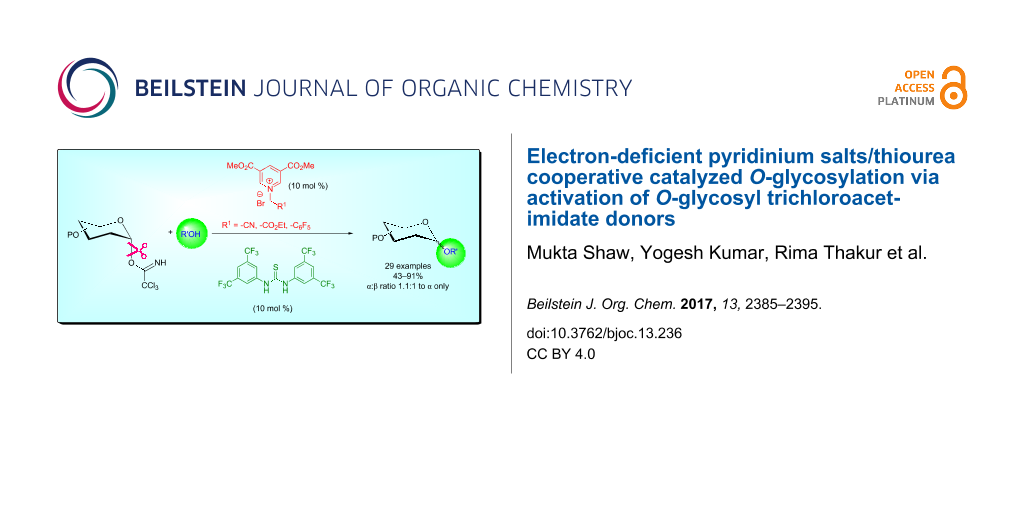
Abstract
The glycosylation of O-glycosyl trichloroacetimidate donors using a synergistic catalytic system of electron-deficient pyridinium salts/aryl thiourea derivatives at room temperature is demonstrated. The acidity of the adduct formed by the 1,2-addition of alcohol to the electron-deficient pyridinium salt is increased in the presence of an aryl thiourea derivative as an hydrogen-bonding cocatalyst. This transformation occurs under mild reaction conditions with a wide range of O-glycosyl trichloroacetimidate donors and glycosyl acceptors to afford the corresponding O-glycosides in moderate to good yields with predictable selectivity. In addition, the optimized method is also utilized for the regioselective O-glycosylation by using a partially protected acceptor.

Graphical Abstract
Introduction
The glycosidic linkage is the principal bond present in a crucial class of biomolecules such as oligosaccharides and glycoconjugates, where one sugar unit is linked with another sugar unit or any other molecules (aglycons) [1-4]. Owing to their high importance, several efficient protocols have been developed for the stereoselective glycosylation in the past few decades [5-10]. However, the synthesis of fundamental glycosidic bonds with high efficiency and selectivity yet remains one of the major challenges for organic chemists, in particular, carbohydrate chemists.
Nature extensively employs small organic molecules as catalysts for the acceleration of many important biochemical reactions, such as glycosyltransferase reactions, hydrolysis of strong amide bonds and others [11-13]. Taking inspiration from nature, in the last few decades chemists around the world have utilized organic molecules to accelerate many imperative organic transformations [14-17]. One of the major applications of organocatalysis lies in the field of enantioselective synthesis, where organocatalysts are considered as fundamental tools in the catalysis toolbox [18-22]. Moreover, the reactivity and selectivity of organocatalysts can be further amplified in the presence of other cocatalysts known as “cooperative catalysis” [23]. In particular, cooperativity between Brønsted acids and hydrogen-bonding cocatalysts such as thiourea derivatives has attracted much interest [24-29]. Despite the broad application of cooperative catalysis, it is still uncommonly employed in the area of carbohydrate chemistry, especially for glycosylation reactions, due to the prerequisite of having both catalysts being compatible under the reaction conditions. The Schmidt group has successfully applied the synergistic catalysts (thiourea derivatives with phosphorus acids) for stereoselective O-glycoside bond formation [30]. Similarly, Galan et al. reported a method for the preparation of 2-deoxyglycosides from glycals under the influence of cooperative catalysis (chiral phosphoric acids/thiourea derivatives) [31]. Encouraged by these reports and our own research interest in developing stereoselective glycosylation methods, we decided to focus our attention on the synthesis of glycosides via cooperative catalysis. A highly reactive glycosyl donor for instance, O-glycosyl trichloroacetimidate, generally requires a pKa value less than 5 for activation at room temperature [32-36]. It is known from the literature that pyridinium salts exhibit pKa values of about 5.2 [37]. Of late, Berkessel et al. disclosed an elegant method, where different electron deficient pyridinium salts (expected pKa values less than 5) were used as a catalyst for the activation of glycals to provide stereoselective 2-deoxyglycosides with high yields [38]. Based on this fact, we anticipated that for electron-deficient pyridinium salts the pKa value would be further diminished in the presence of hydrogen-bonding cocatalysts such as thiourea derivatives. The presence of Schreiner′s thiourea in the reaction medium enhances the acidity of the ammonium salt due to doubling their dual hydrogen bonding ability with the carboxylate and the alkoxy group of the ammonium salt. A thiourea derivative also enhances the nucleophilicity of the glycosyl acceptor by imparting a partial negative charge on it. Hence, the application of the synergistic catalyst system consisting of electron-deficient pyridinium salts/thiourea derivatives for glycosidic bond formation will be an exciting addition to the literature (Scheme 1).
