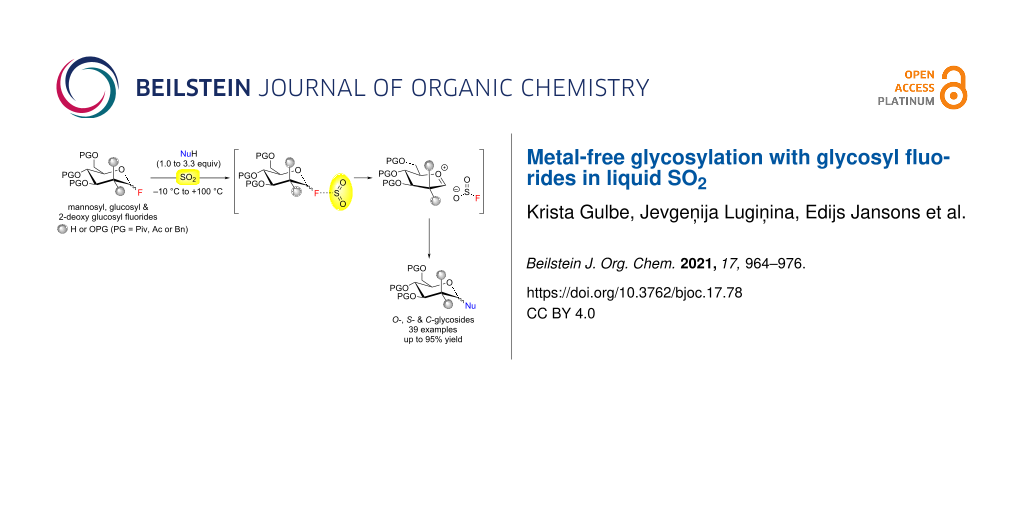
Abstract
Liquid SO2 is a polar solvent that dissolves both covalent and ionic compounds. Sulfur dioxide possesses also Lewis acid properties, including the ability to covalently bind Lewis basic fluoride ions in a relatively stable fluorosulfite anion (FSO2−). Herein we report the application of liquid SO2 as a promoting solvent for glycosylation with glycosyl fluorides without any external additive. By using various temperature regimes, the method is applied for both armed and disarmed glucose and mannose-derived glycosyl fluorides in moderate to excellent yields. A series of pivaloyl-protected O- and S-mannosides, as well as one example of a C-mannoside, are synthesized to demonstrate the scope of the glycosyl acceptors. The formation of the fluorosulfite species during the glycosylation with glycosyl fluorides in liquid SO2 is proved by 19F NMR spectroscopy. A sulfur dioxide-assisted glycosylation mechanism that proceeds via solvent separated ion pairs is proposed, whereas the observed α,β-selectivity is substrate-controlled and depends on the thermodynamic equilibrium.

Graphical Abstract
Introduction
The glycosylation reaction is still one of the most important and basic synthetic strategies in carbohydrate chemistry that provides access to the various types of glycoconjugates [1-4]. Due to the large diversity of glycosyl donors and acceptors there is no general glycosylation method developed so far. To ensure high yielding, as well as regio- and stereoselective glycosidic bond formation, a proper combination of glycosyl donor and acceptor, protecting and leaving groups, promoter, solvent and temperature has to be applied.
In 1981, Mukaiyama et al. introduced glycosyl fluorides [5] as a new class of glycosyl donors [6]. The C–F bond is one of the strongest single bonds in the realm of organic compounds with a bond dissociation energy (BDE) of 570 kJ/mol [7]. Thus, glycosyl fluorides possess a considerably higher thermal and chemical stability compared to the corresponding chlorides (BDE 432 kJ/mol) and bromides (BDE 366 kJ/mol). Due to the advantageous stability during purification, handling and storage, glycosyl fluorides have become widely used glycosyl donors in glycoconjugate synthesis [8,9]. Furthermore, varied reactivity between differentially protected glycosyl fluorides as well as between glycosyl fluorides and other glycosyl donors makes these substrates relevant for more effective glycosylation via orthogonal activation [10,11]. According to the hard–soft acid–base (HSAB) theory the fluoride leaving group is considered to be a hard Lewis base [12,13]. Consequently, a series of fluoride-activating systems containing hard Lewis acidic centers have been published following the first report [7,14-17]. Among these promoters Sn(II) species (SnCl2–AgX, X = ClO4 or B(C6F5)4) [6,18], group IVB metallocenes (Cp2MCl2–AgClO4, M = Zr, Hf, Ti) [19-21], BF3·OEt2 [22] and protic acids (TfOH, HClO4, HB(C6F5)4) [23] are the most frequently used. During the last decade, apart from reports on novel promoters (Hf(OTf)4 [24], InI3 [25], In(OTf)3 [26], B(C6F5)3 [27]) and coupling partners [28], great attention has been paid to a stereoselective glycosylation by sterically fixed glycosyl fluorides as glycosyl donors [29-31]. The enhanced stability of glycosyl fluorides has also allowed to develop a straightforward protecting-group-free strategy towards oligosaccharides and glycopeptides under basic aqueous conditions [32,33]. Nevertheless, most of the conventional conditions for glycosyl fluoride activation have considerable drawbacks in terms of atom efficiency and environmental impact. These methods generally require (1) stoichiometric amounts of promoters, often heavy metals; (2) multiple additives (co-promoter, molecular sieves, acid scavenger) to facilitate the reaction and/or suppress formation of side-products; (3) low temperatures; (4) complex experimental procedures. Additionally, the majority of the methods reported to date have been applied only for the synthesis of O- [4,34,35] and C-glycosides [36] and by employing more reactive armed [1] glycosyl fluorides.
