Abstract
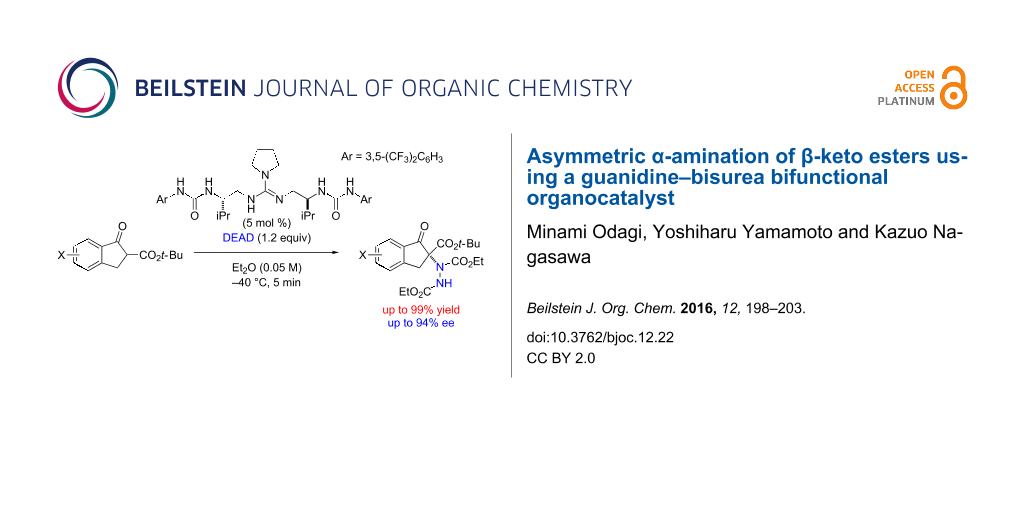
An asymmetric α-amination of β-keto esters with azodicarboxylate in the presence of a guanidine–bisurea bifunctional organocatalyst was investigated. The α-amination products were obtained in up to 99% yield with up to 94% ee.

Graphical Abstract
Introduction
Asymmetric α-amination of β-keto esters is an important synthetic route to optically active α-amino acid derivatives with chiral quaternary stereocenters [1,2]. Since an α-amino acid moiety is frequently found in biologically active compounds, considerable efforts have been made to achieve a stereoselective synthesis of this structure [3,4]. In particular, catalytic asymmetric α-amination of β-keto esters has been widely explored, using both metal catalysts and organocatalysts [5-18].
We have developed a series of guanidine–bis(thio)urea bifunctional organocatalysts, and have used them in a variety of asymmetric reactions [19,20]. Recently, we disclosed an α-hydroxylation of tetralone-derived β-keto esters 2 using guanidine–bisurea bifunctional organocatalyst 1a in the presence of cumene hydroperoxide (CHP) as an oxidant (Figure 1a) [21]. This reaction provides the corresponding α-hydroxylation products 3 in high yield with high enantioselectivity. A computational study of the transition state of this reaction revealed that inter- and intramolecular hydrogen-bonding networks between catalyst and substrate are critical for obtaining high enantioselectivity [22]. Based upon these insights, we expected that guanidine–bisurea bifunctional organocatalyst 1 would be effective in promoting α-amination of β-keto esters as a result of interactions between guanidine and enolate of the β-keto ester, and between urea and azodicarboxylate (Figure 1b). Herein, we describe the catalytic asymmetric α-amination of β-keto esters with azodicarboxylates as a nitrogen source in the presence of 1.
![[1860-5397-12-22-1]](/bjoc/content/figures/1860-5397-12-22-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Asymmetric α-hydroxylation of 2 in the presence of 1a. b) Asymmetric α-amination of 4 explored in this study.
Figure 1: a) Asymmetric α-hydroxylation of 2 in the presence of 1a. b) Asymmetric α-amination of 4 explored i...
