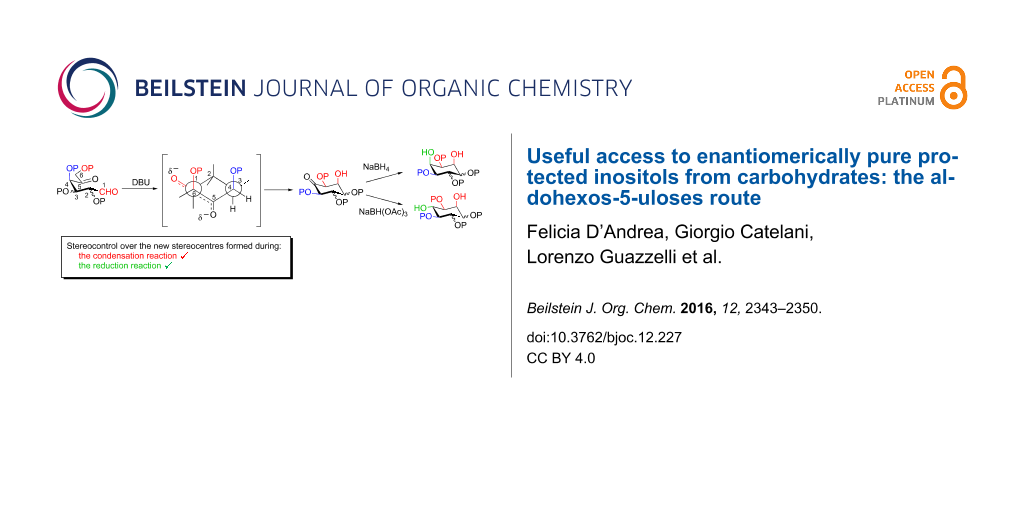
Abstract
The intramolecular aldol condensation of aldohexos-5-ulose derivatives of the D-xylo and L-ribo stereoseries has been studied. Only one of the four possible inososes was isolated from both stereoseries in reasonable yields (30–38%). The results obtained, together with the previous findings for the L-arabino and L-lyxo stereoseries, allowed for the rationalisation of a mechanism of the reaction based on open-transition-state models and electron-withdrawing inductive effects. Complementary reductions of the intermediate inososes were possible by changing the reaction conditions, and two isomeric inositol derivatives were obtained with complete stereoselection from each inosose. The presented approach permits us to control the configuration of three out of the six stereocentres of the inositol frame and gives access to seven of the nine inositols. Noteworthy, for the D-xylo derivative, the two-step sequence (condensation followed by reduction with NaBH(OAc)3) represents the biomimetic synthesis of myo-inositol. Furthermore, the sugar-based pathway leads directly to enantiomerically pure selectively protected inositols and does not require any desymmetrisation procedure which is needed when myo-inositol and other achiral precursors are employed as starting materials. As an example of application of the method, the indirect selective protection of secondary inositols’ hydroxy functions, by placing specific protecting groups on the aldohexos-5-ulose precursor has been presented.

Graphical Abstract
Introduction
Inositols are a family of biologically relevant compounds [1,2] constituted by nine stereoisomeric hexahydroxycyclohexanes (Figure 1): five have been found in nature (myo, scyllo, D-chiro, L-chiro, and neo), while the remaining four are unnatural synthetic products (cis, epi, allo, and muco).
![[1860-5397-12-227-1]](/bjoc/content/figures/1860-5397-12-227-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Several myo-inositol phosphates, for instance D-myo-inositol-1,4,5-triphosphate [D-I(1,4,5)P3] and D-myo-inositol-1,3,4,5-tetrakisphosphate [D-I(1,3,4,5)P4], are ubiquitous in all living organisms. They are involved in different crucial cellular functions which spread from cell growth to intracellular signal transductions events [3-5]. The field investigating inositol phosphates and their involvement in mediating certain aspects of cell biology is continuously broadening. Hence, a deeper understanding of how they act on a molecular level is required.
For this reason, many research efforts were directed toward the investigation of the structure–activity relationship (SAR) between inositol phosphates and biomacromolecules. These studies require various regio- and stereoisomers of inositol phosphates [6,7] and have prompted the development of practical and efficient synthetic methods for stereoselectively accessing all natural and unnatural inositols as well as their derivatives.
There are three general synthetic approaches to prepare inositols: 1) stereoselective elaborations of the inexpensive, commercially available myo-inositol [8-13]; 2) elaboration of the six carbon atom skeleton of either a) tetrahydroxycyclohexene derivatives [14,15] (synthetic or natural conduritols) through stereoselective cis-hydroxylation or epoxidation–hydrolysis of the double bond or b) benzene [16,17] or halo-benzenes [18-21], by microbial oxidation; 3) carbocyclization involving organometallic intermediates (Ferrier II reaction [22] performed on 6-O-acyl-hex-5-enopyranosides followed by reduction [23,24]; trialkylaluminium [25,26] or titanium(IV)-assisted [27] conversion of hex-5-enopyranosides and SmI2-promoted [28-30] pinacol coupling of dialdehyde derivatives).
A strategy related to the latter approach relies on a base-promoted aldol condensation of aldohexos-5-uloses followed by reduction of the carbonyl group, as reported in the retrosynthetic Scheme 1. This method again uses sugars as starting material but differs from previous work in that it is metal-free.