In glycosylation reactions the solvent plays a critical role in terms of stabilizing the oxocarbenium ion intermediate and/or affecting the α,β-selectivity [1]. In 2017, Matheu et al. reported a ″green″ glycosylation procedure by employing supercritical CO2 (scCO2) as a weakly Lewis acidic reaction medium [37]. The method was successfully applied for the synthesis of O-glycosides from disarmed glycosyl chlorides and bromides in the absence of additional promoter. Herein we disclose a related concept by applying liquid SO2. In contrast to scCO2, liquid SO2 is a polar Lewis acidic solvent and due to its relatively high boiling point (bp −10 °C) and low vapor pressure (ca. 3 bar at 20 °C) it can be easily liquefied and handled in its liquid state [38]. Advantages of liquid SO2 over conventional solvents are: (1) it is aprotic solvent with Lewis acid properties; (2) it dissolves both covalent and ionic compounds [39]; (3) it has good price–quality ratio: ≤5 EUR/kg for the high-purity product (99.98%, H2O content ≤50 ppm); (4) it can be easily recycled by changing temperature and/or pressure regimes. The latter approach is used on industrial scale, where processes dealing with a recirculation of SO2 in a closed contour are well known. Since the first report by Walden at the beginning of the 20th century [40], a variety of Lewis acid-mediated chemical transformations [41-45], especially those with carbenium ion intermediates [46-56], have benefited from the use of liquid SO2 as the reaction medium. To the best of our knowledge, there has been only one example where liquid SO2 has been applied as the glycosylation medium to stabilize the oxocarbenium ion formed from glycosyl perchlorate that is generated in situ from glycosyl chloride and AgClO4 [56]. Apart from that, SO2 has considerable affinity to the Lewis basic halide ions [57-59]. Kuhn et al. [60] and later Eisfield and Regitz [61] have published ab initio studies on the stability of halosulfites HalSO2− (Hal = F, Cl, Br or I) that can be formed between halide ions and the SO2 molecule. They disclosed that the formation of fluorosulfite anion (FSO2−) has the highest energy gain and it appears to be stable even in highly polar solvents (ε ≤ 45), while all other halosulfites may dissociate. Thus, we proposed that a plausible formation of the fluorosulfite species and stabilization of the oxocarbenium ion intermediate could facilitate the glycosylation with glycosyl fluorides as glycosyl donors in liquid SO2 without the need of external promoter.
Results and Discussion
We started our study by short screening of the glycosylation conditions in liquid SO2 (Table 1). To avoid a potential cleavage of acid-labile protecting groups and to obtain an easily analyzable reaction mixture, pivaloyl-protected mannosyl fluoride α-1a as a relatively stable disarmed glycosyl donor was selected as a model substrate. Reactions were carried out in a pressure reactor equipped with a glass tube. By employing a slight excess of 2-phenylethanol (2a) as a glycosyl acceptor, the reaction temperature was optimized to 100 °C (Table 1, entry 2). At this temperature full conversion of mannosyl fluoride α-1a was achieved and the desired O-mannoside 3a was isolated in a high yield and α-selectivity. Hemiacetal α-4 was isolated as the only side-product formed via glycosyl donor hydrolysis with the water present in commercial SO2 [62]. To note, at lower temperatures (Table 1, entry 1) no reaction was observed and mannosyl fluoride α-1a was fully recovered. Recently, Pedersen et al. have studied the vessel effect on the C–F bond activation of glucosyl fluorides [63]. They have proposed an autocatalytic glycosylation by SiF4 generated in situ form initially released HF that reacts with silicates of the glassware surface. To clarify the role of a glass vessel in our case, several experiments were carried out in a pressure reactor equipped with a PTFE tube (Table 1, entries 3–5). Under previously optimized reaction conditions (100 °C, Table 1, entry 2), mannoside 3a was isolated in only 8% yield (Table 1, entry 3). The yield was increased to 23% when acceptor 2a was added in an excess (3.0 equiv, Table 1, entry 4). Finally, full conversion of fluoride α-1a and sufficient yield of desired product 3a were reached with 3.0 equiv of nucleophile at 150 °C (Table 1, entry 5). Thus, in contrast to the previous report [63], in our case the reaction was not fully stopped by changing the reaction vessel from glass to PTFE tube. At this point, we can confirm the ability of SO2 to activate the glycosyl fluoride with a probable co-promoting assistance of a glass vessel. Next, in order to increase the yield of mannoside 3b formed from a less reactive secondary alcohol 2b, various additives were tested (Table 1, entries 7–9). Presence of basic molecular sieves (4 Å) as a drying agent led to lower yield and did not suppress the formation of hemiacetal α-4 (Table 1, entry 7), while no reaction was observed when additives containing a fluorophilic silicon center were used (Table 1, entries 8 and 9). The inhibitory effect of basic molecular sieves may point to the presence and contributory role of protic acid (HF or H2SO3) in the course of the reaction [64]. Whereas, silyl additives can react with alcohol yielding silyl ether [65,66] that do not react further with glycosyl fluoride α-1a under our conditions. The formation of silyl ether was detected in a crude reaction mixture by NMR spectroscopy.