![[1860-5397-13-236-i1]](/bjoc/content/inline/1860-5397-13-236-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanistic hypothesis for work.
Scheme 1: Mechanistic hypothesis for work.
Results and Discussion
To check our hypothesis, a series of 1H NMR spectroscopic studies were conducted by selecting commonly used O-glucopyranosyl trichloroacetimidate 1α [39-41] as glycosyl donor and 3,5-di(methoxycarbonyl)-N-(cyanomethyl)pyridinium bromide (3a) as a catalyst. For example, when glycosyl donor 1α was treated with catalyst 3a (10 mol %) at room temperature for 4 h (Table 1, entry 1) in CD2Cl2 solvent, it was observed that there is neither any interaction of 1α with catalyst 3a nor decomposition of 1α (Figure 1b) as the peak position of 1α remained unchanged. The salt remains insoluble in CD2Cl2 and hence did not show any peak in the 1H NMR spectra. However, when the mixture of electron-deficient pyridinium salt 3a and glycosyl acceptor 2a (1:1) was dissolved in CD2Cl2 and investigated by 1H NMR, an upfield shift of the -CH2- peak of 3a from δ 6.17 to δ 4.03 (Figure 2c) was observed. This result clearly supports the possible formation of 1,2-adduct X, on the reaction of 3a with 2a which is eventually responsible for the loss of aromaticity of the pyridinium ring.
![[1860-5397-13-236-1]](/bjoc/content/figures/1860-5397-13-236-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR (a) glycosyl donor 1α and (b) a mixture of 1α and 10 mol % 3a in CD2Cl2 at room temperature.
Figure 1: 1H NMR (a) glycosyl donor 1α and (b) a mixture of 1α and 10 mol % 3a in CD2Cl2 at room temperature.
![[1860-5397-13-236-2]](/bjoc/content/figures/1860-5397-13-236-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR (a) glycosyl acceptor 2a, (b) pyridinium salt 3a (in DMSO-d6) and (c) a mixture of 2a and 3a in 1:1 ratio in CD2Cl2 at room temperature.
Figure 2: 1H NMR (a) glycosyl acceptor 2a, (b) pyridinium salt 3a (in DMSO-d6) and (c) a mixture of 2a and 3a...
Based on the outcomes of 1H NMR spectroscopic studies, we started optimizing the reaction conditions. Upon treatment of glycosyl donor 1α and glycosyl acceptor 2a in 1:1.1 molar ratio with 10 mol % of 3a in dry DCM at room temperature, the desired O-glycoside 5a was isolated in 56% yield and with poor selectivity (Table 1, entry 2). The use of 25 mol % of 3a was required to drive the reaction to completion with 86% yield (Table 1, entry 3). This result, as envisaged, was indeed interesting and encouraging, which clearly indicates the ability of the electron-deficient pyridinium salt to activate the trichloroacetimidate donor. However, it took longer reaction time and required higher catalyst loading (up to 25 mol %). This outcome can be attributed to lower acidity of the ammonium salt formed by 1,2-addition of the acceptor to the pyridinium salt. The conjugate base formed after the release of a proton from the ammonium salt may be quite stable to impart a negative charge to the acceptor oxygen.
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-236-i3.svg?max-width=637&scale=1.0)
|
|||||
| entry | catalyst | cocatalyst 4 | solvent | reaction time | yieldb (α/β ratioc) |
|---|---|---|---|---|---|
| 1d | 3a | – | DCM | 4 h | n.r., n.d.e |
| 2 | 3a | – | DCM | 24 h | 56% (1:1)f |
| 3g | 3a | – | DCM | 4 h | 86% (1.1:1) |
| 4 | 3a | + | DCM | 2 h | 90% (2.2:1) |
| 5 | 3b | + | DCM | 6 h | 72% (2:1) |
| 6 | 3c | + | DCM | 8 h | 64% (1.7:1) |
| 7h | 3a | + | DCM | 5 h | 86% (2.1:1) |
| 8 | 3a | + | ACN | 4 h | 56 % (2.1:1) |
| 9 | 3a | + | THF | 7 h | 37% (1:1) |
| 10 | 3a | + | toluene | 24 h | trace |
| 11 | 3a | + | DCE | 3 h | 80% (1.4:1) |
| 12i | 3a | + | DCM | 5 h | 82%(2:1) |
| 13 | HBr | – | DCM | 8 h | tracej |
| 14k | 3a | – | DCM | 4 h | n.r. |
aReaction conditions: 1α (0.15 mmol), 2a (0.165 mmol), 3a–c (10 mol %), 4 (10 mol %), solvent (3 mL), at room temperature under nitrogen atmosphere. bYield of isolated product. cAnomeric ratios were determined by 1H NMR spectroscopy. d1α was stirred with 10 mol % 3a for 4 h at room temperature. en.r. – no reaction, n.d. – no decomposition. fReaction was not completed. g25 mol % of 3a was used. hPerformed at 0 °C. iInverse addition condition. jA trace amount of glucosyl bromide was also formed. k25 mol % of 2,4,6-trimethylpyridine was added.