Results and Discussion
The reaction conditions for α-amination of β-keto ester 4a in the presence of diethyl azodicarboxylate (DEAD) were optimized as follows. First, we focused on the catalyst structure (Table 1) [23]. Initially, the R3 substituent on the chiral spacer of the catalyst 1 was optimized (Table 1, entries 1–4). The catalyst with a benzyl group at R3 (1a) afforded 5a in excellent yield with moderate enantioselectivity for R configuration (Table 1, entry 1) [24,25]. When R3 was changed to a phenyl group, the enantioselectivity was slightly increased to 59% ee (Table 1, entry 2). In the case of a methyl group, 5a was obtained in 98% yield with 50% ee (Table 1, entry 3). An isopropyl group as R3 group was most effective, affording 5a with 66% ee (Table 1, entry 4). Next, we optimized R1 and R2 on the guanidine moiety (Table 1, entries 5 and 6). A catalyst bearing a six-membered ring at R1 and R2 (1e) gave excellent yield, but with only 27% ee (Table 1, entry 5). Interestingly, catalyst 1f bearing a pyrrolidine ring at R1 and R2 showed the highest selectivity, and 5a was obtained in 99% yield with 80% ee (Table 1, entry 6). Thus, we chose 1f as the optimized catalyst for the reaction [26].
Table 1: Optimization of catalyst structure.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-22-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | catalyst 1 | α-amination product 5a | ||||
|---|---|---|---|---|---|---|
| R1, R2 | R3 | yield (%)b | ee (%)c | |||
| 1 | 1a | H, –(CH2)17CH3 | Bn | 99 | 53 | |
| 2 | 1b | H, –(CH2)17CH3 | Ph | 94 | 59 | |
| 3 | 1c | H, –(CH2)17CH3 | Me | 98 | 50 | |
| 4 | 1d | H, –(CH2)17CH3 | iPr | 97 | 66 | |
| 5 | 1e | –(CH2)5– | iPr | 93 | 27 | |
| 6 | 1f | –(CH2)4– | iPr | 99 | 80 | |
aReaction conditions: 4a (0.1 mmol), DEAD (0.12 mmol) and 1 (5 mol %) in toluene (2.0 mL) at 0 °C. bIsolated yield. cDetermined by HPLC analysis using a chiral stationary phase. DEAD = diethyl azodicarboxylate.
Next, we investigated various solvents, such as ethyl acetate, dichloromethane, acetonitrile and diethyl ether (Table 2, entries 1–5) for the reaction in the presence of catalyst 1f (Table 2). The best result was obtained with diethyl ether, and 5a was isolated in 95% yield with 85% ee (Table 2, entry 5). The enantioselectivity was improved to 90% ee by decreasing the reaction temperature to −40 °C without decrease in the yield (Table 2, entry 6). When the reaction was performed at −78 °C, the yield of 5a was dropped to 91% (Table 2, entry 7).
Table 2: Investigation of solvent effect.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-22-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | time (min) | temp (°C) | α-amination product 5a | |
|---|---|---|---|---|---|
| yield (%)b | ee (%)c | ||||
| 1 | toluene | 5 | 0 | 99 | 80 |
| 2 | EtOAc | 5 | 0 | 99 | 78 |
| 3 | DCM | 30 | 0 | 99 | 75 |
| 4 | MeCN | 30 | 0 | 97 | 58 |
| 5 | Et2O | 5 | 0 | 95 | 85 |
| 6 | Et2O | 5 | −40 | 99 | 90 |
| 7 | Et2O | 30 | −78 | 91 | 89 |
aReaction conditions: 4a (0.1 mmol), DEAD (0.12 mmol) and 1f (5 mol %) in solvent (2.0 mL). bIsolated yield. cDetermined by HPLC analysis using a chiral stationary phase. DEAD = diethyl azodicarboxylate. EtOAc = ethyl acetate. DCM = dichloromethane. MeCN = acetonitrile. Et2O = diethyl ether.