Table 1: Screening of conditions for glycosylation in liquid SO2.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-78-i6.svg?max-width=637&scale=1.0)
|
|||||||
| entry | NuH | (equiv) | T (°C) | additive (equiv) | α/β ratiob | yield 3 (%)c | yield α-4 (%)c |
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-78-i7.svg?max-width=637&scale=1.0)
2a |
1.1 | 30 to 80 | – | NR | ||
| 2 | 1.1 | 100 | – | 97:3 | 3a, 87 | 12 | |
| 3d,e | 1.1 | 100 | – | 94:6 | 3a, 8 | 40 | |
| 4d,f | 3.0 | 100 | – | 96:4 | 3a, 23 | 35 | |
| 5d | 3.0 | 150 | – | 97:3 | 3a, 69 | 30 | |
| 6 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-78-i8.svg?max-width=637&scale=1.0)
2b |
1.0 | 100 | – | α-only | 3b, 67 | 27 |
| 7 | 1.7 | 100 | 4 Å MS | α-only | 3b, 34 | 15 | |
| 8 | 1.1 | 100 | HMDSO (1.1) | NR | |||
| 9 | 1.7 | 100 | allyl-TMS (2.2) | NR | |||
aUnless otherwise stated, reactions were carried out by using 0.193–0.771 mmol of α-1a and 25 ± 5 g of liquid SO2 in a pressure reactor containing a glass tube. bDetermined by 1H NMR analysis of the crude reaction mixture. cYield of isolated product. dReaction carried out in a pressure reactor containing a PTFE tube. e53% of α-1a was recovered. f48% of α-1a was recovered. NR = no reaction; MS = molecular sieves; HMDSO = hexamethyldisiloxane; TMS = trimethylsilyl.
When the optimized model reaction (Table 1, entry 2) between mannosyl fluoride α-1a and 2-phenylethanol (2a) was carried out in pure conventional solvents (MeCN, THF, toluene or DCM) often used for glycosylation, no reaction was observed (Table 2, entries 1, 4, 6 and 9). Only traces of mannoside 3a and/or hemiacetal α-4 were detected by NMR spectroscopy in the presence of H3PO4 as a protic acid additive having a similar pKa value to that of H2SO3 that is likely to be present in liquid SO2 (Table 2, entries 3 and 8) [55]. Thus, the previously considered probable contributory effect of H2SO3 can be ruled out. Further, in combination with polar aprotic Lewis basic solvents (MeCN, THF) [67] sulfur dioxide was deactivated (Table 2, entries 2 and 5), while in less polar solvents (toluene, DCM) the presence of sulfur dioxide was clearly advantageous and glycoside 3a was isolated in good yields (Table 2, entries 7 and 10).
Table 2: Comparison with conventional solvents.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-78-i9.svg?max-width=637&scale=1.0)
|
||
| entry | solvent |
yield (%)b
(α/β ratioc) |
| 0 | liquid SO2 | 3a, 87 (97:3) |
| 1 | MeCN | NR |
| 2d | MeCN + SO2 | NR |
| 3 | MeCN + conc.H3PO4e | traces of 3a & α-4 |
| 4 | THF | NR |
| 5d | THF + SO2 | NR |
| 6 | toluene | NR |
| 7d | toluene + SO2 | 3a, 62% (α-only) |
| 8 | toluene + conc.H3PO4f | traces of α-4 |
| 9 | DCM | NR |
| 10d | DCM + SO2 |
3a, 65% (98:2)
α-4, 32% |
aReactions were carried out in (a) a pressure reactor containing a glass tube for entries 2, 5, 7, 9, and 10; (b) a glass pressure tube for entries 1, 3, 4, 6, and 8. bYield of isolated product. cDetermined by 1H NMR analysis of a crude reaction mixture. dSolutions were prepared by bubbling SO2 through the selected solvent for 10 min. e1.2 equiv. f1.4 equiv.
Next, the reactivity of various mannosyl halides α-1a–c towards O- and S-nucleophiles were compared under optimized reaction conditions (Table 3). In the case of 2-phenylethanol (2a) as an O-nucleophile, a similar reactivity, yield of mannoside 3a and α-selectivity were observed (Table 3, entries 1–3) among all the halides α-1a–c, although mannosyl chloride α-1b and bromide α-1c were not fully consumed. The superior reactivity of glycosyl fluoride α-1a in liquid SO2 compared to other halides was clearly demonstrated when thiol 2c was used as an acceptor (Table 3, entries 4–6). S-Mannoside 3c was isolated from mannosyl fluoride α-1a in twice as high yield as from the corresponding chloride α-1b or bromide α-1c. The stability of the latter in liquid SO2 at such a high temperature was unexpected due to their generally established labile nature. Additionally, when competitive glycosylation reactions in the presence of both O- and S-nucleophiles were performed, all mannosyl halides α-1a–c gave O-mannoside 3a as the major product in 58 to 71% yield, while overall yield of products 3a,c varied from 77% for α-1b to quant. for α-1a (Table S1, Supporting Information File 1).