The acidity of the ammonium salt may be enhanced by the introduction of a cocatalyst such as an aryl thiourea derivative, which has the ability to form a dual hydrogen bond with the carboxylate and the alkoxy group of the ammonium salt [42-44]. To ensure our postulation, a 1H NMR spectroscopic study was carried out with a mixture of glycosyl acceptor 2a, pyridinium salt 3a (10 mol %) and aryl thiourea 4 (10 mol %) in CD2Cl2 at room temperature (Figure 3). The -Me peak of -CO2Me of the ammonium salt shifted from δ 4.09 to δ 3.97, which indirectly confirms the presence of hydrogen bonding between -NH of the thiourea and the carbonyl carbon of -CO2Me of 3a (Figure 3d). Also, the magnification of the nucleophilicity of the glycosyl acceptor by the thiourea derivative results in shifting of the -OH peak from δ 2.70 to δ 3.62 (Figure 3d).
![[1860-5397-13-236-3]](/bjoc/content/figures/1860-5397-13-236-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR (a) glycosyl acceptor 2a, (b) pyridinium salt 3a (in DMSO-d6), (c) aryl thiourea and (d) a mixture of 2a, 3a (10 mol %) and 4 (10 mol %) in CD2Cl2 at room temperature.
Figure 3: 1H NMR (a) glycosyl acceptor 2a, (b) pyridinium salt 3a (in DMSO-d6), (c) aryl thiourea and (d) a m...
Once this understanding was gained from 1H NMR studies, aryl thiourea cocatalyst 4 (10 mol %) was added with 10 mol % of 3a to the reaction mixture of 1α and 2a and pleasingly the reaction was completed within a short span of time, glycoside 5a was obtained with improved yield (90%) and selectivity (α:β 2.2:1 ratio, Table 1, entry 4). Furthermore, several other catalysts were also screened for glycosylation, such as 3,5-di(methoxycarbonyl)-N-[(ethoxycarbonyl)methyl]pyridinium bromide (3b, Table 1, entry 5), 3,5-di(methoxycarbonyl)-N-[(pentafluorophenyl)methyl]pyridinium bromide (3c, Table 1, entry 6). However, the results were not up to our expectation and the desired glycoside was obtained in low yield. Once, we fixed the catalyst for glycosylation, further parameters were also optimized. Lowering the reaction temperature (room temperature to 0 °C), the product formation rate slowed down and the selectivity remains unchanged (Table 1, entry 7). Similarly, changing the solvent system to acetonitrile, tetrahydrofuran, toluene, and dichloroethane had an adverse effect on the reaction rate, yield and selectivity (Table 1, entries 8–11). Performing the reaction under inverse addition conditions had no impact on the selectivity of glycoside formation (Table 1, entry 12) [39,40]. Aware of the fact that HBr, which is eventually generated in situ during the course of reaction from ammonium salt X, might be the potential catalyst for the activation of the glycosyl trichloroacetimidate donor, we conducted an additional experiment. Glycosyl donor 1α was treated with 25 mol % of HBr instead of pyridinium salt 3a (Table 1, entry 13) where only a trace of glycoside 5a and some glycosyl bromide were formed along with the hydrolyzed product. Hence, it could be concluded that HBr is not the real catalyst in this cooperative catalysis. Further, the addition of acid scavenger such as 2,4,6-trimethylpyridine inhibits the formation of the glycoside, which indirectly supports that the pyridinium salt is a decisive catalyst for the activation of the glycosyl trichloroacetimidate donor (Table 1, entry 14). Therefore, the optimal reaction conditions for O-glycosylation are the following: the use of 3,5-di(methoxycarbonyl)-N-(cyanomethyl)pyridinium bromide (3a) as catalyst (10 mol %), thiourea derivative 4 as cocatalyst (10 mol %) and dichloromethane as solvent at room temperature (Table 1, entry 4).
With the optimized conditions in hand, our emphasis was then focused on the exploration of the synergistic catalytic system on glycosylation of 1α with several acceptors (Table 2). In all cases, reactions proceeded smoothly within 2–6 h and in good yields with moderate to good selectivity, as determined by the 1H and 13C NMR spectra. Glycosylation with less sterically hindered primary alcohols, e.g., allyl alcohol (2b), benzyl alcohol (2c), 4-methoxy benzyl alcohol (2d), and secondary alcohols, e.g., cyclohexanol (2e), cyclopentanol (2f) produced their corresponding glucosides 5b–f in 78–88% yields and with moderate α-selectivity (Table 2, entries 1–5). It is important to note that, when a halogenated primary alcohol such as 2-bromoethanol (2g) was treated with glycosyl donor 1α, it gave exclusively β-glycoside 5g in 72% yield (Table 2, entry 6). However, 3-chloropropanol (2h) as acceptor procured glucoside 5h in 70% yield with marginal selectivity (Table 2, entry 7). Similarly, the reaction with the bulky primary alcohol, 1-adamantanemethanol (2i) produced the desired glucoside 5i in 75% yield and moderate selectivity (Table 2, entry 8). Further, reactions with hindered acceptors, for instance, 1-adamantanol (2j), (+)-menthol (2k), (–)-menthol (2l), cholesterol (2m) required longer reaction times with high catalyst loading and produced the desired glucosides 5j–m, respectively, in moderate to good yields (43–67%) and with good α-selectivity (Table 2, entries 9–12). As expected, on treatment with a sugar-based acceptor such as 1,2:3,4-di-O-isopropylidene-D-galactose (2n), the corresponding glycoside 5n was produced in 72% yield with moderate selectivity (Table 2, entry 13) and the acid sensitive group survived well [34].