As a further investigation, we optimized the ester moiety of the azodicarboxylate (Table 3). In addition to the ethyl ester (Table 3, entry 1), we examined benzyl, isopropyl, and tert-butyl ester as azodicarboxylate (Table 3, entries 2–4). By changing the ethyl ester to a benzyl or isopropyl ester, the amination products 6a and 7a were obtained in excellent yield, but the enantioselectivity was dropped to 64 and 79% ee, respectively (Table 3, entries 2 and 3). In the case of the tert-butyl ester, the reactivity of the azodicarboxylate was drastically decreased, and the reaction has not been completed after 48 h. The enantioselectivity of 8a was also poor (Table 3, entry 4).
Table 3: Optimization of the ester moiety of azodicarboxylate.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-22-i5.svg?max-width=637&scale=1.0)
|
|||||
| entry | azodicarboxylate | time | α-amination product | ||
|---|---|---|---|---|---|
| R | yield (%)b | ee (%)c | |||
| 1 | Et | 5 min | 5a | 99 | 90 |
| 2 | Bn | 5 min | 6a | 98 | 64 |
| 3 | iPr | 30 min | 7a | 98 | 79 |
| 4 | t-Bu | 48 h | 8a | 58 | 44 |
aReaction conditions: 4a (0.1 mmol), azodicarboxylate (0.12 mmol) and 1f (5 mol %) in Et2O (2.0 mL) at −40 °C. bIsolated yield. cDetermined by HPLC analysis using a chiral stationary phase.
With the optimal reaction conditions in hand (Table 2, entry 6), we investigated the substrate scope for α-amination of β-keto esters (Scheme 1). First, various indanone-derived β-keto esters were examined. With electron-donating substituents such as methoxy and methyl, the corresponding amination products 5b–f were obtained in high yield (72–99%) with high enantioselectivity (77–94% ee). In the case of substrates bearing electron-withdrawing groups, such as halogen atoms, the amination products 5g–j were obtained with high enantioselectivity (73–86% ee). On the other hand, in the case of tetralone derivative 4k and cyclopentanone derivative 4l, the enantioselectivity of the products 5k and 5l was moderate to low (61% ee and 38% ee, respectively).
![[1860-5397-12-22-i1]](/bjoc/content/inline/1860-5397-12-22-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Substrate scope of α-amination.
Scheme 1: Substrate scope of α-amination.
To get insight into the transition state of the reaction, we performed a nonlinear effect (NLE) study (Figure 2) [27]. We found a linear relationship between % ee of 1f and 5a in the reaction. This result suggests that the stereoselectivity is controlled by the monomeric structure of 1f [28-31]. Furthermore, to confirm the requirement of bifunctionality in catalyst 1, we performed the α-amination reaction in the presence of carbamate 9 or triurea 10 as a catalyst (Scheme 2). In both cases, the enantioselectivity of the α-amination product 3a was drastically decreased. These results clearly show that the guanidine and urea moieties in the catalyst 1f are mandatory for obtaining high enantioselectivity, presumably interacting with the enolate of 4a and DEAD, respectively.
![[1860-5397-12-22-2]](/bjoc/content/figures/1860-5397-12-22-2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-22-i2]](/bjoc/content/inline/1860-5397-12-22-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: α-Amination of 4a using 9 or 10 as catalyst.
Scheme 2: α-Amination of 4a using 9 or 10 as catalyst.
Conclusion
In conclusion, we have developed an asymmetric α-amination of β-keto esters 4 by using guanidine–bisurea bifunctional organocatalyst 1f in the presence of diethyl azodicarboxylate (DEAD). The α-amination of various indanone-derived β-keto esters proceeded in high yield (up to 99% yield) and with high enantioselectivity (up to 94% ee).
Supporting Information
| Supporting Information File 1: Experimental procedures, copies of NMR spectra and HPLC chromatograms. | ||
| Format: PDF | Size: 3.6 MB | Download |
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” (no. 23105013) from The Ministry of Education, Culture, Sports, Science and Technology. M.O. thanks the Japan Society for the Promotion of Science (JSPS) for a fellowship (no. 201506486).