Table 3: Reactivity comparison of mannosyl halides α-1a–c in liquid SO2.a
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-78-i10.svg?max-width=637&scale=1.0)
|
||||||||
| entry | α-1 | 2 | composition of a crude reaction mixture (mol %)b | α:β ratiob | yield 3 (%)c | |||
| α-1 | α-3 | β-3 | α-4 | |||||
| 1 | a |
2a
(Y = O) |
ND | 86 | 3 | 11 | 97:3 | 3a, 87 |
| 2 | b | 4 | 85 | 2 | 9 | 98:2 | 3a, 91 | |
| 3 | c | 14 | 80 | 2 | 4 | 98:2 | 3a, 81 | |
| 4 | a |
2c
(Y = S) |
ND | 82 | 18 | ND | 82:18 | 3c, 95 |
| 5 | b | 46 | 44 | 2 | 8 | 96:4 | 3c, 46 | |
| 6 | c | 42 | 42 | 10 | 6 | 81:19 | 3c, 49 | |
aReactions were carried out by using 0.173–0.771 mmol of α-1 and 25 ±5 g of liquid SO2. bDetermined by 1H NMR analysis of a crude reaction mixture. cYield of isolated product.
Pivaloyl-protected mannosyl fluoride α-1a was further applied for the synthesis of various O-, S- and C-glycosides to demonstrate the scope of acceptors compatible with our glycosylation conditions (Scheme 1). Most of the primary alcohols (2a, 2d–3f) were glycosylated in high yields (up to 91%). In the case of less reactive secondary alcohols (2b, 2h, 2j, 2k) and phenol (2l) better yields were obtained when 3.0 equiv of nucleophile were used. For example, the yield of mannoside 3l was increased from 34% to 79% when the amount of phenol (2l) was changed from 1.0 to 3.0 equiv. Similar reactivity relationships were observed in a series of thiols (2c, 2m–p), but the glycosylation yields comparing to the corresponding alcohols were slightly higher (up to 95%). By employing 2-phenylethanethiol (2c), a gram-scale synthesis of mannoside 3c was successfully demonstrated. Diminished reactivity towards glycosylation of some alcohols (2g, 2i, 2r) in liquid SO2 can be explained by possible formation of stable carbocation species [52]. Thus, in contrast to the other primary alcohols, an excess of 3.0 equiv was required to provide a moderate 62% yield of mannoside 3g when benzyl alcohol (2g) was used as an acceptor. Next, the formation of tertiary carbenium ion from 3-methyl-butan-2-ol (2i) via 1,2-hydrogen shift in an initial formed secondary carbocation [52] explains the relatively low yield of mannoside 3i. The same problem was observed when 1-adamantanol (S5) was used as a glycosyl acceptor and the desired mannoside was formed in only 6% NMR yield (Figure S1, Supporting Information File 1). Finally, a mixture of mannosides α-3r and α-3s was obtained when cyclopropylmethanol (2r) was applied. The cyclopropylmethyl carbocation (C4H7+), which is generated in liquid SO2 medium, can undergo a rearrangement to form a cyclobutyl carbocation [68]. The latter can be trapped by a water molecule forming cyclobutanol (2s) that further reacts with the glycosyl donor. Additionally, our glycosylation approach in liquid SO2 was applied for the synthesis of C-glycoside 3q by employing electron-rich 1,2,3-trimethoxybenzene (2q). Also binucleophiles 7a and 7b were glycosylated with a slight excess of mannosyl fluoride α-1a to form bis-mannosides α-8 in good yields (Scheme 2). In a series of pivaloyl-protected mannosides 3 a substrate-controlled α-selectivity due to the favoring effect of both neighboring ester-type protecting groups and the anomeric effect was observed [3].
![[1860-5397-17-78-i1]](/bjoc/content/inline/1860-5397-17-78-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Scope of glycosyl acceptors for glycosylation with pivaloyl-protected mannosyl fluoride α-1a in liquid SO2. aUnless otherwise stated, reactions were carried out by using 0.193–0.771 mmol of α-1a and 25 ± 5 g of liquid SO2; α/β ratios were determined by 1H NMR analysis of the crude reaction mixture. b56% yield when 1.1 equiv NuH was used. c67% yield when 1.0 equiv NuH was used. d34% yield when 1.0 equiv NuH was used. eReaction conditions: 1.2 equiv NuH, 43 g liquid SO2; α/β = 83:17. f42% yield when 1.1 equiv NuH was used. g59% yield when 1.0 equiv NuH was used.
