 (36 bar). The UV–vis absorption was monitored at 275 nm.
Synthesis of 2-(methyldisulfanyl)benzothiazole (41)
1,2-Bis(benzothiazol-2-yl)disulfane (43, 1.00 g, 3.00 mmol, 1 equiv) and dimethyl sulfide (44, 0.28 g, 3.00 mmol, 1 equiv) were dissolved in dry CH3NO2 (10 mL) and dry CH2Cl2 (10 mL). The solution was cooled to 0 °C and then treated with BF3·Et2O (43 mg, 0.3 mmol, 0.1 equiv). After stirring at 0 °C for 3 hours and at room temperature overnight, the reaction was quenched by the addition of water (10 mL) and extracted with ethyl acetate (3 × 50 mL). The combined extracts were dried with MgSO4 and concentrated. The residue was purified by column chromatography (cyclohexane/ethyl acetate 1:1) to give 41 as a colorless solid (0.82 g, 3.85 mmol, 64%). Rf 0.60 (cyclohexane/ethyl acetate 5:1; TLC visualized with UV illumination at 366 nm); GC (HP-5MS): I = 1854; IR (diamond-ATR) ν̃: 3060 (s), 2916 (s), 1425 (w), 1310 (s), 1236 (s), 1005 (w), 756 (w), 431 (s) cm−1; 1H NMR (500 MHz, CDCl3, 298 K) δ 7.88 (ddd, J = 8.1, 1.2, 0.7 Hz, 1H, CH), 7.87 (ddd, J = 7.9, 1.2, 0.6 Hz, 1H, CH), 7.43 (ddd, J = 8.3, 7.3, 1.2 Hz, 1H, CH), 7.33 (ddd, J = 8.2, 7.2, 1.2 Hz, 1H, CH), 2.67 (s, 3H, CH3) ppm; 13C NMR (125 MHz, CDCl3, 298 K) δ 172.50 (C), 155.17 (C), 135.90 (C), 126.37 (CH), 124.70 (CH), 122.24 (CH), 121.27 (CH), 23.62 (CH3) ppm.
Synthesis of ethyl (Z)-3-(methylsulfanyl)acrylate ((Z)-42) and ethyl (E)-3-(methylsulfanyl)acrylate ((E)-42)
Ethyl propiolate (45, 70 mg, 0.71 mmol, 1 equiv) was dissolved in distilled water (5 mL) followed by the addition of sodium methanethiolate (50 mg, 0.71 mmol, 1 equiv). The solution was stirred for 30 minutes at room temperature. Water (5 mL) was added and the product was extracted with ethyl acetate (3 × 10 mL). The combined extracts were dried over MgSO4 and concentrated to afford the crude product. Purification by column chromatography (cyclohexane/ethyl acetate 99:1) gave a mixture of stereoisomers (Z)-42 and (E)-42 as pale yellow oil (96 mg, 0.65 mmol, 92%, dr 94:6 by 1H NMR). The product mixture was separated by preparative HPLC to give pure (Z)-42 (73 mg, 0.50 mmol, 70%) and (E)-42 (6 mg, 0.04 mmol, 6%).
(Z)-42. Rf 0.74 (cyclohexane/ethyl acetate 1:1); GC (HP-5MS): I = 1200; IR (diamond-ATR) ν̃: 2982 (w), 2927 (w),1695 (m), 1569 (m), 1434 (w), 1374 (w), 1300 (w), 1266 (w), 1213 (m), 1166 (s), 1095 (w), 1033 (w), 986 (w), 961 (w), 800 (w), 727 (w), 687 (w) cm−1; 1H NMR (700 MHz, CDCl3, 298 K) δ 7.04 (d, J = 10.14 Hz, 1H, CH), 5.83 (d, J = 10.14 Hz, 1H, CH), 4.20 (q, J = 7.15 Hz, 2H, CH2), 2.39 (s, 3H, CH3), 1.29 (t, J = 7.17 Hz, 3H, CH3) ppm; 13C NMR (175 MHz, CDCl3, 298 K) δ 166.75 (C), 151.84 (CH), 113.18 (CH), 60.17 (CH2), 19.28 (CH3), 14.44 (CH3) ppm.