Table 2: Acceptor scope in glycosylation reaction with donor 1αa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-236-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry | ROH | product | time (h) | yieldb | α/β ratioc |
|---|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-236-i5.svg?max-width=637&scale=1.0)
2b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-236-i6.svg?max-width=637&scale=1.0)
5b |
2 | 88% | 1.2:1 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-236-i7.svg?max-width=637&scale=1.0)
2c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-236-i8.svg?max-width=637&scale=1.0)
5c |
2 | 88% | 2:1 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-236-i9.svg?max-width=637&scale=1.0)
2d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-236-i10.svg?max-width=637&scale=1.0)
5d |
2 | 82% | 3.3:1 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-236-i11.svg?max-width=637&scale=1.0)
2e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-236-i12.svg?max-width=637&scale=1.0)
5e |
2 | 78% | 2.1:1 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-236-i13.svg?max-width=637&scale=1.0)
2f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-236-i14.svg?max-width=637&scale=1.0)
5f |
2 | 85% | 2.5:1 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-236-i15.svg?max-width=637&scale=1.0)
2g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-236-i16.svg?max-width=637&scale=1.0)
5g |
2 | 72% | β only |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-236-i17.svg?max-width=637&scale=1.0)
2h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-236-i18.svg?max-width=637&scale=1.0)
5h |
2 | 70% | 1.1:1 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-236-i19.svg?max-width=637&scale=1.0)
2i |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-236-i20.svg?max-width=637&scale=1.0)
5i |
6 | 75% | 3.3:1 |
| 9d |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-236-i21.svg?max-width=637&scale=1.0)
2j |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-236-i22.svg?max-width=637&scale=1.0)
5j |
5 | 43% | 2:1 |
| 10d |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-236-i23.svg?max-width=637&scale=1.0)
2k |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-236-i24.svg?max-width=637&scale=1.0)
5k |
4 | 61% | α only |
| 11d |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-236-i25.svg?max-width=637&scale=1.0)
2l |
![[Graphic 24]](/bjoc/content/inline/1860-5397-13-236-i26.svg?max-width=637&scale=1.0)
5l |
4 | 67% | 5:1 |
| 12d |
![[Graphic 25]](/bjoc/content/inline/1860-5397-13-236-i27.svg?max-width=637&scale=1.0)
2m |
![[Graphic 26]](/bjoc/content/inline/1860-5397-13-236-i28.svg?max-width=637&scale=1.0)
5m |
6 | 60% | 10.1:1 |
| 13 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-13-236-i29.svg?max-width=637&scale=1.0)
2n |
![[Graphic 28]](/bjoc/content/inline/1860-5397-13-236-i30.svg?max-width=637&scale=1.0)
5n |
3 | 72% | 1:1.3 |
aReaction conditions: 1α (0.15 mmol), 2a–n (0.165 mmol), 3a (10 mol %), 4 (10 mol %), solvent (3 mL), at room temperature under nitrogen atmosphere. bYield of isolated product. cAnomeric ratios were determined by 1H NMR spectroscopy. d20 mol % of 3a and 20 mol % of 4 was used.
To further demonstrate the efficacy of this method other important glycosides were synthesized with different donors as tabulated in Table 3. Glycosylation of D-galactopyranosyl trichloroacetimidate 6α with a variety of glycosyl acceptors, e.g., 2c, 2g, 2h, 2i and 2n under the optimized reaction conditions gave their corresponding galactosides 9–13, respectively, with moderate selectivity (Table 3, entry 1–5) [40,41]. Similarly, the reaction of D-mannopyranosyl trichloroacetimidate 7α with glycosyl acceptors 2c, 2g, 2h, and 2n produces their corresponding mannosides 14–18 (61–78% yields) with moderate selectivity (Table 3, entries 6–10) [40,41]. Gratifyingly, the highest stereoselective outcome was observed when 4,6-O-benzylidine-2,3-di-O-benzyl-α-D-glucopyranosyl trichloroacetimidate 8α was used as a glycosyl donor. For example, when 8α was treated with different donors such as 2i, 2m, 2n under the optimized conditions, glucosides 19–21 were procured in good yields with excellent α-selectivity (Table 3, entry 11–13) [45].
Table 3: Glycosylation of donors 6α–8α with variety of acceptorsa.