Scheme 1: Scope of glycosyl acceptors for glycosylation with pivaloyl-protected mannosyl fluoride α-1a in liq...
![[1860-5397-17-78-i2]](/bjoc/content/inline/1860-5397-17-78-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Glycosylation of binucleophiles 7a,b in liquid SO2.
Scheme 2: Glycosylation of binucleophiles 7a,b in liquid SO2.
On the other hand, mixing of glycosyl donor α-1a and 1-O-methyl glucoside α-5 under the developed glycosylation conditions did not provide the expected disaccharide (Scheme 1). Instead, the formation of 1,6-anhydroglucose β-6 via intramolecular attack [69,70] was detected, while fluoride α-1a stayed unchanged. By employing fully protected 1-O-methyl glucoside α-S9 as a glycosyl donor, we have demonstrated that methoxide can act as a mediocre leaving group in liquid SO2 (Scheme S1, Supporting Information File 1). Other limitations for the glycosylation with mannosyl fluoride α-1a include steric hindrance and the presence of a Lewis basic nitrogen or fluorophilic trimethylsilyl group in the molecule of the glycosyl acceptor (Figure S1, Supporting Information File 1).
To our delight, no cleavage of the pivaloyl protecting groups in liquid SO2 medium was observed and the main side-product formed in the series of mannosides 3 was the previously mentioned tetra-O-pivaloyl mannopyranose α-4. In some experiments traces of 1,1'-mannoside α,α-S14 formed in the reaction between hemiacetal α-4 and glycosyl donor α-1a were detected (see Supporting Information File 1) [71].
Further, we turned our attention to the reactivity of other glycosyl fluorides in liquid SO2. We continued with pivaloyl-protected glucosyl fluoride β-9 (Scheme 3). The reaction conditions were optimized to 100 °C and 3.0 equiv of nucleophile (Table S3, Supporting Information File 1). The target glucosides 10 were obtained in a moderate yield and β-selectivity induced through the neighboring ester type protecting group assistance. At lower temperatures the glycosylation yield was lower, although full conversion of glucosyl fluoride β-9 was still observed. Compared to the analogue mannose derivative α-1a, glucose β-9 turned out to be less stable and more prone to various side-reactions. A series of side-products formed by hydrolysis and protecting group migrations were detected and their structures are proposed (see Supporting Information File 1).
![[1860-5397-17-78-i3]](/bjoc/content/inline/1860-5397-17-78-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Pivaloyl-protected glucosyl fluoride β-9 as a glycosyl donor in liquid SO2.
Scheme 3: Pivaloyl-protected glucosyl fluoride β-9 as a glycosyl donor in liquid SO2.
Next, glycosyl fluorides α-11 and α-12 containing more acid-sensitive acetyl protecting groups were applied for the glycosylation of 2-phenylethanol (2a) and 2-phenylethanethiol (2c) in liquid SO2 (Table 4). A temperature screening was performed to identify optimal reaction conditions (Table S4, Supporting Information File 1). The acetyl-protected mannosyl fluoride α-11 gave the desired mannosides 13 in a moderate yield and α-selectivity. The latter was comparable to the selectivity observed for the pivaloyl-protected mannosides 3. This time a couple of mono-deprotected side-products was observed (see Supporting Information File 1). The reactivity of acetyl-protected glucosyl fluoride α-12 was similar to that of mannose α-11. Glucosides 14 were isolated in a moderate yield, but without any α,β-selectivity due to the mismatched interaction between the anomeric effect and neighboring protecting group assistance. The diminished selectivity compared to the series of pivaloyl-protected glucosides 10 leads to the conclusion that the Lewis basic carbonyl oxygen of the acetyl group is more coordinated and less nucleophilic in liquid SO2 than the carbonyl oxygen of the pivaloyl group. The profile of side-products in this glucose series was similar to that observed for fluoride β-9 (see Supporting Information File 1).
Table 4: Acetyl protected manno- and glucopyranosyl fluorides α-11 and α-12 as glycosyl donors in liquid SO2.a
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-78-i11.svg?max-width=637&scale=1.0)
|
||||
| entry | glycosyl fluoride | Y (2) | α/β ratiob | yield (%)c |
| 1 | α-11 | O | 91:9 | 13a, 55 |
| 2 | S | 78:22 | 13b, 67 | |
| 3 | α-12 | O | 54:46 | 14a, 43 |
| 4 | S | 48:52 | 14b, 76 | |
aReactions were carried out in a scale of 0.277–0.300 mmol (α-11 or α-12). bDetermined by 1H NMR analysis of a crude reaction mixture. cYield of isolated product.