(E)-42. Rf 0.76 (cyclohexane/ethyl acetate 1:1); GC (HP-5MS): I = 1177; IR (diamond-ATR) ν̃: 2980 (w), 2925 (w), 1701 (s), 1578 (s), 1444 (w), 1366 (w), 1322 (w), 1297 (m), 1251 (s), 1161 (s), 1095 (w), 1037 (m), 945 (m), 886 (w), 832 (w), 799 (w), 702 (w) cm−1; 1H NMR (700 MHz, CDCl3, 298 K) δ 7.76 (d, J = 14.93 Hz, 1H, CH), 5.68 (d, J = 14.90 Hz, 1H, CH), 4.21 (q, J = 7.14 Hz, 2H, CH2), 2.35 (s, 3H, CH3), 1.31 (t, J = 7.13 Hz, 3H, CH3) ppm; 13C NMR (175 MHz, CDCl3, 297 K) δ 165.59 (C), 147.21 (CH),113.56 (CH), 60.55 (CH2), 27.26 (CH3), 14.67 (CH3) ppm.
Supporting Information
| Supporting Information File 1: DMSP demethylation pathway in Celeribacter spp. and copies of spectra. | ||
| Format: PDF | Size: 452.3 KB | Download |
References
-
Giovannoni, S. J.; Stingl, U. Nature 2005, 437, 343–348. doi:10.1038/nature04158
Return to citation in text: [1] -
González, J. M.; Moran, M. A. Appl. Environ. Microbiol. 1997, 63, 4237–4242. doi:10.1128/aem.63.11.4237-4242.1997
Return to citation in text: [1] -
Selje, N.; Simon, M.; Brinkhoff, T. Nature 2004, 427, 445–448. doi:10.1038/nature02272
Return to citation in text: [1] -
Brinkhoff, T.; Giebel, H.-A.; Simon, M. Arch. Microbiol. 2008, 189, 531–539. doi:10.1007/s00203-008-0353-y
Return to citation in text: [1] -
Lee, O. O.; Tsoi, M. M. Y.; Li, X.; Wong, P.-K.; Qian, P.-Y. Int. J. Syst. Evol. Microbiol. 2007, 57, 1919–1924. doi:10.1099/ijs.0.64801-0
Return to citation in text: [1] -
Ruiz-Ponte, C.; Cilia, V.; Lambert, C.; Nicolas, J. L. Int. J. Syst. Bacteriol. 1998, 48, 537–542. doi:10.1099/00207713-48-2-537
Return to citation in text: [1] -
Shiba, T. Syst. Appl. Microbiol. 1991, 14, 140–145. doi:10.1016/s0723-2020(11)80292-4
Return to citation in text: [1] -
Biebl, H.; Allgaier, M.; Tindall, B. J.; Koblizek, M.; Lünsdorf, H.; Pukall, R.; Wagner-Döbler, I. Int. J. Syst. Evol. Microbiol. 2005, 55, 1089–1096. doi:10.1099/ijs.0.63511-0
Return to citation in text: [1] -
Lafay, B.; Ruimy, R.; Rausch de Traubenberg, C.; Breittmayer, V.; Gauthier, M. J.; Christen, R. Int. J. Syst. Bacteriol. 1995, 45, 290–296. doi:10.1099/00207713-45-2-290
Return to citation in text: [1] -
González, J. M.; Simó, R.; Massana, R.; Covert, J. S.; Casamayor, E. O.; Pedrós-Alió, C.; Moran, M. A. Appl. Environ. Microbiol. 2000, 66, 4237–4246. doi:10.1128/aem.66.10.4237-4246.2000
Return to citation in text: [1] -
Dickschat, J. S.; Rabe, P.; Citron, C. A. Org. Biomol. Chem. 2015, 13, 1954–1968. doi:10.1039/c4ob02407a
Return to citation in text: [1] -
Reisch, C. R.; Stoudemayer, M. J.; Varaljay, V. A.; Amster, I. J.; Moran, M. A.; Whitman, W. B. Nature 2011, 473, 208–211. doi:10.1038/nature10078
Return to citation in text: [1] -
Todd, J. D.; Rogers, R.; Li, Y. G.; Wexler, M.; Bond, P. L.; Sun, L.; Curson, A. R. J.; Malin, G.; Steinke, M.; Johnston, A. W. B. Science 2007, 315, 666–669. doi:10.1126/science.1135370
Return to citation in text: [1] -
Todd, J. D.; Kirkwood, M.; Newton-Payne, S.; Johnston, A. W. B. ISME J. 2012, 6, 223–226. doi:10.1038/ismej.2011.79
Return to citation in text: [1] -
Kirkwood, M.; Le Brun, N. E.; Todd, J. D.; Johnston, A. W. B. Microbiology (London, U. K.) 2010, 156, 1900–1906. doi:10.1099/mic.0.038927-0