![[1860-5397-12-227-i1]](/bjoc/content/inline/1860-5397-12-227-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic approach to inositols from aldohexos-5-uloses.
Scheme 1: Retrosynthetic approach to inositols from aldohexos-5-uloses.
The first application of this approach was described by Kiely [31,32] who obtained a sample of myo-inositol in a mixture with other non characterised stereoisomers by treating D-xylo-hexos-5-ulose [31] and its 6-phosphate [32] with 0.1 N aqueous NaOH followed by NaBH4 reduction of the crude inosose mixture.
Although these pioneering results were not of synthetic significance, they elucidated for the first time the biosynthetic correlation between D-glucose and myo-inositol through the intermediate formation of D-xylo-hexos-5-ulose. A more synthetically useful result was obtained when the aldol condensation of aldohexos-5-uloses derivatives was run in organic solvents in the presence of an organic base. The reaction occurred in a completely stereocontrolled manner and led to a single inosose intermediate [33-35]. The subsequent stereoselective reduction gave single inositols in good yields: epi-inositol starting from L-arabino-hexos-5-ulose [33] and D-chiro-inositol starting from the L-lyxo stereoisomeric dicarbonyl precursor [34].
Taking these considerations into account, we have recently completed the preparation of at least one enantioform of the four diastereoisomeric aldohexos-5-ulose series. A general approach was developed starting from β-D-galactopyranosides, including the commercially and cheaply available lactose [36-38].
Presented here is the extension of the above synthetic route to inositols using aldohexos-5-uloses derivatives of the D-xylo and L-ribo series, respectively C-4 and C-2 epimers of the L-arabino series. Based on these aldol condensations together with the work previously performed on the L-arabino [33] and L-lyxo [34] series, a working mechanism is presented to explain the complete stereocontrol over the formation of the two new stereogenic centres (red in Scheme 1). Furthermore, the stereochemical outcome of the intermediate inososes reduction by using different reagents, namely the stereocontrol over a third stereogenic centre (green in Scheme 1), is also reported.
Results and Discussion
The aldol condensation reactions were performed at room temperature by treating aldohexos-5-ulose derivatives with catalytic amounts of DBU (0.15 equiv) as an organic base promoting agent in toluene, a 1:1 mixture of toluene–CH2Cl2, or CH2Cl2 to circumvent solubility problems (Table 1, see Supporting Information File 1 for full experimental data).
Table 1: Preparation of inosose derivatives 5–8 and 11–12 by intramolecular aldol condensation of aldohexos-5-uloses derivatives 1–4 and 9–10.
| Compound | Conditionsa | Products (% yield) |
|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-227-i3.svg?max-width=637&scale=1.0)
|
5% DBU solution, dry toluene, rt, 6 h |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-227-i4.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-227-i5.svg?max-width=637&scale=1.0)
|
5% DBU solution, dry CH2Cl2, 0 °C, 1.5 h |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-227-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-227-i7.svg?max-width=637&scale=1.0)
|
5% DBU solution, dry 1:1 toluene/CH2Cl2, rt, 2.5 h |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-227-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-227-i9.svg?max-width=637&scale=1.0)
|
5% DBU solution, dry CH2Cl2, rt, 2.0 h |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-227-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-227-i11.svg?max-width=637&scale=1.0)
|
5% DBU solution, dry CH2Cl2, rt, 1 h |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-227-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-227-i13.svg?max-width=637&scale=1.0)
|
5% DBU solution, dry 1:1 toluene/CH2Cl2, rt, 3 h |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-227-i14.svg?max-width=637&scale=1.0)
|
aIntramolecular cyclizations of 1 [33], 3 [34], and 4 [35] have been reported in previous works.
In all the cases studied, only one of the four possible inososes was isolated. This was characterised by a cis axial–equatorial arrangement of the two oxygenated substituents on the new stereocentres (2 and 3) which were also cis to the other substituent α to the keto group (position 6). This 2,3,6-cis arrangement of the aldol products was observed irrespective to the stereochemistry of the other two positions (4 and 5), corresponding to the C-2 and C-3 of the parent dicarbonyl derivative.
To determine the effect of additional protecting groups on the reaction, the intramolecular aldol condensation was also performed on aldohexos-5-uloses of the L-lyxo and D-xylo series bearing a methyl protection on position 4 or a benzyl protection on position 3 (compounds 4 [35] and 10 [38], respectively). The stereo-outcome and the yields confirmed the cyclisation results obtained for dibenzyl derivatives.
Considering the results obtained, the intramolecular aldol condensation of the lyxo and arabino stereoseries affords the inososes in fairly good yields (54–60%) [33-35], while the same reaction run on the ribo and xylo stereoseries gives the cyclization products only in moderate yields (30–38%). In these latter cases, the colour of the reaction mixture changed over time from light yellow to dark orange. NMR analysis of the crude mixture verified the formation of a single inosose, which was the one isolated after chromatographic purification, together with indistinct side products possibly deriving by polycondensations or eliminations after the aldol condensation.
The structure and stereochemistry of inososes 5–8, 11, and 12 were confirmed by 1H, 13C and 2D NMR experiments. For example, the conformation of inosose 6 was established by the similar value of the vicinal proton coupling constants and by the presence of long-range coupling between H-2 and H-6 (J2,6 = 1.4 Hz), and H-3 and H-5 (J3,5 = 2.6 Hz). For 11 and 12, the high value of the J5,6 (9.4 Hz and 8.5 Hz, respectively) and J4,5 (9.7 Hz and 9.3 Hz), and the low value of the J2,3 (2.4 Hz and 2.7 Hz) and J3,4 (2.2 Hz and 2.3 Hz) agree with four substituents in the equatorial position for the preferred conformer.