References
-
Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e
Return to citation in text: [1] -
Nájera, C.; Sansano, J. C. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o
Return to citation in text: [1] -
Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6
Return to citation in text: [1] -
Yet, L. Angew. Chem., Int. Ed. 2001, 40, 875–877. doi:10.1002/1521-3773(20010302)40:5<875::AID-ANIE875>3.0.CO;2-C
Return to citation in text: [1] -
Greck, C.; Drouillat, B.; Thomassigny, C. Eur. J. Org. Chem. 2004, 1377–1385. doi:10.1002/ejoc.200300657
Return to citation in text: [1] -
Janey, J. M. Angew. Chem., Int. Ed. 2005, 44, 4292–4300. doi:10.1002/anie.200462314
Return to citation in text: [1] -
Vilaivan, T.; Bhanthumnavin, W. Molecules 2010, 15, 917–958. doi:10.3390/molecules15020917
Return to citation in text: [1] -
Smith, A. M. R.; Hii, K. K. Chem. Rev. 2011, 111, 1637–1656. doi:10.1021/cr100197z
Return to citation in text: [1] -
Marigo, M.; Juhl, K.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2003, 42, 1367–1369. doi:10.1002/anie.200390350
Return to citation in text: [1] -
Terada, M.; Nakano, M.; Ube, H. J. Am. Chem. Soc. 2006, 128, 16044–16045. doi:10.1021/ja066808m
Return to citation in text: [1] -
Huber, D. P.; Stanek, K.; Togni, A. Tetrahedron: Asymmetry 2006, 17, 658–664. doi:10.1016/j.tetasy.2006.01.035
Return to citation in text: [1] -
Kang, Y. K.; Kim, D. Y. Tetrahedron Lett. 2006, 47, 4565–4568. doi:10.1016/j.tetlet.2006.05.003
Return to citation in text: [1] -
He, R.; Wang, X.; Hashimoto, T.; Maruoka, K. Angew. Chem., Int. Ed. 2008, 47, 9466–9468. doi:10.1002/anie.200804140
Return to citation in text: [1] -
Jung, S. H.; Kim, D. Y. Tetrahedron Lett. 2008, 49, 5527–5530. doi:10.1016/j.tetlet.2008.07.041
Return to citation in text: [1] -
Lan, Q.; Wang, X.; He, R.; Ding, C.; Maruoka, K. Tetrahedron Lett. 2009, 50, 3280–3282. doi:10.1016/j.tetlet.2009.02.041
Return to citation in text: [1] -
Konishi, H.; Lam, T. Y.; Malerich, J. P.; Rawal, V. H. Org. Lett. 2010, 12, 2028–2031. doi:10.1021/ol1005104
Return to citation in text: [1] -
Chosh, S.; Nandakumar, M. V.; Krautscheid, H.; Schneider, C. Tetrahedron Lett. 2010, 51, 1860–1862. doi:10.1016/j.tetlet.2010.02.007
Return to citation in text: [1] -
Azuma, T.; Kobayashi, Y.; Sakata, K.; Sasamori, T.; Tokitoh, N.; Takemoto, Y. J. Org. Chem. 2014, 79, 1805–1817. doi:10.1021/jo4028775
Return to citation in text: [1] -
Sohtome, Y.; Nagasawa, K. Synlett 2010, 1–22. doi:10.1055/s-0029-1218542
Return to citation in text: [1] -
Sohtome, Y.; Nagasawa, K. Chem. Commun. 2012, 48, 7777–7789. doi:10.1039/c2cc31846f
Return to citation in text: [1] -
Odagi, M.; Furukori, K.; Watanabe, T.; Nagasawa, K. Chem. – Eur. J. 2013, 19, 16740–16745. doi:10.1002/chem.201303006
Return to citation in text: [1] -
Odagi, M.; Furukori, K.; Yamamoto, Y.; Sato, M.; Iida, K.; Yamanaka, M.; Nagasawa, K. J. Am. Chem. Soc. 2015, 137, 1909–1915. doi:10.1021/ja511149y
Return to citation in text: [1] -
Guanidine–bisthiourea bifunctional organocatalyst was not suitable for the reaction. For details, see Tables S2 and S3 in Supporting Information File 1.