Return to citation in text: [1] -
Todd, J. D.; Curson, A. R. J.; Kirkwood, M.; Sullivan, M. J.; Green, R. T.; Johnston, A. W. B. Environ. Microbiol. 2011, 13, 427–438. doi:10.1111/j.1462-2920.2010.02348.x
Return to citation in text: [1] -
Curson, A. R. J.; Rogers, R.; Todd, J. D.; Brearley, C. A.; Johnston, A. W. B. Environ. Microbiol. 2008, 10, 757–767. doi:10.1111/j.1462-2920.2007.01499.x
Return to citation in text: [1] -
Curson, A. R. J.; Sullivan, M. J.; Todd, J. D.; Johnston, A. W. B. ISME J. 2011, 5, 1191–1200. doi:10.1038/ismej.2010.203
Return to citation in text: [1] -
Sun, J.; Todd, J. D.; Thrash, J. C.; Qian, Y.; Qian, M. C.; Temperton, B.; Guo, J.; Fowler, E. K.; Aldrich, J. T.; Nicora, C. D.; Lipton, M. S.; Smith, R. D.; De Leenheer, P.; Payne, S. H.; Johnston, A. W. B.; Davie-Martin, C. L.; Halsey, K. H.; Giovannoni, S. J. Nat. Microbiol. 2016, 1, 16065. doi:10.1038/nmicrobiol.2016.65
Return to citation in text: [1] -
Lovelock, J. E.; Maggs, R. J.; Rasmussen, R. A. Nature 1972, 237, 452–453. doi:10.1038/237452a0
Return to citation in text: [1] -
Charlson, R. J.; Lovelock, J. E.; Andreae, M. O.; Warren, S. G. Nature 1987, 326, 655–661. doi:10.1038/326655a0
Return to citation in text: [1] -
Dickschat, J. S.; Zell, C.; Brock, N. L. ChemBioChem 2010, 11, 417–425. doi:10.1002/cbic.200900668
Return to citation in text: [1] -
Brock, N. L.; Citron, C. A.; Zell, C.; Berger, M.; Wagner-Döbler, I.; Petersen, J.; Brinkhoff, T.; Simon, M.; Dickschat, J. S. Beilstein J. Org. Chem. 2013, 9, 942–950. doi:10.3762/bjoc.9.108
Return to citation in text: [1] -
Brock, N. L.; Menke, M.; Klapschinski, T. A.; Dickschat, J. S. Org. Biomol. Chem. 2014, 12, 4318–4323. doi:10.1039/c4ob00719k
Return to citation in text: [1] -
Celik, E.; Maczka, M.; Bergen, N.; Brinkhoff, T.; Schulz, S.; Dickschat, J. S. Org. Biomol. Chem. 2017, 15, 2919–2922. doi:10.1039/c7ob00357a
Return to citation in text: [1] -
Durham, B. P.; Sharma, S.; Luo, H.; Smith, C. B.; Amin, S. A.; Bender, S. J.; Dearth, S. P.; Van Mooy, B. A. S.; Campagna, S. R.; Kujawinski, E. B.; Armbrust, E. V.; Moran, M. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 453–457. doi:10.1073/pnas.1413137112
Return to citation in text: [1] -
Mönnich, J.; Tebben, J.; Bergemann, J.; Case, R.; Wohlrab, S.; Harder, T. ISME J. 2020, 14, 1614–1625. doi:10.1038/s41396-020-0631-5
Return to citation in text: [1] -
Bruhn, J. B.; Nielsen, K. F.; Hjelm, M.; Hansen, M.; Bresciani, J.; Schulz, S.; Gram, L. Appl. Environ. Microbiol. 2005, 71, 7263–7270. doi:10.1128/aem.71.11.7263-7270.2005
Return to citation in text: [1] -
Greer, E. M.; Aebisher, D.; Greer, A.; Bentley, R. J. Org. Chem. 2008, 73, 280–283. doi:10.1021/jo7018416
Return to citation in text: [1] -
Kintaka, K.; Ono, H.; Tsubotani, S.; Harada, S.; Okazaki, H. J. Antibiot. 1984, 37, 1294–1300. doi:10.7164/antibiotics.37.1294
Return to citation in text: [1] -
Geng, H.; Bruhn, J. B.; Nielsen, K. F.; Gram, L.; Belas, R. Appl. Environ. Microbiol. 2008, 74, 1535–1545. doi:10.1128/aem.02339-07
Return to citation in text: [1] -