A comparable example of intramolecular aldol condensation of a 1,5-dicarbonyl derivative has been reported by Tadano et al. [39]. Almost a sole aldol product was obtained in a good yield and was characterised by the same 2,3,6-cis relationship observed in our experiments. In this case, the authors focused their attention only on the new formed chiral centres and on the cis-orientation of their substituents. To rationalise the result obtained, they referred to the topological rules, which were proposed by Seebach and Golinski [40] to explain the preferred formation of the threo-configuration in intermolecular Michael additions. Unfortunately, not all these rules are applicable to the intramolecular reaction under investigation and, even more disappointing, they do not give any correlation between the two new chiral centres and the chiral centre α to the keto group which was already present on the parent dicarbonyl compound. Indeed, this appears as the most relevant and unexpected result of the present work. We performed the intramolecular aldol condensation on 4- and 6-deoxy-aldohexos-5-uloses of the L-arabino series (not shown). In the latter case, no reaction was observed even after longer reaction times. On the contrary, the C-4 deoxygenated compound afforded an inseparable complex mixture of products, possibly derived from a first carbocyclisation followed by elimination reactions.
In an attempt to find an explanation of the stereochemical outcome of the reaction, we directed our attention toward the open-transition-state models. These have been proposed to explain the prevalent formation of syn products, irrespective of the enolate geometry [41], in aldol reactions performed in the absence of a coordinating metal center (for instance, in the case of tin and zirconium enolates, and of “naked” enolates generated from enolsilanes [42]).
In the open-transition-state model, the enolate and the carbonyl group are orientated in an antiperiplanar fashion, maximazing the distance between the negatively charged oxygen atoms.
On these bases, a possible transition state for the intramolecular reaction, which considers also the 1,4,6-cis relationship (numbering referred to the dicarbonyl compound), is reported in Figure 2.
![[1860-5397-12-227-2]](/bjoc/content/figures/1860-5397-12-227-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Hypothesis of the preferred transition state.
Figure 2: Hypothesis of the preferred transition state.
A working hypothesis is that the axial arrangement of electron-withdrawing substituents in positions 4 and 6 diminishes the negative charge density on the 5-oxygen atom and decreases in this way the overall energy of the transition state, thereby favouring the intramolecular reaction. These kind of inductive effects in cyclic system has been reported before [43,44].
The structure of every inosose obtained allows for complementary stereoselective reductions by choosing the proper reaction conditions. In fact (Table 1), the substituents α to the keto function (2 and 6) and the axial OH in position 3 have always a cis orientation which would favour an equatorially directed anti reduction using NaBH4 as a reducing reagent in an alcoholic solvent (condition A). In particular, it has been proven that the axial OH group β to the keto function plays a fundamental role in directing the nucleophilic attack [45-47]. It has also to be mentioned that by changing the experimental conditions (temperature and alcoholic solvent) a different diastereoselectivity has been reported in the reduction of the same protected inosose [45,48]. This highlights how subtle changes in the inosose structure and/or in the experimental conditions affect the stereo-outcome of the reduction.
Also, the same free axial hydroxy function, deriving from the aldehyde which is beta to the carbonyl group (position 3), would allow for the complementary axial directed syn reduction using NaBH(OAc)3 in an AcOH–CH3CN mixture through an intramolecular hydride transfer as reported by Evans [49] (condition B). The axial OH function would be able to coordinate the reducing agent and thus give an internal hydride transfer from the same side through a six membered cyclic transition state (Figure 3).
![[1860-5397-12-227-3]](/bjoc/content/figures/1860-5397-12-227-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Stereoselective reduction of inosose intermediate.
Figure 3: Stereoselective reduction of inosose intermediate.
In Table 2, the results of the complementary reductions (conditions A and B) are summarised (see Supporting Information File 1 for full experimental data). Seven different inositol derivatives 13–19 were obtained from the four inososes 5–7 and 11 in a stereoselective way, thereby controlling the stereochemistry of a third stereocentre of the inositol frame. The results of the reduction reactions are in good agreement with what has been reported before on similar compounds [45]. The structure and stereochemistry of inositols 13–19 were confirmed by 1H, 13C and 2D NMR experiments directly or after per-acetylation of the reduction product and/or through comparison with previously reported compounds.
Table 2: Stereoselective complementary reductions (conditions A and B) of inososes 5–7 and 11a,b.