Return to citation in text: [1] -
The absolute stereochemistry of 5a was assigned by comparison with a known compound (ref. [17]).
Return to citation in text: [1] -
Based on previously reported transition states (Figure 1a), we expected that the α-amination product would be the S conformer. However, the reaction afforded the R conformer. This result suggests that the reaction proceeds through a different transition state from previously reported reactions. Further investigation of the transition state is on-going.
Return to citation in text: [1] -
The results of optimization of substituents on the aromatic ring are summarized in Table S1 in Supporting Information File 1.
Return to citation in text: [1] -
Satyanarayana, T.; Abraham, S.; Kagan, H. B. Angew. Chem., Int. Ed. 2009, 48, 456–494. doi:10.1002/anie.200705241
Return to citation in text: [1] -
Sohtome, Y.; Takemura, N.; Takada, K.; Takagi, R.; Iguchi, T.; Nagasawa, K. Chem. – Asian J. 2007, 2, 1150–1160. doi:10.1002/asia.200700145
Return to citation in text: [1] -
Sohtome, Y.; Shin, B.; Horitsugi, N.; Takagi, R.; Noguchi, K.; Nagasawa, K. Angew. Chem., Int. Ed. 2010, 49, 7299–7303. doi:10.1002/anie.201003172
Return to citation in text: [1] -
Sohtome, Y.; Tanaka, S.; Takada, K.; Yamaguchi, T.; Nagasawa, K. Angew. Chem., Int. Ed. 2010, 49, 9254–9257. doi:10.1002/anie.201005109
Return to citation in text: [1] -
Sohtome, Y.; Shin, B.; Horitsugi, N.; Noguchi, K.; Nagasawa, K. Chem. – Asian J. 2011, 6, 2463–2470. doi:10.1002/asia.201100363
Return to citation in text: [1]
| 1. | Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e |
| 2. | Nájera, C.; Sansano, J. C. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o |
| 21. | Odagi, M.; Furukori, K.; Watanabe, T.; Nagasawa, K. Chem. – Eur. J. 2013, 19, 16740–16745. doi:10.1002/chem.201303006 |
| 19. | Sohtome, Y.; Nagasawa, K. Synlett 2010, 1–22. doi:10.1055/s-0029-1218542 |
| 20. | Sohtome, Y.; Nagasawa, K. Chem. Commun. 2012, 48, 7777–7789. doi:10.1039/c2cc31846f |
| 5. | Greck, C.; Drouillat, B.; Thomassigny, C. Eur. J. Org. Chem. 2004, 1377–1385. doi:10.1002/ejoc.200300657 |
| 6. | Janey, J. M. Angew. Chem., Int. Ed. 2005, 44, 4292–4300. doi:10.1002/anie.200462314 |
| 7. | Vilaivan, T.; Bhanthumnavin, W. Molecules 2010, 15, 917–958. doi:10.3390/molecules15020917 |
| 8. | Smith, A. M. R.; Hii, K. K. Chem. Rev. 2011, 111, 1637–1656. doi:10.1021/cr100197z |
| 9. | Marigo, M.; Juhl, K.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2003, 42, 1367–1369. doi:10.1002/anie.200390350 |
| 10. | Terada, M.; Nakano, M.; Ube, H. J. Am. Chem. Soc. 2006, 128, 16044–16045. doi:10.1021/ja066808m |
| 11. | Huber, D. P.; Stanek, K.; Togni, A. Tetrahedron: Asymmetry 2006, 17, 658–664. doi:10.1016/j.tetasy.2006.01.035 |
| 12. | Kang, Y. K.; Kim, D. Y. Tetrahedron Lett. 2006, 47, 4565–4568. doi:10.1016/j.tetlet.2006.05.003 |