The armed benzyl-protected glycosyl fluorides α-15 and 16 were more reactive than their acylated analogues and the corresponding glycosides 17 and 18 were obtained at lower temperatures (Scheme 4). The reaction temperature for mannosyl fluoride α-15 was optimized to 30 °C (Table S5, Supporting Information File 1). At higher temperature desired mannoside 17a was not observed, whereas at −10 °C its yield was decreased. Under optimal conditions mannosides 17a–e were obtained in good yields and α-selectivity. Importantly, due to increased reactivity of glycosyl donor α-15 at lower temperature, we have also managed to obtain disaccharide 17f, albeit in low yield. Interestingly, a different temperature regime was adopted for benzyl-protected glucosyl fluoride 16 depending on its anomeric ratio (Table S6, Supporting Information File 1). Thus, the glycosylation temperature for the glucosyl fluoride containing an excess of β-anomer β-16 was optimized to 60 °C, while for the more reactive α-anomer α-16 it was decreased to 30 °C. Regardless of the anomeric ratio, the desired O- and S-glucosides 18a–f were isolated in good yields. Besides, glycosylation of primary nucleophiles with benzyl-protected glucosyl fluoride gave better yields (18a and 18d) than with the corresponding acylated analogues β-9 and α-11 described above. It was also found that the glycosylation stereoselectivity with glucosyl fluoride 16 did not depend on the anomeric ratio of glucosyl fluoride 16: both anomers of 16 yielded glucosides 18 in similar anomeric ratios with excess of the α-anomer. As expected, in the absence of an ester type protecting group at C2 position, for both series of benzyl protected glycosides α-selectivity was observed solely due to the anomeric effect.
![[1860-5397-17-78-i4]](/bjoc/content/inline/1860-5397-17-78-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Benzyl protected manno- and glucopyranosyl fluorides α-15 and 16 as glycosyl donors in liquid SO2. Reactions were carried out at 30 °C for mannosyl fluoride α-15 and glucosyl fluoride α-16; at 60 °C for glucosyl fluoride β-16. Anomeric ratios were determined by HPLC analysis.
Scheme 4: Benzyl protected manno- and glucopyranosyl fluorides α-15 and 16 as glycosyl donors in liquid SO2. ...
The Lewis acidic medium of liquid SO2 was also facilitating for the synthesis of 2-deoxy glucoside 20 from corresponding fluoride α-19 in 91% yield and good α-selectivity at −10 °C (Scheme 5). Due to the absence of a neighboring group at C2 position, the stereoselective synthesis of 2-deoxy glycosides is challenging [72-74]. We hypothesize that the stabilization of the oxocarbenium ion intermediate in a form of a dioxolenium ion by the remote protecting group in C3 or C6 position could be the reason for such a good α-selectivity in liquid SO2 [75].
![[1860-5397-17-78-i5]](/bjoc/content/inline/1860-5397-17-78-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: 2-Deoxy glycosyl fluoride α-19 as a glycosyl donor in liquid SO2.
Scheme 5: 2-Deoxy glycosyl fluoride α-19 as a glycosyl donor in liquid SO2.
Within this study, several experiments were also carried out to test the reactivity of peracylated manno- and glucopyranoses in liquid SO2 (Table S7, Supporting Information File 1). Most of these glycosyl donors were not fully consumed at 100 °C and formed a complex mixture of monosaccharides.
Finally, in order to make our glycosylation procedure more attractive and more accessible to the synthetic community we have demonstrated an application of saturated solutions of SO2 in conventional solvents that do not require a specific equipment, but can be performed in widely available glass pressure tubes (Table 5). In this context it has technically a similarity with ammonia solutions in organic solvents. We prepared saturated SO2 solutions in toluene and DCM. The concentration of SO2 in saturated solutions was determined by iodometric titration. As shown in Table 5, higher yields were obtained in DCM solutions. The yield of O-mannoside 3a was even higher when the glycosylation between mannosyl fluoride α-1a and 2-phenylethanol (2a) was performed in a gram-scale by applying a solvent/substrate ratio of 10:1 (mL/g) (Table 5, entries 2 and 5). A diminished yield was observed for S-mannoside 3c when the glycosylation was carried out in saturated DCM solution (64%) instead of pure SO2 (95%) (Table 5, entry 6). No difference was observed between the yields of thioglucoside 18d in liquid SO2 or its solution in DCM (Table 5, entry 7).
Table 5: Glycosylation with mannosyl fluoride α-1a and glucosyl fluoride α-16 in saturated SO2 solutions.a
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-78-i12.svg?max-width=637&scale=1.0)
|
|||||||
| entry | glycosyl fluoride | scale (g) | solvent | conc. (mL/g) | NuH | α/β ratiob | yield (%)c |
| 1 | α-1a | 0.2 |
2.7 M SO2
in toluene |
75 | 2a | >99:1 | 3a, 66 |
| 2 | 1.5 | 10 | 2a | 95:5 | 3a, 75 | ||
| 3 | 0.1 | 75 | 2c | 90:10 | 3c, 32 | ||
| 4 | α-1a | 0.2 |
2.0 M SO2
in DCM |
75 | 2a | 98:2 | 3a, 84 |
| 5 | 1.5 | 10 | 2a | 96:4 | 3a, 94 | ||
| 6 | 0.1 | 75 | 2c | 86:14 | 3c, 64 | ||
| 7 | α-16 | 0.1 | 20 | 2m | 64:36 | 18d, 84 | |
aReactions were carried out in glass pressure tubes; reaction conditions: (entries 1–6) 1.2 equiv NuH, at 100 °C; (entry 7) 2.0 equiv NuH, at 30 °C. bDetermined by 1H NMR (entries 1–6) or HPLC (entry 7) analysis of a crude reaction mixture. cYield of isolated product.