| Compound | Reduction conditions | Inositol derivatives | Configuration |
|---|---|---|---|
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-227-i15.svg?max-width=637&scale=1.0)
|
A: NaBH4, EtOH,
−78 °C to 15 °C |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-227-i16.svg?max-width=637&scale=1.0)
|
epi |
| B: NaBH(OAc)3, AcOH, CH3CN, rt |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-227-i17.svg?max-width=637&scale=1.0)
|
muco | |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-227-i18.svg?max-width=637&scale=1.0)
|
A: 1) NaBH4, MeOH, 0 °C; 2) Ac2O, pyridine |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-227-i19.svg?max-width=637&scale=1.0)
|
cis |
| B: NaBH(OAc)3, AcOH, CH3CN, rt |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-227-i20.svg?max-width=637&scale=1.0)
|
epi | |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-227-i21.svg?max-width=637&scale=1.0)
|
A: NaBH4, EtOH,
−78 °C to 15 °C |
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-227-i22.svg?max-width=637&scale=1.0)
|
allo |
| B: NaBH(OAc)3, AcOH, CH3CN, rt |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-227-i23.svg?max-width=637&scale=1.0)
|
D-chiro | |
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-227-i24.svg?max-width=637&scale=1.0)
|
A: NaBH4, MeOH,
0 °C |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-227-i25.svg?max-width=637&scale=1.0)
|
epi |
| B: NaBH(OAc)3, AcOH, CH3CN, rt |
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-227-i26.svg?max-width=637&scale=1.0)
|
myo | |
aInositol derivatives in the table are represented in the way they are obtained as a result of the stereoselective reduction and the preferred conformations can be deduced from the experimental section. bPreparation of 13 [33], 13a [33], 17a [34] and 18a [34] has been described in previous works.
Noteworthy, the two step sequence performed on the D-xylo derivative 11 leads to myo-inositol derivative 19 when NaBH(OAc)3 is employed in the stereoselective reduction of the inosose and represents the biomimetic conversion of D-xylo-aldohexos-5-ulose derivatives into myo-inositol derivatives.
As well as the stereoselective synthesis of enantiopure inositol derivatives, also the possibility to pick and tag specific positions of the inositol ring, installing the protective groups earlier on the carbohydrate frame through the well-known sugar chemistry, is of extreme interest. Although some attractive findings have been reported so far [46,47], the regioselective protection of inositols is still a troublesome area due to the comparable reactivity of the secondary hydroxy functions. Therefore, the carbocyclization of a selectively protected dicarbonyl sugar, namely the L-lyxo derivative 20, whose synthesis will be presented in a forthcoming paper, has been investigated (Scheme 2).
![[1860-5397-12-227-i2]](/bjoc/content/inline/1860-5397-12-227-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Intramolecular cyclization of an orthogonally protected L-lyxo-aldohexos-5-ulose derivative.
Scheme 2: Intramolecular cyclization of an orthogonally protected L-lyxo-aldohexos-5-ulose derivative.
The expected inosose 21 was obtained under standard conditions and was then submitted to the reduction step with NaBH(OAc)3. The D-chiro derivative 22 was isolated in a 47% yield over two steps and with complete stereoselectivity in accordance with the predicted stereo-outcome.
In this way, the orthogonal protecting groups (allyl and naphthalenylmethyl) installed on aldohexos-5-ulose 20, differentiated the two 1,2-cis hydroxy couples (2,3 and 4,5 positions) of 22 which can be easily transformed further in a selective manner.
Conclusion
The intramolecular aldol condensation of aldohexos-5-ulose derivatives of the four stereoseries has been studied. By obtaining the inosose derivatives shown, a better understanding of the relative influence of each position on the stereochemical course of the reaction was gained. These findings allowed us to formulate a plausible rationalization of the mechanism based on open-transition-state models and on inductive effects of axial anti arranged electron-withdrawing substituents.
The overall process, condensation followed by reduction, which involves the formation of three new stereocentres, represents a general access to inositol derivatives. In particular, in a complete stereoselective fashion, inositols of six different configurations characterised by the 2,3,6-cis arrangement of three substituents were obtained from aldohexos-5-uloses. Furthermore, the formal synthesis of L-chiro-inositol could be considered as achieved starting from known D-lyxo-aldohexos-5-ulose derivatives [37,50]. Even if in some cases yields of isolated compounds are not too high, this sugar-based approach gives access directly to enantiomerically pure inositol derivatives. This avoids time and cost-consuming desymmetrisation procedures required when myo-inositol is used as starting material in the preparation of inositol derivatives.