| 13. | He, R.; Wang, X.; Hashimoto, T.; Maruoka, K. Angew. Chem., Int. Ed. 2008, 47, 9466–9468. doi:10.1002/anie.200804140 |
| 14. | Jung, S. H.; Kim, D. Y. Tetrahedron Lett. 2008, 49, 5527–5530. doi:10.1016/j.tetlet.2008.07.041 |
| 15. | Lan, Q.; Wang, X.; He, R.; Ding, C.; Maruoka, K. Tetrahedron Lett. 2009, 50, 3280–3282. doi:10.1016/j.tetlet.2009.02.041 |
| 16. | Konishi, H.; Lam, T. Y.; Malerich, J. P.; Rawal, V. H. Org. Lett. 2010, 12, 2028–2031. doi:10.1021/ol1005104 |
| 17. | Chosh, S.; Nandakumar, M. V.; Krautscheid, H.; Schneider, C. Tetrahedron Lett. 2010, 51, 1860–1862. doi:10.1016/j.tetlet.2010.02.007 |
| 18. | Azuma, T.; Kobayashi, Y.; Sakata, K.; Sasamori, T.; Tokitoh, N.; Takemoto, Y. J. Org. Chem. 2014, 79, 1805–1817. doi:10.1021/jo4028775 |
| 3. | Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6 |
| 4. | Yet, L. Angew. Chem., Int. Ed. 2001, 40, 875–877. doi:10.1002/1521-3773(20010302)40:5<875::AID-ANIE875>3.0.CO;2-C |
| 26. | The results of optimization of substituents on the aromatic ring are summarized in Table S1 in Supporting Information File 1. |
| 28. | Sohtome, Y.; Takemura, N.; Takada, K.; Takagi, R.; Iguchi, T.; Nagasawa, K. Chem. – Asian J. 2007, 2, 1150–1160. doi:10.1002/asia.200700145 |
| 29. | Sohtome, Y.; Shin, B.; Horitsugi, N.; Takagi, R.; Noguchi, K.; Nagasawa, K. Angew. Chem., Int. Ed. 2010, 49, 7299–7303. doi:10.1002/anie.201003172 |
| 30. | Sohtome, Y.; Tanaka, S.; Takada, K.; Yamaguchi, T.; Nagasawa, K. Angew. Chem., Int. Ed. 2010, 49, 9254–9257. doi:10.1002/anie.201005109 |
| 31. | Sohtome, Y.; Shin, B.; Horitsugi, N.; Noguchi, K.; Nagasawa, K. Chem. – Asian J. 2011, 6, 2463–2470. doi:10.1002/asia.201100363 |
| 24. | The absolute stereochemistry of 5a was assigned by comparison with a known compound (ref. [17]). |
| 25. | Based on previously reported transition states (Figure 1a), we expected that the α-amination product would be the S conformer. However, the reaction afforded the R conformer. This result suggests that the reaction proceeds through a different transition state from previously reported reactions. Further investigation of the transition state is on-going. |
| 17. | Chosh, S.; Nandakumar, M. V.; Krautscheid, H.; Schneider, C. Tetrahedron Lett. 2010, 51, 1860–1862. doi:10.1016/j.tetlet.2010.02.007 |
| 23. | Guanidine–bisthiourea bifunctional organocatalyst was not suitable for the reaction. For details, see Tables S2 and S3 in Supporting Information File 1. |
| 22. | Odagi, M.; Furukori, K.; Yamamoto, Y.; Sato, M.; Iida, K.; Yamanaka, M.; Nagasawa, K. J. Am. Chem. Soc. 2015, 137, 1909–1915. doi:10.1021/ja511149y |
| 27. | Satyanarayana, T.; Abraham, S.; Kagan, H. B. Angew. Chem., Int. Ed. 2009, 48, 456–494. doi:10.1002/anie.200705241 |
© 2016 Odagi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)