![[Graphic 29]](/bjoc/content/inline/1860-5397-13-236-i31.svg?max-width=637&scale=1.0)
|
||||||
| entry | donor | acceptor | product | time (h) | yieldb | α/β ratioc |
|---|---|---|---|---|---|---|
| 1 | 6α | 2c | 9 | 2 | 91% | 1.7:1 |
| 2 | 2g | 10 | 2 | 74% | 2.1:1 | |
| 3 | 2h | 11 | 2 | 81% | 1:4.6 | |
| 4 | 2i | 12 | 6 | 73% | 2.6:1 | |
| 5 | 2n | 13 | 3 | 62% | 1.4:1 | |
| 6 | 7α | 2c | 14 | 2 | 74% | 1.2:1 |
| 7 | 2g | 15 | 2 | 78% | 3:1 | |
| 8 | 2h | 16 | 2 | 77% | 1.9:1 | |
| 9d | 2m | 17 | 7 | 68% | 1:1.9 | |
| 10 | 2n | 18 | 3 | 61% | α only | |
| 11 | 8α | 2i | 19 | 6 | 61% | α only |
| 12d | 2m | 20 | 8 | 54% | α only | |
| 13 | 2n | 21 | 4 | 61% | α only | |
aReaction conditions: donor (0.15 mmol), acceptor (0.165 mmol), 3a (10 mol %), 4 (10 mol %), solvent (3 mL), at room temperature under nitrogen atmosphere. bYield of isolated product. cAnomeric ratios were determined by 1H NMR spectroscopy. d20 mol % of 3a and 20 mol% of 4 was used.
The regioselective glycosylation is an important aspect in carbohydrate chemistry. It is pleasing to note that on reaction with partially protected acceptor 22 with glycosyl donors 1α and 7α under the optimized conditions lead to the regioisomeric products 23 and 24 [46,47] in moderate yields with good selectivity (Scheme 2).
![[1860-5397-13-236-i2]](/bjoc/content/inline/1860-5397-13-236-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synergistic electron-deficient pyridinium salt/aryl thiourea-catalyzed regioselective O-glycosylation.
Scheme 2: Synergistic electron-deficient pyridinium salt/aryl thiourea-catalyzed regioselective O-glycosylati...
Plausible mechanism
To confirm the reaction pathway, few additional control experiments were carried out (for details see Supporting Information File 1). When the reaction was performed with 1.0 equiv of 3a and acceptor 2a under standard inverse procedure conditions, the desired O-glycoside 5a was procured in 36% yield and 56% of 1α was recovered through column chromatography. Therefore, we conclude that the reaction would have followed an intermolecular glycosylation reaction through an oxocarbenium ion. Combining all of these observations and results from earlier literature reports, a plausible reaction mechanism for the electron-deficient pyridinium salt/thiourea cocatalyzed glycosidation is outlined in Figure 4. It is presumed that at first electron-deficient pyridinium salt 3a undergoes 1,2-addition with the acceptor to produce ammonium salt X. The addition of the thiourea derivative as hydrogen-bonding cocatalyst could activate the glycosyl donor by increasing the acidity of ammonium salt X to form an oxocarbenium intermediate B. Further, the nucleophilic attack of the acceptor to the oxocarbenium ion B would produce the desired glycoside 5. Higher α-selectivity may be attributed to the anomeric effect.
![[1860-5397-13-236-4]](/bjoc/content/figures/1860-5397-13-236-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have disclosed an efficient and general protocol for the glycosylation of trichloroacetimidate glycosyl donors using the concept of cooperativity between an electron-deficient pyridinium salt and an aryl thiourea derivative. 1H NMR studies divulge that a 1,2-adduct formation between the electron-deficient pyridinium salt and the glycosyl acceptor plays a crucial role for the activation of the trichloroacetimidate donors. The presence of thiourea derivatives further enhances the reaction rate and selectivity due to its dual hydrogen bonding ability. The reaction proceeds smoothly at room temperature with good to excellent yields and α-selectivity and is applicable to a wide range of glycosyl donors as well as acceptors. The advantage of this methodology lies in the usage of an environmentally benign catalyst, mild reaction conditions and the regioselective formation of O-glycosides.
Supporting Information
| Supporting Information File 1: Experimental procedures and analytical data. | ||
| Format: PDF | Size: 9.1 MB | Download |
References
-
Peng, P.; Schmidt, R. R. Acc. Chem. Res. 2017, 50, 1171–1183. doi:10.1021/acs.accounts.6b00518
Return to citation in text: [1] -
Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036
Return to citation in text: [1] -
Varki, A. Glycobiology 1993, 3, 97–130. doi:10.1093/glycob/3.2.97
Return to citation in text: [1] -
Doores, K. J.; Gamblin, D. P.; Davis, B. G. Chem. – Eur. J. 2006, 12, 656–665. doi:10.1002/chem.200500557
Return to citation in text: [1] -
Park, Y.; Harper, K. C.; Kuhl, N.; Kwan, E. E.; Liu, R. Y.; Jacobsen, E. N. Science 2017, 355, 162–166. doi:10.1126/science.aal1875
Return to citation in text: [1] -
Seeberger, P. H. Acc. Chem. Res. 2015, 48, 1450–1463. doi:10.1021/ar5004362
Return to citation in text: [1] -
Nigudkar, S. S.; Demchenko, A. V. Chem. Sci. 2015, 6, 2687–2704. doi:10.1039/C5SC00280J
Return to citation in text: [1] -
Shaw, M.; Kumar, A.; Thakur, R. Trends Carbohydr. Res. 2017, 9, 1–28.