In addition, introducing a suitable pattern of protecting groups in the sugar frame, which will then be present in the inositol ring, has been shown. This allows for an indirect regioselective differentiation of the inositols’ secondary hydroxy groups, which is difficult to achieve by common chemical means.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data of new compounds and 1H and 13C NMR spectra of compounds 6, 11, 12, 14–16, 16a, 18, 19 and 21, 22. | ||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Kılbaş, B.; Balci, M. Tetrahedron 2011, 67, 2355–2389. doi:10.1016/j.tet.2011.01.012
Return to citation in text: [1] -
Duchek, J.; Adams, D. R.; Hudlicky, T. Chem. Rev. 2011, 111, 4223–4258. doi:10.1021/cr1004138
Return to citation in text: [1] -
Irvine, R. F.; Schell, M. J. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. doi:10.1038/35073015
Return to citation in text: [1] -
Chi, T. H.; Crabtree, G. R. Science 2000, 287, 1937–1939. doi:10.1126/science.287.5460.1937
Return to citation in text: [1] -
Odom, A. R.; Stahlberg, A.; Wente, S. R.; York, J. D. Science 2000, 287, 2026–2029. doi:10.1126/science.287.5460.2026
Return to citation in text: [1] -
Chung, S.-K.; Shin, B.-G.; Chang, Y.-T.; Suh, B.-C.; Kim, K.-T. Bioorg. Med. Chem. Lett. 1998, 8, 659–662. doi:10.1016/S0960-894X(98)00081-X
Return to citation in text: [1] -
Hank, T.; Stricker, R.; Krishna, U. M.; Falck, J. R.; Chang, Y. T.; Chung, S. K.; Reiser, G. Eur. J. Biochem. 1999, 261, 577–584. doi:10.1046/j.1432-1327.1999.00326.x
Return to citation in text: [1] -
Tagliaferri, F.; Wang, S.-N.; Berlin, W. K.; Outten, R. A.; Shen, T. Y. Tetrahedron Lett. 1990, 31, 1105–1108. doi:10.1016/S0040-4039(00)88737-7
Return to citation in text: [1] -
Guidot, J. P.; Le Gall, T. Tetrahedron Lett. 1993, 34, 4647–4650. doi:10.1016/S0040-4039(00)60646-9
Return to citation in text: [1] -
Gigg, J.; Gigg, R. Carbohydr. Res. 1997, 299, 77–83. doi:10.1016/S0008-6215(96)00330-8
Return to citation in text: [1] -
Riley, A. M.; Jenkins, D. J.; Potter, B. V. L. Carbohydr. Res. 1998, 314, 277–281. doi:10.1016/S0008-6215(98)00317-6
Return to citation in text: [1] -
Husson, C.; Odier, L.; Vottéro, P. J. A. Carbohydr. Res. 1998, 307, 163–165. doi:10.1016/S0008-6215(98)00003-2
Return to citation in text: [1] -
Gurale, B. P.; Sardessai, R. S.; Shashidhar, M. S. Carbohydr. Res. 2014, 399, 8–14. doi:10.1016/j.carres.2014.08.010
Return to citation in text: [1] -
Kwon, Y.-U.; Lee, C.; Chung, S.-K. J. Org. Chem. 2002, 67, 3327–3338. doi:10.1021/jo016237a
Return to citation in text: [1] -
Podeschwa, M.; Plettenburg, O.; vom Brocke, J.; Block, O.; Adelt, S.; Altenbach, H.-J. Eur. J. Org. Chem. 2003, 1958–1972. doi:10.1002/ejoc.200200572
Return to citation in text: [1] -
Carless, H. A. J.; Busia, K. Tetrahedron Lett. 1990, 31, 1617–1620. doi:10.1016/0040-4039(90)80032-H
Return to citation in text: [1] -
Motherwell, W. B.; Williams, A. S. Angew. Chem., Int. Ed. 1995, 34, 2031–2033. doi:10.1002/anie.199520311
Return to citation in text: [1] -
Mandel, M.; Hudlicky, T. J. Chem. Soc., Perkin Trans. 1 1993, 741–743. doi:10.1039/P19930000741
Return to citation in text: [1] -
Hudlicky, T.; Mandel, M.; Rouden, J.; Lee, R. S.; Bachmann, B.; Dudding, T.; Yost, K. J.; Merola, J. S. J. Chem. Soc., Perkin Trans. 1 1994, 1553–1567. doi:10.1039/P19940001553
Return to citation in text: [1] -
Desjardins, M.; Brammer, L. E., Jr.; Hudlicky, T. Carbohydr. Res. 1997, 304, 39–42. doi:10.1016/S0008-6215(97)00186-9
Return to citation in text: [1] -
Brammer, L. E., Jr.; Hudlicky, T. Tetrahedron: Asymmetry 1998, 9, 2011–2014. doi:10.1016/S0957-4166(98)00182-7
Return to citation in text: [1] -
Ferrier, R. J.; Middleton, S. Chem. Rev. 1993, 93, 2779–2831. doi:10.1021/cr00024a008
Return to citation in text: [1] -
Takahashi, H.; Kittaka, H.; Ikegami, S. Tetrahedron Lett. 1998, 39, 9703–9706. doi:10.1016/S0040-4039(98)02231-X
Return to citation in text: [1] -