Berger, M.; Brock, N. L.; Liesegang, H.; Dogs, M.; Preuth, I.; Simon, M.; Dickschat, J. S.; Brinkhoff, T. Appl. Environ. Microbiol. 2012, 78, 3539–3551. doi:10.1128/aem.07657-11
Return to citation in text: [1] -
Brock, N. L.; Nikolay, A.; Dickschat, J. S. Chem. Commun. 2014, 50, 5487–5489. doi:10.1039/c4cc01924e
Return to citation in text: [1] -
Beyersmann, P. G.; Tomasch, J.; Son, K.; Stocker, R.; Göker, M.; Wagner-Döbler, I.; Simon, M.; Brinkhoff, T. Sci. Rep. 2017, 7, 730. doi:10.1038/s41598-017-00784-7
Return to citation in text: [1] -
Thiel, V.; Brinkhoff, T.; Dickschat, J. S.; Wickel, S.; Grunenberg, J.; Wagner-Döbler, I.; Simon, M.; Schulz, S. Org. Biomol. Chem. 2010, 8, 234–246. doi:10.1039/b909133e
Return to citation in text: [1] -
Seyedsayamdost, M. R.; Case, R. J.; Kolter, R.; Clardy, J. Nat. Chem. 2011, 3, 331–335. doi:10.1038/nchem.1002
Return to citation in text: [1] [2] -
Grob, K.; Zürcher, F. J. Chromatogr. 1976, 117, 285–294. doi:10.1016/0021-9673(76)80005-2
Return to citation in text: [1] [2] -
Kováts, E. Helv. Chim. Acta 1958, 41, 1915–1932. doi:10.1002/hlca.19580410703
Return to citation in text: [1] -
Elmore, J. S.; Mottram, D. S.; Enser, M.; Wood, J. D. Meat Sci. 2000, 55, 149–159. doi:10.1016/s0309-1740(99)00137-0
Return to citation in text: [1] [2] -
Dickschat, J. S.; Wickel, S.; Bolten, C. J.; Nawrath, T.; Schulz, S.; Wittmann, C. Eur. J. Org. Chem. 2010, 2687–2695. doi:10.1002/ejoc.201000155
Return to citation in text: [1] [2] [3] -
Cerny, C.; Guntz-Dubini, R. J. Agric. Food Chem. 2006, 54, 574–577. doi:10.1021/jf052222s
Return to citation in text: [1] -
Spadone, J.-C.; Takeoka, G.; Liardon, R. J. Agric. Food Chem. 1990, 38, 226–233. doi:10.1021/jf00091a050
Return to citation in text: [1] -
Lee, S.-R.; Macku, C.; Shibamoto, T. J. Agric. Food Chem. 1991, 39, 1972–1975. doi:10.1021/jf00011a017
Return to citation in text: [1] -
Pino, J. A.; Mesa, J.; Muñoz, Y.; Martí, M. P.; Marbot, R. J. Agric. Food Chem. 2005, 53, 2213–2223. doi:10.1021/jf0402633
Return to citation in text: [1] -
Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] -
Ansorena, D.; Gimeno, O.; Astiasarán, I.; Bello, J. Food Res. Int. 2001, 34, 67–75. doi:10.1016/s0963-9969(00)00133-2
Return to citation in text: [1] -
Citron, C. A.; Rabe, P.; Dickschat, J. S. J. Nat. Prod. 2012, 75, 1765–1776. doi:10.1021/np300468h
Return to citation in text: [1] -
Owens, J. D.; Allagheny, N.; Kipping, G.; Ames, J. M. J. Sci. Food Agric. 1997, 74, 132–140. doi:10.1002/(sici)1097-0010(199705)74:1<132::aid-jsfa779>3.0.co;2-8
Return to citation in text: [1] -
Cha, Y. J.; Cadwallader, K. R. J. Agric. Food Chem. 1998, 46, 1123–1128. doi:10.1021/jf970380g
Return to citation in text: [1] -
Wickel, S. M.; Citron, C. A.; Dickschat, J. S. Eur. J. Org. Chem. 2013, 2906–2913. doi:10.1002/ejoc.201300049
Return to citation in text: [1] -
Kubec, R.; Drhová, V.; Velíšek, J. J. Agric. Food Chem. 1998, 46, 4334–4340. doi:10.1021/jf980379x
Return to citation in text: [1] -
Ferhat, M. A.; Tigrine-Kordjani, N.; Chemat, S.; Meklati, B. Y.; Chemat, F. Chromatographia 2007, 65, 217–222. doi:10.1365/s10337-006-0130-5
Return to citation in text: [1] -