Return to citation in text: [1] -
Toshima, K.; Tatsuta, K. Chem. Rev. 1993, 93, 1503–1531. doi:10.1021/cr00020a006
Return to citation in text: [1] -
Davis, B. G. J. Chem. Soc., Perkin Trans. 1 2000, 2137–2160. doi:10.1039/A809774G
Return to citation in text: [1] -
Crout, D. H. G.; Vic, G. Curr. Opin. Chem. Biol. 1998, 2, 98–111. doi:10.1016/S1367-5931(98)80041-0
Return to citation in text: [1] -
Withers, S. G. Carbohydr. Polym. 2001, 44, 325–337. doi:10.1016/S0144-8617(00)00249-6
Return to citation in text: [1] -
Lopez, X.; Mujika, J. I.; Blackburn, G. M.; Karplus, M. J. Phys. Chem. A 2003, 107, 2304–2315. doi:10.1021/jp022014s
Return to citation in text: [1] -
MacMillan, D. W. C. Nature 2008, 455, 304–308. doi:10.1038/nature07367
Return to citation in text: [1] -
Gröger, H.; Wilken, J. Angew. Chem., Int. Ed. 2001, 40, 529–532. doi:10.1002/1521-3773(20010202)40:3<529::AID-ANIE529>3.0.CO;2-X
Return to citation in text: [1] -
Balmond, E. I.; Galan, M. C.; McGarrigle, E. M. Synlett 2013, 2335–2339. doi:10.1055/s-0033-1338970
Return to citation in text: [1] -
Vedachalam, S.; Tan, S. M.; Teo, H. P.; Cai, S.; Liu, X.-W. Org. Lett. 2012, 14, 174–177. doi:10.1021/ol202959y
Return to citation in text: [1] -
Berkessel, A.; Groeger, H. Asymmetric Organocatalysis: from Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH, 2005.
ISBN: 978-3-527-30517-9.
Return to citation in text: [1] -
Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Adv. Synth. Catal. 2015, 357, 253–281. doi:10.1002/adsc.201401003
Return to citation in text: [1] -
Tu, Y.; Wang, Z.-X.; Shi, Y. J. Am. Chem. Soc. 1996, 118, 9806–9807. doi:10.1021/ja962345g
Return to citation in text: [1] -
Sigman, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 4901–4902. doi:10.1021/ja980139y
Return to citation in text: [1] -
Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157–160. doi:10.1021/ol990623l
Return to citation in text: [1] -
Raup, D. E. A.; Cardinal-David, B.; Holte, D.; Scheidt, K. A. Nat. Chem. 2010, 2, 766–771. doi:10.1038/nchem.727
Return to citation in text: [1] -
Weil, T.; Kotke, M.; Kleiner, C. M.; Schreiner, P. R. Org. Lett. 2008, 10, 1513–1516. doi:10.1021/ol800149y
Return to citation in text: [1] -
Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. doi:10.1039/B801793J
Return to citation in text: [1] -
Zhang, Z.; Lippert, K. M.; Hausmann, H.; Kotke, M.; Schreiner, P. R. J. Org. Chem. 2011, 76, 9764–9776. doi:10.1021/jo201864e
Return to citation in text: [1] -
Klausen, R. S.; Jacobsen, E. N. Org. Lett. 2009, 11, 887–890. doi:10.1021/ol802887h
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Ooi, T. Science 2009, 326, 120–123. doi:10.1126/science.1176758
Return to citation in text: [1] -
Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Chem. Rev. 2016, 116, 4006–4123. doi:10.1021/acs.chemrev.5b00676
Return to citation in text: [1] -
Geng, Y.; Kumar, A.; Faidallah, H. M.; Albar, H. A.; Mhkalid, I. A.; Schmidt, R. R. Angew. Chem., Int. Ed. 2013, 52, 10089–10092. doi:10.1002/anie.201302158
Return to citation in text: [1] -
Palo-Nieto, C.; Sau, A.; Williams, R.; Galan, M. C. J. Org. Chem. 2017, 82, 407–414. doi:10.1021/acs.joc.6b02498
Return to citation in text: [1] -
Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1986, 25, 212–235. doi:10.1002/anie.198602121
Return to citation in text: [1] -
Kimura, T.; Eto, T.; Takahashi, D.; Toshima, K. Org. Lett. 2016, 18, 3190–3193. doi:10.1021/acs.orglett.6b01404
Return to citation in text: [1] -
Mensah, E. A.; Azzarelli, J. M.; Nguyen, H. M. J. Org. Chem. 2009, 74, 1650–1657. doi:10.1021/jo802468p
Return to citation in text: [1] [2] -
Roy, R.; Palanivel, A. K.; Mallick, A.; Vankar, Y. D. Eur. J. Org. Chem. 2015, 4000–4005. doi:10.1002/ejoc.201500137
Return to citation in text: [1] -
Kumar, A.; Kumar, V.; Dere, R. T.; Schmidt, R. R. Org. Lett. 2011, 13, 3612–3615. doi:10.1021/ol201231v