By employing benzyl-protected glucosyl fluoride 16 with different anomeric ratios, we have demonstrated that the stereochemical outcome of the glycosylation in liquid SO2 does not depend on the configuration of the anomeric center of the glycosyl donor. This observation points to the formation of a solvent-separated ion pair (SSIP) between the oxocarbenium ion and a counteranion, for example, fluorosulfite. At the same time, according to the Lewis base properties characterized by lithium cation basicity (LiCB) liquid SO2 (76.3) is similar to DCM (83) [67]. Thus, liquid SO2 could be classified as a non-coordinating solvent that unlikely coordinates to the oxocarbenium ion intermediate and affects the glycosylation stereoselectivity [1]. As a result, we can conclude that the stereoselectivity of the glycosylation in liquid SO2 is substrate-controlled and approaches a thermodynamic equilibrium determined by the anomeric effect or interference of both the anomeric effect and the assistance of the neighboring ester-type protecting group.
Next, we have also observed that the anomerization of the glycosylated products towards their thermodynamic equilibrium is promoted by the species formed during the glycosylation reaction [76]. Thus, when anomerically pure thiomannoside β-3c was subjected to the glycosylation conditions (100 °C, 16 h) in liquid SO2 without any additives, no anomerization was observed and the tested substrate β-3c was almost fully recovered. In contrast, when the same thiomannoside β-3c was added to the glycosylation mixture containing 1-dodecanethiol (2m) and mannosyl fluoride α-1a or bromide α-1c (Table 6), anomerization occurred approaching the anomeric ratio observed initially for mannoside 3c (α/β = 82:18, Scheme 1).
Table 6: Anomerization of thiomannoside β-3c under glycosylation conditions.
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-78-i13.svg?max-width=637&scale=1.0)
|
|||||||
| entry | α-1 | composition of a crude reaction mixture (mol %)a | α/β ratioa | ||||
| 3c | 3m | α-1 | α-4 | 3c | 3m | ||
| 1 |
a
(X = F) |
48 | 35 | ND | 17 | 71:29 | 82:18 |
| 2 |
c
(X = Br) |
39 | 48 | 13 | ND | 81:19 | 82:18 |
aDetermined by 1H NMR analysis of a crude reaction mixture.
Finally, we proved the formation of the fluorosulfite species by employing 19F NMR spectroscopy (Figure 1). Glycosylation of the reaction mixture was treated with Et3N to stabilize the possibly formed fluorosulfite anions in form of an ammonium salt. The 19F NMR spectra of the water-soluble components was than compared to the standard obtained from the reaction between TBAF and SO2. The peak that corresponds to the FSO2− anion was observed at 38.34 ppm (TFA as an external standard, −76.55 ppm) [77].
![[1860-5397-17-78-1]](/bjoc/content/figures/1860-5397-17-78-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Detection of the FSO2− species by 19F NMR (471 MHz, D2O).
Figure 1: Detection of the FSO2− species by 19F NMR (471 MHz, D2O).
Also DFT calculations were performed on the model reaction α-11 + MeOH → α-13c to elucidate the influence of SO2 on the dissociation of the glycosidic C–F bond [78] (Figure 2). Indeed, it was found that the coordination of the Lewis acidic SO2 to the fluoride (transition state TS-A≠ versus TS-A(SO2)≠) decreases the C–F bond dissociation energy (ΔΔG) by 10.6 kcal/mol. The formation of the neighboring group stabilized the oxocarbenium ion (dioxolenium ion) and its reaction with alcohol leads to the experimentally observed glycosides and the FSO2H adduct. We assume that due to the formation of the latter also substrates, which do not possess the participating group at C2 position, still react through the oxocarbenium ion intermediate.
![[1860-5397-17-78-2]](/bjoc/content/figures/1860-5397-17-78-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Computational study of reaction mechanism α-11 + MeOH → α-13c in the presence of and in absence of SO2 (Gaussian 09, Revision D.01; Gaussian, Inc.; m052x method and the 6-31+g(d) basis set). Enthalpy and Gibbs free energy values referenced against the starting value for the substrates and catalyst are given in kcal/mol.
Figure 2: Computational study of reaction mechanism α-11 + MeOH → α-13c in the presence of and in absence of ...
Conclusion
In summary, novel sulfur dioxide-assisted and metal-free glycosylation conditions by employing a combination of glycosyl fluoride as the glycosyl donor and liquid SO2 as both solvent and promoter have been developed. Due to the absence of any external additive, the presented method is considered to be an atom efficient and environmentally friendly synthetic approach. The glycosylation conditions in liquid SO2 have been optimized for both disarmed and armed mannose- and glucose-derived glycosyl fluorides, and novel conditions have been successfully applied for the synthesis of O-, S- and C-glycosides in moderate to excellent yields. The glycosylation in liquid SO2 is proposed to proceed via a solvent-separated ion pair and with stereoselectivity that is substrate-controlled and presents a thermodynamic equilibrium. The latter was clearly demonstrated when the more challenging 2-deoxyglucosyl fluoride was used as a glycosyl donor and the assistance of a remote acyl-protecting group provided good α-selectivity. The initially proposed formation of the fluorosulfite species during the glycosylation in liquid SO2 was proved by employing 19F NMR spectroscopy and DFT calculations. Finally, a more conventional experimental procedure has been provided for the application of saturated SO2 solution in DCM or toluene. This protocol does not require specific equipment and the reactions can be performed in widely available glass pressure tubes.