Takeoka, G. R.; Buttery, R. G.; Teranishi, R.; Flath, R. A.; Güntert, M. J. Agric. Food Chem. 1991, 39, 1848–1851. doi:10.1021/jf00010a032
Return to citation in text: [1] [2] [3] -
Nawrath, T.; Mgode, G. F.; Weetjens, B.; Kaufmann, S. H. E.; Schulz, S. Beilstein J. Org. Chem. 2012, 8, 290–299. doi:10.3762/bjoc.8.31
Return to citation in text: [1] -
Citron, C. A.; Barra, L.; Wink, J.; Dickschat, J. S. Org. Biomol. Chem. 2015, 13, 2673–2683. doi:10.1039/c4ob02609h
Return to citation in text: [1] -
Schulz, S.; Dickschat, J. S. Nat. Prod. Rep. 2007, 24, 814–842. doi:10.1039/b507392h
Return to citation in text: [1] -
Dickschat, J. S. Nat. Prod. Rep. 2017, 34, 310–328. doi:10.1039/c7np00003k
Return to citation in text: [1] -
Ueda, D.; Matsugane, S.; Okamoto, W.; Hashimoto, M.; Sato, T. Angew. Chem., Int. Ed. 2018, 57, 10347–10351. doi:10.1002/anie.201805383
Return to citation in text: [1] -
Werz, D. B.; Staeb, T. H.; Benisch, C.; Rausch, B. J.; Rominger, F.; Gleiter, R. Org. Lett. 2002, 4, 339–342. doi:10.1021/ol016953z
Return to citation in text: [1] -
Leverett, C. A.; Purohit, V. C.; Romo, D. Angew. Chem., Int. Ed. 2010, 49, 9479–9483. doi:10.1002/anie.201004671
Return to citation in text: [1] -
Dickschat, J. S. Nat. Prod. Rep. 2014, 31, 838–861. doi:10.1039/c3np70080a
Return to citation in text: [1] -
Takeoka, G. R.; Buttery, R. G.; Flath, R. A. J. Agric. Food Chem. 1992, 40, 1925–1929. doi:10.1021/jf00022a040
Return to citation in text: [1] -
Werkhoff, P.; Güntert, M.; Krammer, G.; Sommer, H.; Kaulen, J. J. Agric. Food Chem. 1998, 46, 1076–1093. doi:10.1021/jf970655s
Return to citation in text: [1] -
Ferreira, L.; Perestrelo, R.; Caldeira, M.; Câmara, J. S. J. Sep. Sci. 2009, 32, 1875–1888. doi:10.1002/jssc.200900024
Return to citation in text: [1] -
Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. doi:10.1021/om100106e
Return to citation in text: [1]
| 34. | Beyersmann, P. G.; Tomasch, J.; Son, K.; Stocker, R.; Göker, M.; Wagner-Döbler, I.; Simon, M.; Brinkhoff, T. Sci. Rep. 2017, 7, 730. doi:10.1038/s41598-017-00784-7 |
| 35. | Thiel, V.; Brinkhoff, T.; Dickschat, J. S.; Wickel, S.; Grunenberg, J.; Wagner-Döbler, I.; Simon, M.; Schulz, S. Org. Biomol. Chem. 2010, 8, 234–246. doi:10.1039/b909133e |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 36. | Seyedsayamdost, M. R.; Case, R. J.; Kolter, R.; Clardy, J. Nat. Chem. 2011, 3, 331–335. doi:10.1038/nchem.1002 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 41. | Cerny, C.; Guntz-Dubini, R. J. Agric. Food Chem. 2006, 54, 574–577. doi:10.1021/jf052222s |
| 42. | Spadone, J.-C.; Takeoka, G.; Liardon, R. J. Agric. Food Chem. 1990, 38, 226–233. doi:10.1021/jf00091a050 |
| 39. | Elmore, J. S.; Mottram, D. S.; Enser, M.; Wood, J. D. Meat Sci. 2000, 55, 149–159. doi:10.1016/s0309-1740(99)00137-0 |
| 59. | Werz, D. B.; Staeb, T. H.; Benisch, C.; Rausch, B. J.; Rominger, F.; Gleiter, R. Org. Lett. 2002, 4, 339–342. doi:10.1021/ol016953z |
| 40. | Dickschat, J. S.; Wickel, S.; Bolten, C. J.; Nawrath, T.; Schulz, S.; Wittmann, C. Eur. J. Org. Chem. 2010, 2687–2695. doi:10.1002/ejoc.201000155 |
| 38. | Kováts, E. Helv. Chim. Acta 1958, 41, 1915–1932. doi:10.1002/hlca.19580410703 |