Return to citation in text: [1] -
Portenkirchner, E.; Enengl, C.; Enengl, S.; Hinterberger, G.; Schlager, S.; Apaydin, D.; Neugebauer, H.; Knör, G.; Sariciftci, N. S. ChemElectroChem 2014, 1, 1543–1548. doi:10.1002/celc.201402132
Return to citation in text: [1] -
Das, S.; Pekel, D.; Neudörfl, J.-M.; Berkessel, A. Angew. Chem., Int. Ed. 2015, 54, 12479–12483. doi:10.1002/anie.201503156
Return to citation in text: [1] -
Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311
Return to citation in text: [1] [2] -
Schmidt, R. R.; Kinzy, W. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21–123. doi:10.1016/S0065-2318(08)60150-X
Return to citation in text: [1] [2] [3] [4] -
Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353–3356. doi:10.1016/S0040-4039(00)92704-7
Return to citation in text: [1] [2] [3] -
Fan, E.; Van Arman, S. A.; Kincaid, S.; Hamilton, A. D. J. Am. Chem. Soc. 1993, 115, 369–370. doi:10.1021/ja00054a066
Return to citation in text: [1] -
Albert, J. S.; Hamilton, A. D. Tetrahedron Lett. 1993, 34, 7363–7366. doi:10.1016/S0040-4039(00)60126-0
Return to citation in text: [1] -
Raposo, C.; Crego, M.; Mussons, M. L.; Caballero, M. C.; Morán, J. R. Tetrahedron Lett. 1994, 35, 3409–3410. doi:10.1016/S0040-4039(00)76921-8
Return to citation in text: [1] -
Cancogni, D.; Lay, L. Synlett 2014, 25, 2873–2878. doi:10.1055/s-0034-1379471
Return to citation in text: [1] -
Michigami, K.; Terauchi, M.; Hayashi, M. Synthesis 2013, 1519–1523. doi:10.1055/s-0033-1338469
Return to citation in text: [1] -
Peng, P.; Schmidt, R. R. J. Am. Chem. Soc. 2015, 137, 12653–12659. doi:10.1021/jacs.5b07895
Return to citation in text: [1]
| 45. | Cancogni, D.; Lay, L. Synlett 2014, 25, 2873–2878. doi:10.1055/s-0034-1379471 |
| 40. | Schmidt, R. R.; Kinzy, W. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21–123. doi:10.1016/S0065-2318(08)60150-X |
| 41. | Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353–3356. doi:10.1016/S0040-4039(00)92704-7 |
| 40. | Schmidt, R. R.; Kinzy, W. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21–123. doi:10.1016/S0065-2318(08)60150-X |
| 41. | Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353–3356. doi:10.1016/S0040-4039(00)92704-7 |
| 1. | Peng, P.; Schmidt, R. R. Acc. Chem. Res. 2017, 50, 1171–1183. doi:10.1021/acs.accounts.6b00518 |
| 2. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036 |
| 3. | Varki, A. Glycobiology 1993, 3, 97–130. doi:10.1093/glycob/3.2.97 |
| 4. | Doores, K. J.; Gamblin, D. P.; Davis, B. G. Chem. – Eur. J. 2006, 12, 656–665. doi:10.1002/chem.200500557 |
| 18. |
Berkessel, A.; Groeger, H. Asymmetric Organocatalysis: from Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH, 2005.
ISBN: 978-3-527-30517-9. |
| 19. | Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Adv. Synth. Catal. 2015, 357, 253–281. doi:10.1002/adsc.201401003 |
| 20. | Tu, Y.; Wang, Z.-X.; Shi, Y. J. Am. Chem. Soc. 1996, 118, 9806–9807. doi:10.1021/ja962345g |
| 21. | Sigman, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 4901–4902. doi:10.1021/ja980139y |
| 22. | Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157–160. doi:10.1021/ol990623l |
| 39. | Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311 |
| 40. | Schmidt, R. R.; Kinzy, W. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21–123. doi:10.1016/S0065-2318(08)60150-X |
| 14. | MacMillan, D. W. C. Nature 2008, 455, 304–308. doi:10.1038/nature07367 |
| 15. | Gröger, H.; Wilken, J. Angew. Chem., Int. Ed. 2001, 40, 529–532. doi:10.1002/1521-3773(20010202)40:3<529::AID-ANIE529>3.0.CO;2-X |
| 16. | Balmond, E. I.; Galan, M. C.; McGarrigle, E. M. Synlett 2013, 2335–2339. doi:10.1055/s-0033-1338970 |
| 17. | Vedachalam, S.; Tan, S. M.; Teo, H. P.; Cai, S.; Liu, X.-W. Org. Lett. 2012, 14, 174–177. doi:10.1021/ol202959y |
| 34. | Mensah, E. A.; Azzarelli, J. M.; Nguyen, H. M. J. Org. Chem. 2009, 74, 1650–1657. doi:10.1021/jo802468p |