Takahashi, H.; Kittaka, H.; Ikegami, S. J. Org. Chem. 2001, 66, 2705–2716. doi:10.1021/jo001575h
Return to citation in text: [1] -
Das, S. K.; Mallet, J.-M.; Sinaÿ, P. Angew. Chem., Int. Ed. Engl. 1997, 36, 493–496. doi:10.1002/anie.199704931
Return to citation in text: [1] -
Jia, C.; Pearce, A. J.; Blériot, Y.; Zhang, Y.; Zhang, L.-H.; Sollogoub, M.; Sinaÿ, P. Tetrahedron: Asymmetry 2004, 15, 699–703. doi:10.1016/j.tetasy.2003.11.029
Return to citation in text: [1] -
Sollogoub, M.; Mallet, J.-M.; Sinaÿ, P. Tetrahedron Lett. 1998, 39, 3471–3472. doi:10.1016/S0040-4039(98)00529-2
Return to citation in text: [1] -
Chiara, J. L.; Martin-Lomas, M. Tetrahedron Lett. 1994, 35, 2969–2972. doi:10.1016/S0040-4039(00)76674-3
Return to citation in text: [1] -
Guidot, J. P.; Le Gall, T.; Mioskowski, C. Tetrahedron Lett. 1994, 35, 6671–6672. doi:10.1016/S0040-4039(00)73464-2
Return to citation in text: [1] -
Carpintero, M.; Fernández-Mayoralas, A.; Jaramillo, C. J. Org. Chem. 1997, 62, 1916–1917. doi:10.1021/jo9623898
Return to citation in text: [1] -
Kiely, D. E.; Fletcher, H. G., Jr. J. Org. Chem. 1969, 34, 1386–1390. doi:10.1021/jo01257a042
Return to citation in text: [1] [2] -
Kiely, D. E.; Sherman, R. S. J. Am. Chem. Soc. 1975, 97, 6810–6814. doi:10.1021/ja00856a035
Return to citation in text: [1] [2] -
Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Catelani, G.; D'Andrea, F.; Griselli, A.; Guazzelli, L.; Legnani, L.; Toma, L. Tetrahedron Lett. 2008, 49, 4534–4536. doi:10.1016/j.tetlet.2008.05.040
Return to citation in text: [1] [2] [3] [4] -
Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V.; Vittorino, E. Carbohydr. Res. 2003, 338, 2349–2358. doi:10.1016/j.carres.2003.08.001
Return to citation in text: [1] -
Guazzelli, L.; Catelani, G.; D'Andrea, F. Carbohydr. Res. 2010, 345, 369–376. doi:10.1016/j.carres.2009.11.027
Return to citation in text: [1] [2] -
Catelani, G.; D'Andrea, F.; Guazzelli, L.; Pistarà, V. Carbohydr. Res. 2009, 344, 717–724. doi:10.1016/j.carres.2009.01.014
Return to citation in text: [1] [2] -
Tadano, K.; Kanazawa, S.; Takao, K.; Ogawa, S. Tetrahedron 1992, 48, 4283–4300. doi:10.1016/S0040-4020(01)80440-3
Return to citation in text: [1] -
Seebach, D.; Goliński, J. Helv. Chim. Acta 1981, 64, 1413–1423. doi:10.1002/hlca.19810640518
Return to citation in text: [1] -
Yamamoto, Y.; Yatagai, H.; Naruta, Y.; Maruyama, K. J. Am. Chem. Soc. 1980, 102, 7107–7109. doi:10.1021/ja00543a040
Return to citation in text: [1] -
Murata, S.; Suzuki, M.; Noyori, R. J. Am. Chem. Soc. 1980, 102, 3248–3249. doi:10.1021/ja00529a062
Return to citation in text: [1] -
Jensen, H. H.; Lyngbye, L.; Jensen, A.; Bols, M. Chem. – Eur. J. 2002, 8, 1218–1226. doi:10.1002/1521-3765(20020301)8:5<1218::AID-CHEM1218>3.0.CO;2-X
Return to citation in text: [1] -
Jensen, H. H.; Bols, M. Acc. Chem. Res. 2006, 39, 259–265. doi:10.1021/ar050189p
Return to citation in text: [1] -
Jagdhane, R. C.; Patil, M. T.; Krishnaswamy, S.; Shashidhar, M. S. Tetrahedron 2013, 69, 5144–5151. doi:10.1016/j.tet.2013.04.081
Return to citation in text: [1] [2] [3] -
Jagdhane, R. C.; Shashidhar, M. S. Eur. J. Org. Chem. 2010, 2945–2953. doi:10.1002/ejoc.201000009
Return to citation in text: [1] [2] -
Sureshan, K. M.; Shashidhar, M. S.; Praveen, T.; Das, T. Chem. Rev. 2003, 103, 4477–4504. doi:10.1021/cr0200724
Return to citation in text: [1] [2] -
Collet, C.; Chrétien, F.; Chapleur, Y.; Lamandé-Langle, S. Beilstein J. Org. Chem. 2016, 12, 353–361. doi:10.3762/bjoc.12.39
Return to citation in text: [1] -
Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035
Return to citation in text: [1] -
Adinolfi, M.; Barone, G.; Iadonisi, A.; Mangoni, L. Tetrahedron Lett. 1998, 39, 2021–2024. doi:10.1016/S0040-4039(98)00161-0
Return to citation in text: [1]
| 39. | Tadano, K.; Kanazawa, S.; Takao, K.; Ogawa, S. Tetrahedron 1992, 48, 4283–4300. doi:10.1016/S0040-4020(01)80440-3 |
| 40. | Seebach, D.; Goliński, J. Helv. Chim. Acta 1981, 64, 1413–1423. doi:10.1002/hlca.19810640518 |