| 58. | Ueda, D.; Matsugane, S.; Okamoto, W.; Hashimoto, M.; Sato, T. Angew. Chem., Int. Ed. 2018, 57, 10347–10351. doi:10.1002/anie.201805383 |
| 39. | Elmore, J. S.; Mottram, D. S.; Enser, M.; Wood, J. D. Meat Sci. 2000, 55, 149–159. doi:10.1016/s0309-1740(99)00137-0 |
| 53. | Takeoka, G. R.; Buttery, R. G.; Teranishi, R.; Flath, R. A.; Güntert, M. J. Agric. Food Chem. 1991, 39, 1848–1851. doi:10.1021/jf00010a032 |
| 36. | Seyedsayamdost, M. R.; Case, R. J.; Kolter, R.; Clardy, J. Nat. Chem. 2011, 3, 331–335. doi:10.1038/nchem.1002 |
| 56. | Schulz, S.; Dickschat, J. S. Nat. Prod. Rep. 2007, 24, 814–842. doi:10.1039/b507392h |
| 57. | Dickschat, J. S. Nat. Prod. Rep. 2017, 34, 310–328. doi:10.1039/c7np00003k |
| 37. | Grob, K.; Zürcher, F. J. Chromatogr. 1976, 117, 285–294. doi:10.1016/0021-9673(76)80005-2 |
| 40. | Dickschat, J. S.; Wickel, S.; Bolten, C. J.; Nawrath, T.; Schulz, S.; Wittmann, C. Eur. J. Org. Chem. 2010, 2687–2695. doi:10.1002/ejoc.201000155 |
| 43. | Lee, S.-R.; Macku, C.; Shibamoto, T. J. Agric. Food Chem. 1991, 39, 1972–1975. doi:10.1021/jf00011a017 |
| 44. | Pino, J. A.; Mesa, J.; Muñoz, Y.; Martí, M. P.; Marbot, R. J. Agric. Food Chem. 2005, 53, 2213–2223. doi:10.1021/jf0402633 |
| 63. | Werkhoff, P.; Güntert, M.; Krammer, G.; Sommer, H.; Kaulen, J. J. Agric. Food Chem. 1998, 46, 1076–1093. doi:10.1021/jf970655s |
| 40. | Dickschat, J. S.; Wickel, S.; Bolten, C. J.; Nawrath, T.; Schulz, S.; Wittmann, C. Eur. J. Org. Chem. 2010, 2687–2695. doi:10.1002/ejoc.201000155 |
| 64. | Ferreira, L.; Perestrelo, R.; Caldeira, M.; Câmara, J. S. J. Sep. Sci. 2009, 32, 1875–1888. doi:10.1002/jssc.200900024 |
| 53. | Takeoka, G. R.; Buttery, R. G.; Teranishi, R.; Flath, R. A.; Güntert, M. J. Agric. Food Chem. 1991, 39, 1848–1851. doi:10.1021/jf00010a032 |
| 62. | Takeoka, G. R.; Buttery, R. G.; Flath, R. A. J. Agric. Food Chem. 1992, 40, 1925–1929. doi:10.1021/jf00022a040 |
| 60. | Leverett, C. A.; Purohit, V. C.; Romo, D. Angew. Chem., Int. Ed. 2010, 49, 9479–9483. doi:10.1002/anie.201004671 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 49. | Cha, Y. J.; Cadwallader, K. R. J. Agric. Food Chem. 1998, 46, 1123–1128. doi:10.1021/jf970380g |
| 47. | Citron, C. A.; Rabe, P.; Dickschat, J. S. J. Nat. Prod. 2012, 75, 1765–1776. doi:10.1021/np300468h |
| 48. | Owens, J. D.; Allagheny, N.; Kipping, G.; Ames, J. M. J. Sci. Food Agric. 1997, 74, 132–140. doi:10.1002/(sici)1097-0010(199705)74:1<132::aid-jsfa779>3.0.co;2-8 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 46. | Ansorena, D.; Gimeno, O.; Astiasarán, I.; Bello, J. Food Res. Int. 2001, 34, 67–75. doi:10.1016/s0963-9969(00)00133-2 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 37. | Grob, K.; Zürcher, F. J. Chromatogr. 1976, 117, 285–294. doi:10.1016/0021-9673(76)80005-2 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 65. | Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. doi:10.1021/om100106e |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 1. | Giovannoni, S. J.; Stingl, U. Nature 2005, 437, 343–348. doi:10.1038/nature04158 |
| 2. | González, J. M.; Moran, M. A. Appl. Environ. Microbiol. 1997, 63, 4237–4242. doi:10.1128/aem.63.11.4237-4242.1997 |