| 11. | Crout, D. H. G.; Vic, G. Curr. Opin. Chem. Biol. 1998, 2, 98–111. doi:10.1016/S1367-5931(98)80041-0 |
| 12. | Withers, S. G. Carbohydr. Polym. 2001, 44, 325–337. doi:10.1016/S0144-8617(00)00249-6 |
| 13. | Lopez, X.; Mujika, J. I.; Blackburn, G. M.; Karplus, M. J. Phys. Chem. A 2003, 107, 2304–2315. doi:10.1021/jp022014s |
| 39. | Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311 |
| 40. | Schmidt, R. R.; Kinzy, W. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21–123. doi:10.1016/S0065-2318(08)60150-X |
| 41. | Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353–3356. doi:10.1016/S0040-4039(00)92704-7 |
| 5. | Park, Y.; Harper, K. C.; Kuhl, N.; Kwan, E. E.; Liu, R. Y.; Jacobsen, E. N. Science 2017, 355, 162–166. doi:10.1126/science.aal1875 |
| 6. | Seeberger, P. H. Acc. Chem. Res. 2015, 48, 1450–1463. doi:10.1021/ar5004362 |
| 7. | Nigudkar, S. S.; Demchenko, A. V. Chem. Sci. 2015, 6, 2687–2704. doi:10.1039/C5SC00280J |
| 8. | Shaw, M.; Kumar, A.; Thakur, R. Trends Carbohydr. Res. 2017, 9, 1–28. |
| 9. | Toshima, K.; Tatsuta, K. Chem. Rev. 1993, 93, 1503–1531. doi:10.1021/cr00020a006 |
| 10. | Davis, B. G. J. Chem. Soc., Perkin Trans. 1 2000, 2137–2160. doi:10.1039/A809774G |
| 42. | Fan, E.; Van Arman, S. A.; Kincaid, S.; Hamilton, A. D. J. Am. Chem. Soc. 1993, 115, 369–370. doi:10.1021/ja00054a066 |
| 43. | Albert, J. S.; Hamilton, A. D. Tetrahedron Lett. 1993, 34, 7363–7366. doi:10.1016/S0040-4039(00)60126-0 |
| 44. | Raposo, C.; Crego, M.; Mussons, M. L.; Caballero, M. C.; Morán, J. R. Tetrahedron Lett. 1994, 35, 3409–3410. doi:10.1016/S0040-4039(00)76921-8 |
| 31. | Palo-Nieto, C.; Sau, A.; Williams, R.; Galan, M. C. J. Org. Chem. 2017, 82, 407–414. doi:10.1021/acs.joc.6b02498 |
| 37. | Portenkirchner, E.; Enengl, C.; Enengl, S.; Hinterberger, G.; Schlager, S.; Apaydin, D.; Neugebauer, H.; Knör, G.; Sariciftci, N. S. ChemElectroChem 2014, 1, 1543–1548. doi:10.1002/celc.201402132 |
| 30. | Geng, Y.; Kumar, A.; Faidallah, H. M.; Albar, H. A.; Mhkalid, I. A.; Schmidt, R. R. Angew. Chem., Int. Ed. 2013, 52, 10089–10092. doi:10.1002/anie.201302158 |
| 38. | Das, S.; Pekel, D.; Neudörfl, J.-M.; Berkessel, A. Angew. Chem., Int. Ed. 2015, 54, 12479–12483. doi:10.1002/anie.201503156 |
| 24. | Weil, T.; Kotke, M.; Kleiner, C. M.; Schreiner, P. R. Org. Lett. 2008, 10, 1513–1516. doi:10.1021/ol800149y |
| 25. | Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. doi:10.1039/B801793J |
| 26. | Zhang, Z.; Lippert, K. M.; Hausmann, H.; Kotke, M.; Schreiner, P. R. J. Org. Chem. 2011, 76, 9764–9776. doi:10.1021/jo201864e |
| 27. | Klausen, R. S.; Jacobsen, E. N. Org. Lett. 2009, 11, 887–890. doi:10.1021/ol802887h |
| 28. | Uraguchi, D.; Ueki, Y.; Ooi, T. Science 2009, 326, 120–123. doi:10.1126/science.1176758 |
| 29. | Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Chem. Rev. 2016, 116, 4006–4123. doi:10.1021/acs.chemrev.5b00676 |
| 46. | Michigami, K.; Terauchi, M.; Hayashi, M. Synthesis 2013, 1519–1523. doi:10.1055/s-0033-1338469 |
| 47. | Peng, P.; Schmidt, R. R. J. Am. Chem. Soc. 2015, 137, 12653–12659. doi:10.1021/jacs.5b07895 |
| 23. | Raup, D. E. A.; Cardinal-David, B.; Holte, D.; Scheidt, K. A. Nat. Chem. 2010, 2, 766–771. doi:10.1038/nchem.727 |
| 32. | Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1986, 25, 212–235. doi:10.1002/anie.198602121 |
| 33. | Kimura, T.; Eto, T.; Takahashi, D.; Toshima, K. Org. Lett. 2016, 18, 3190–3193. doi:10.1021/acs.orglett.6b01404 |
| 34. | Mensah, E. A.; Azzarelli, J. M.; Nguyen, H. M. J. Org. Chem. 2009, 74, 1650–1657. doi:10.1021/jo802468p |
| 35. | Roy, R.; Palanivel, A. K.; Mallick, A.; Vankar, Y. D. Eur. J. Org. Chem. 2015, 4000–4005. doi:10.1002/ejoc.201500137 |
| 36. | Kumar, A.; Kumar, V.; Dere, R. T.; Schmidt, R. R. Org. Lett. 2011, 13, 3612–3615. doi:10.1021/ol201231v |
© 2017 Shaw et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)