| 41. | Yamamoto, Y.; Yatagai, H.; Naruta, Y.; Maruyama, K. J. Am. Chem. Soc. 1980, 102, 7107–7109. doi:10.1021/ja00543a040 |
| 1. | Kılbaş, B.; Balci, M. Tetrahedron 2011, 67, 2355–2389. doi:10.1016/j.tet.2011.01.012 |
| 2. | Duchek, J.; Adams, D. R.; Hudlicky, T. Chem. Rev. 2011, 111, 4223–4258. doi:10.1021/cr1004138 |
| 14. | Kwon, Y.-U.; Lee, C.; Chung, S.-K. J. Org. Chem. 2002, 67, 3327–3338. doi:10.1021/jo016237a |
| 15. | Podeschwa, M.; Plettenburg, O.; vom Brocke, J.; Block, O.; Adelt, S.; Altenbach, H.-J. Eur. J. Org. Chem. 2003, 1958–1972. doi:10.1002/ejoc.200200572 |
| 32. | Kiely, D. E.; Sherman, R. S. J. Am. Chem. Soc. 1975, 97, 6810–6814. doi:10.1021/ja00856a035 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 8. | Tagliaferri, F.; Wang, S.-N.; Berlin, W. K.; Outten, R. A.; Shen, T. Y. Tetrahedron Lett. 1990, 31, 1105–1108. doi:10.1016/S0040-4039(00)88737-7 |
| 9. | Guidot, J. P.; Le Gall, T. Tetrahedron Lett. 1993, 34, 4647–4650. doi:10.1016/S0040-4039(00)60646-9 |
| 10. | Gigg, J.; Gigg, R. Carbohydr. Res. 1997, 299, 77–83. doi:10.1016/S0008-6215(96)00330-8 |
| 11. | Riley, A. M.; Jenkins, D. J.; Potter, B. V. L. Carbohydr. Res. 1998, 314, 277–281. doi:10.1016/S0008-6215(98)00317-6 |
| 12. | Husson, C.; Odier, L.; Vottéro, P. J. A. Carbohydr. Res. 1998, 307, 163–165. doi:10.1016/S0008-6215(98)00003-2 |
| 13. | Gurale, B. P.; Sardessai, R. S.; Shashidhar, M. S. Carbohydr. Res. 2014, 399, 8–14. doi:10.1016/j.carres.2014.08.010 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
| 35. | Catelani, G.; D'Andrea, F.; Griselli, A.; Guazzelli, L.; Legnani, L.; Toma, L. Tetrahedron Lett. 2008, 49, 4534–4536. doi:10.1016/j.tetlet.2008.05.040 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 6. | Chung, S.-K.; Shin, B.-G.; Chang, Y.-T.; Suh, B.-C.; Kim, K.-T. Bioorg. Med. Chem. Lett. 1998, 8, 659–662. doi:10.1016/S0960-894X(98)00081-X |
| 7. | Hank, T.; Stricker, R.; Krishna, U. M.; Falck, J. R.; Chang, Y. T.; Chung, S. K.; Reiser, G. Eur. J. Biochem. 1999, 261, 577–584. doi:10.1046/j.1432-1327.1999.00326.x |
| 31. | Kiely, D. E.; Fletcher, H. G., Jr. J. Org. Chem. 1969, 34, 1386–1390. doi:10.1021/jo01257a042 |
| 32. | Kiely, D. E.; Sherman, R. S. J. Am. Chem. Soc. 1975, 97, 6810–6814. doi:10.1021/ja00856a035 |
| 49. | Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035 |
| 3. | Irvine, R. F.; Schell, M. J. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. doi:10.1038/35073015 |
| 4. | Chi, T. H.; Crabtree, G. R. Science 2000, 287, 1937–1939. doi:10.1126/science.287.5460.1937 |
| 5. | Odom, A. R.; Stahlberg, A.; Wente, S. R.; York, J. D. Science 2000, 287, 2026–2029. doi:10.1126/science.287.5460.2026 |
| 31. | Kiely, D. E.; Fletcher, H. G., Jr. J. Org. Chem. 1969, 34, 1386–1390. doi:10.1021/jo01257a042 |
| 45. | Jagdhane, R. C.; Patil, M. T.; Krishnaswamy, S.; Shashidhar, M. S. Tetrahedron 2013, 69, 5144–5151. doi:10.1016/j.tet.2013.04.081 |
| 23. | Takahashi, H.; Kittaka, H.; Ikegami, S. Tetrahedron Lett. 1998, 39, 9703–9706. doi:10.1016/S0040-4039(98)02231-X |
| 24. | Takahashi, H.; Kittaka, H.; Ikegami, S. J. Org. Chem. 2001, 66, 2705–2716. doi:10.1021/jo001575h |
| 27. | Sollogoub, M.; Mallet, J.-M.; Sinaÿ, P. Tetrahedron Lett. 1998, 39, 3471–3472. doi:10.1016/S0040-4039(98)00529-2 |
| 45. | Jagdhane, R. C.; Patil, M. T.; Krishnaswamy, S.; Shashidhar, M. S. Tetrahedron 2013, 69, 5144–5151. doi:10.1016/j.tet.2013.04.081 |
| 46. | Jagdhane, R. C.; Shashidhar, M. S. Eur. J. Org. Chem. 2010, 2945–2953. doi:10.1002/ejoc.201000009 |
| 47. | Sureshan, K. M.; Shashidhar, M. S.; Praveen, T.; Das, T. Chem. Rev. 2003, 103, 4477–4504. doi:10.1021/cr0200724 |
| 22. | Ferrier, R. J.; Middleton, S. Chem. Rev. 1993, 93, 2779–2831. doi:10.1021/cr00024a008 |
| 28. | Chiara, J. L.; Martin-Lomas, M. Tetrahedron Lett. 1994, 35, 2969–2972. doi:10.1016/S0040-4039(00)76674-3 |