| 7. | Shiba, T. Syst. Appl. Microbiol. 1991, 14, 140–145. doi:10.1016/s0723-2020(11)80292-4 |
| 18. | Curson, A. R. J.; Sullivan, M. J.; Todd, J. D.; Johnston, A. W. B. ISME J. 2011, 5, 1191–1200. doi:10.1038/ismej.2010.203 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 6. | Ruiz-Ponte, C.; Cilia, V.; Lambert, C.; Nicolas, J. L. Int. J. Syst. Bacteriol. 1998, 48, 537–542. doi:10.1099/00207713-48-2-537 |
| 19. | Sun, J.; Todd, J. D.; Thrash, J. C.; Qian, Y.; Qian, M. C.; Temperton, B.; Guo, J.; Fowler, E. K.; Aldrich, J. T.; Nicora, C. D.; Lipton, M. S.; Smith, R. D.; De Leenheer, P.; Payne, S. H.; Johnston, A. W. B.; Davie-Martin, C. L.; Halsey, K. H.; Giovannoni, S. J. Nat. Microbiol. 2016, 1, 16065. doi:10.1038/nmicrobiol.2016.65 |
| 5. | Lee, O. O.; Tsoi, M. M. Y.; Li, X.; Wong, P.-K.; Qian, P.-Y. Int. J. Syst. Evol. Microbiol. 2007, 57, 1919–1924. doi:10.1099/ijs.0.64801-0 |
| 16. | Todd, J. D.; Curson, A. R. J.; Kirkwood, M.; Sullivan, M. J.; Green, R. T.; Johnston, A. W. B. Environ. Microbiol. 2011, 13, 427–438. doi:10.1111/j.1462-2920.2010.02348.x |
| 51. | Kubec, R.; Drhová, V.; Velíšek, J. J. Agric. Food Chem. 1998, 46, 4334–4340. doi:10.1021/jf980379x |
| 3. | Selje, N.; Simon, M.; Brinkhoff, T. Nature 2004, 427, 445–448. doi:10.1038/nature02272 |
| 4. | Brinkhoff, T.; Giebel, H.-A.; Simon, M. Arch. Microbiol. 2008, 189, 531–539. doi:10.1007/s00203-008-0353-y |
| 17. | Curson, A. R. J.; Rogers, R.; Todd, J. D.; Brearley, C. A.; Johnston, A. W. B. Environ. Microbiol. 2008, 10, 757–767. doi:10.1111/j.1462-2920.2007.01499.x |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 12. | Reisch, C. R.; Stoudemayer, M. J.; Varaljay, V. A.; Amster, I. J.; Moran, M. A.; Whitman, W. B. Nature 2011, 473, 208–211. doi:10.1038/nature10078 |
| 14. | Todd, J. D.; Kirkwood, M.; Newton-Payne, S.; Johnston, A. W. B. ISME J. 2012, 6, 223–226. doi:10.1038/ismej.2011.79 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 11. | Dickschat, J. S.; Rabe, P.; Citron, C. A. Org. Biomol. Chem. 2015, 13, 1954–1968. doi:10.1039/c4ob02407a |
| 15. | Kirkwood, M.; Le Brun, N. E.; Todd, J. D.; Johnston, A. W. B. Microbiology (London, U. K.) 2010, 156, 1900–1906. doi:10.1099/mic.0.038927-0 |
| 50. | Wickel, S. M.; Citron, C. A.; Dickschat, J. S. Eur. J. Org. Chem. 2013, 2906–2913. doi:10.1002/ejoc.201300049 |
| 10. | González, J. M.; Simó, R.; Massana, R.; Covert, J. S.; Casamayor, E. O.; Pedrós-Alió, C.; Moran, M. A. Appl. Environ. Microbiol. 2000, 66, 4237–4246. doi:10.1128/aem.66.10.4237-4246.2000 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 8. | Biebl, H.; Allgaier, M.; Tindall, B. J.; Koblizek, M.; Lünsdorf, H.; Pukall, R.; Wagner-Döbler, I. Int. J. Syst. Evol. Microbiol. 2005, 55, 1089–1096. doi:10.1099/ijs.0.63511-0 |
| 9. | Lafay, B.; Ruimy, R.; Rausch de Traubenberg, C.; Breittmayer, V.; Gauthier, M. J.; Christen, R. Int. J. Syst. Bacteriol. 1995, 45, 290–296. doi:10.1099/00207713-45-2-290 |