| 29. | Guidot, J. P.; Le Gall, T.; Mioskowski, C. Tetrahedron Lett. 1994, 35, 6671–6672. doi:10.1016/S0040-4039(00)73464-2 |
| 30. | Carpintero, M.; Fernández-Mayoralas, A.; Jaramillo, C. J. Org. Chem. 1997, 62, 1916–1917. doi:10.1021/jo9623898 |
| 45. | Jagdhane, R. C.; Patil, M. T.; Krishnaswamy, S.; Shashidhar, M. S. Tetrahedron 2013, 69, 5144–5151. doi:10.1016/j.tet.2013.04.081 |
| 48. | Collet, C.; Chrétien, F.; Chapleur, Y.; Lamandé-Langle, S. Beilstein J. Org. Chem. 2016, 12, 353–361. doi:10.3762/bjoc.12.39 |
| 18. | Mandel, M.; Hudlicky, T. J. Chem. Soc., Perkin Trans. 1 1993, 741–743. doi:10.1039/P19930000741 |
| 19. | Hudlicky, T.; Mandel, M.; Rouden, J.; Lee, R. S.; Bachmann, B.; Dudding, T.; Yost, K. J.; Merola, J. S. J. Chem. Soc., Perkin Trans. 1 1994, 1553–1567. doi:10.1039/P19940001553 |
| 20. | Desjardins, M.; Brammer, L. E., Jr.; Hudlicky, T. Carbohydr. Res. 1997, 304, 39–42. doi:10.1016/S0008-6215(97)00186-9 |
| 21. | Brammer, L. E., Jr.; Hudlicky, T. Tetrahedron: Asymmetry 1998, 9, 2011–2014. doi:10.1016/S0957-4166(98)00182-7 |
| 42. | Murata, S.; Suzuki, M.; Noyori, R. J. Am. Chem. Soc. 1980, 102, 3248–3249. doi:10.1021/ja00529a062 |
| 16. | Carless, H. A. J.; Busia, K. Tetrahedron Lett. 1990, 31, 1617–1620. doi:10.1016/0040-4039(90)80032-H |
| 17. | Motherwell, W. B.; Williams, A. S. Angew. Chem., Int. Ed. 1995, 34, 2031–2033. doi:10.1002/anie.199520311 |
| 25. | Das, S. K.; Mallet, J.-M.; Sinaÿ, P. Angew. Chem., Int. Ed. Engl. 1997, 36, 493–496. doi:10.1002/anie.199704931 |
| 26. | Jia, C.; Pearce, A. J.; Blériot, Y.; Zhang, Y.; Zhang, L.-H.; Sollogoub, M.; Sinaÿ, P. Tetrahedron: Asymmetry 2004, 15, 699–703. doi:10.1016/j.tetasy.2003.11.029 |
| 43. | Jensen, H. H.; Lyngbye, L.; Jensen, A.; Bols, M. Chem. – Eur. J. 2002, 8, 1218–1226. doi:10.1002/1521-3765(20020301)8:5<1218::AID-CHEM1218>3.0.CO;2-X |
| 44. | Jensen, H. H.; Bols, M. Acc. Chem. Res. 2006, 39, 259–265. doi:10.1021/ar050189p |
| 36. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V.; Vittorino, E. Carbohydr. Res. 2003, 338, 2349–2358. doi:10.1016/j.carres.2003.08.001 |
| 37. | Guazzelli, L.; Catelani, G.; D'Andrea, F. Carbohydr. Res. 2010, 345, 369–376. doi:10.1016/j.carres.2009.11.027 |
| 38. | Catelani, G.; D'Andrea, F.; Guazzelli, L.; Pistarà, V. Carbohydr. Res. 2009, 344, 717–724. doi:10.1016/j.carres.2009.01.014 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
| 46. | Jagdhane, R. C.; Shashidhar, M. S. Eur. J. Org. Chem. 2010, 2945–2953. doi:10.1002/ejoc.201000009 |
| 47. | Sureshan, K. M.; Shashidhar, M. S.; Praveen, T.; Das, T. Chem. Rev. 2003, 103, 4477–4504. doi:10.1021/cr0200724 |
| 38. | Catelani, G.; D'Andrea, F.; Guazzelli, L.; Pistarà, V. Carbohydr. Res. 2009, 344, 717–724. doi:10.1016/j.carres.2009.01.014 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
| 35. | Catelani, G.; D'Andrea, F.; Griselli, A.; Guazzelli, L.; Legnani, L.; Toma, L. Tetrahedron Lett. 2008, 49, 4534–4536. doi:10.1016/j.tetlet.2008.05.040 |
| 35. | Catelani, G.; D'Andrea, F.; Griselli, A.; Guazzelli, L.; Legnani, L.; Toma, L. Tetrahedron Lett. 2008, 49, 4534–4536. doi:10.1016/j.tetlet.2008.05.040 |
| 35. | Catelani, G.; D'Andrea, F.; Griselli, A.; Guazzelli, L.; Legnani, L.; Toma, L. Tetrahedron Lett. 2008, 49, 4534–4536. doi:10.1016/j.tetlet.2008.05.040 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
| 33. | Pistarà, V.; Barili, P. L.; Catelani, G.; Corsaro, A.; D'Andrea, F.; Fisichella, S. Tetrahedron Lett. 2000, 41, 3253–3256. doi:10.1016/S0040-4039(00)00360-9 |
| 37. | Guazzelli, L.; Catelani, G.; D'Andrea, F. Carbohydr. Res. 2010, 345, 369–376. doi:10.1016/j.carres.2009.11.027 |
| 50. | Adinolfi, M.; Barone, G.; Iadonisi, A.; Mangoni, L. Tetrahedron Lett. 1998, 39, 2021–2024. doi:10.1016/S0040-4039(98)00161-0 |
| 34. | Catelani, G.; Corsaro, A.; D'Andrea, F.; Mariani, M.; Pistarà, V. Bioorg. Med. Chem. Lett. 2002, 12, 3313–3315. doi:10.1016/S0960-894X(02)00692-3 |
© 2016 D’Andrea et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)