| 13. | Todd, J. D.; Rogers, R.; Li, Y. G.; Wexler, M.; Bond, P. L.; Sun, L.; Curson, A. R. J.; Malin, G.; Steinke, M.; Johnston, A. W. B. Science 2007, 315, 666–669. doi:10.1126/science.1135370 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 22. | Dickschat, J. S.; Zell, C.; Brock, N. L. ChemBioChem 2010, 11, 417–425. doi:10.1002/cbic.200900668 |
| 23. | Brock, N. L.; Citron, C. A.; Zell, C.; Berger, M.; Wagner-Döbler, I.; Petersen, J.; Brinkhoff, T.; Simon, M.; Dickschat, J. S. Beilstein J. Org. Chem. 2013, 9, 942–950. doi:10.3762/bjoc.9.108 |
| 20. | Lovelock, J. E.; Maggs, R. J.; Rasmussen, R. A. Nature 1972, 237, 452–453. doi:10.1038/237452a0 |
| 21. | Charlson, R. J.; Lovelock, J. E.; Andreae, M. O.; Warren, S. G. Nature 1987, 326, 655–661. doi:10.1038/326655a0 |
| 53. | Takeoka, G. R.; Buttery, R. G.; Teranishi, R.; Flath, R. A.; Güntert, M. J. Agric. Food Chem. 1991, 39, 1848–1851. doi:10.1021/jf00010a032 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 52. | Ferhat, M. A.; Tigrine-Kordjani, N.; Chemat, S.; Meklati, B. Y.; Chemat, F. Chromatographia 2007, 65, 217–222. doi:10.1365/s10337-006-0130-5 |
| 31. | Geng, H.; Bruhn, J. B.; Nielsen, K. F.; Gram, L.; Belas, R. Appl. Environ. Microbiol. 2008, 74, 1535–1545. doi:10.1128/aem.02339-07 |
| 32. | Berger, M.; Brock, N. L.; Liesegang, H.; Dogs, M.; Preuth, I.; Simon, M.; Dickschat, J. S.; Brinkhoff, T. Appl. Environ. Microbiol. 2012, 78, 3539–3551. doi:10.1128/aem.07657-11 |
| 33. | Brock, N. L.; Nikolay, A.; Dickschat, J. S. Chem. Commun. 2014, 50, 5487–5489. doi:10.1039/c4cc01924e |
| 29. | Greer, E. M.; Aebisher, D.; Greer, A.; Bentley, R. J. Org. Chem. 2008, 73, 280–283. doi:10.1021/jo7018416 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 30. | Kintaka, K.; Ono, H.; Tsubotani, S.; Harada, S.; Okazaki, H. J. Antibiot. 1984, 37, 1294–1300. doi:10.7164/antibiotics.37.1294 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 27. | Mönnich, J.; Tebben, J.; Bergemann, J.; Case, R.; Wohlrab, S.; Harder, T. ISME J. 2020, 14, 1614–1625. doi:10.1038/s41396-020-0631-5 |
| 55. | Citron, C. A.; Barra, L.; Wink, J.; Dickschat, J. S. Org. Biomol. Chem. 2015, 13, 2673–2683. doi:10.1039/c4ob02609h |
| 28. | Bruhn, J. B.; Nielsen, K. F.; Hjelm, M.; Hansen, M.; Bresciani, J.; Schulz, S.; Gram, L. Appl. Environ. Microbiol. 2005, 71, 7263–7270. doi:10.1128/aem.71.11.7263-7270.2005 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
| 24. | Brock, N. L.; Menke, M.; Klapschinski, T. A.; Dickschat, J. S. Org. Biomol. Chem. 2014, 12, 4318–4323. doi:10.1039/c4ob00719k |
| 25. | Celik, E.; Maczka, M.; Bergen, N.; Brinkhoff, T.; Schulz, S.; Dickschat, J. S. Org. Biomol. Chem. 2017, 15, 2919–2922. doi:10.1039/c7ob00357a |
| 54. | Nawrath, T.; Mgode, G. F.; Weetjens, B.; Kaufmann, S. H. E.; Schulz, S. Beilstein J. Org. Chem. 2012, 8, 290–299. doi:10.3762/bjoc.8.31 |
| 26. | Durham, B. P.; Sharma, S.; Luo, H.; Smith, C. B.; Amin, S. A.; Bender, S. J.; Dearth, S. P.; Van Mooy, B. A. S.; Campagna, S. R.; Kujawinski, E. B.; Armbrust, E. V.; Moran, M. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 453–457. doi:10.1073/pnas.1413137112 |
| 45. | Adams, R. P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Pub Corp.: Carol Stream, IL, 2009. |
© 2021 Chhalodia et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)