Supporting Information
| Supporting Information File 1: Experimental procedures; experimental data for synthesized compounds; competitive glycosylation of O- and S-nucleophiles; problematic glycosyl acceptors; reaction optimization data; reactivity of other glycosyl donors; proposed structures of side-products; detailed description of 19F NMR studies; stability tests for various glycosyl donors. | ||
| Format: PDF | Size: 2.6 MB | Download |
| Supporting Information File 2: Copies of NMR spectra. | ||
| Format: PDF | Size: 21.3 MB | Download |
| Supporting Information File 3: DFT calculations. | ||
| Format: PDF | Size: 1.0 MB | Download |
References
-
Bennett, C. S., Ed. Selective Glycosylations: Synthetic Methods and Catalysts; Wiley-VCH: Weinheim, Germany, 2017. doi:10.1002/9783527696239
Return to citation in text: [1] [2] [3] [4] -
Fraser-Reid, B. O.; Tatsuta, K.; Thiem, J.; Coté, G. L.; Flitsch, S.; Ito, Y.; Kondo, H.; Nishimura, S.-i.; Yu, B., Eds. Glycoscience: Chemistry and Chemical Biology; Springer: Berlin, Germany, 2008.
Return to citation in text: [1] -
Demchenko, A. V., Ed. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527621644
Return to citation in text: [1] [2] -
Nielsen, M. M.; Pedersen, C. M. Chem. Rev. 2018, 118, 8285–8358. doi:10.1021/acs.chemrev.8b00144
Return to citation in text: [1] [2] -
Yokoyama, M. Carbohydr. Res. 2000, 327, 5–14. doi:10.1016/s0008-6215(99)00324-9
Return to citation in text: [1] -
Mukaiyama, T.; Murai, Y.; Shoda, S.-i. Chem. Lett. 1981, 10, 431–432. doi:10.1246/cl.1981.431
Return to citation in text: [1] [2] -
Shoda, S.-I.; Kulkarni, S. S.; Gervay-Hague, J. Glycoside Synthesis from Anomeric Halides. In Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Demchenko, A. V., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 29–93. doi:10.1002/9783527621644.ch2
Return to citation in text: [1] [2] -
Shimizu, M.; Togo, H.; Yokoyama, M. Synthesis 1998, 799–822. doi:10.1055/s-1998-2070
Return to citation in text: [1] -
Williams, S. J.; Withers, S. G. Carbohydr. Res. 2000, 327, 27–46. doi:10.1016/s0008-6215(00)00041-0
Return to citation in text: [1] -
Kaeothip, S.; Demchenko, A. V. Carbohydr. Res. 2011, 346, 1371–1388. doi:10.1016/j.carres.2011.05.004
Return to citation in text: [1] -
Kulkarni, S. S.; Wang, C.-C.; Sabbavarapu, N. M.; Podilapu, A. R.; Liao, P.-H.; Hung, S.-C. Chem. Rev. 2018, 118, 8025–8104. doi:10.1021/acs.chemrev.8b00036
Return to citation in text: [1] -
Pearson, R. G. J. Am. Chem. Soc. 1963, 85, 3533–3539. doi:10.1021/ja00905a001
Return to citation in text: [1] -
Pearson, R. G.; Songstad, J. J. Am. Chem. Soc. 1967, 89, 1827–1836. doi:10.1021/ja00984a014
Return to citation in text: [1] -
Toshima, K. Carbohydr. Res. 2000, 327, 15–26. doi:10.1016/s0008-6215(99)00325-0
Return to citation in text: [1] -
Toshima, K. Glycosyl Halides. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid, B. O.; Tatsuta, K.; Thiem, J.; Coté, G. L.; Flitsch, S.; Ito, Y.; Kondo, H.; Nishimura, S.-i.; Yu, B., Eds.; Springer: Berlin, Germany, 2008; pp 429–449.
Return to citation in text: [1] -
Mukaiyama, T.; Jona, H. Proc. Jpn. Acad., Ser. B 2002, 78, 73–83. doi:10.2183/pjab.78.73
Return to citation in text: [1] -
Mukaiyama, T. Angew. Chem., Int. Ed. 2004, 43, 5590–5614. doi:10.1002/anie.200300641
Return to citation in text: [1] -
Mukaiyama, T.; Maeshima, H.; Jona, H. Chem. Lett. 2001, 30, 388–389. doi:10.1246/cl.2001.388
Return to citation in text: [1] -
Matsumoto, T.; Maeta, H.; Suzuki, K.; Tsuchihashi, l. G.-i. Tetrahedron Lett. 1988, 29, 3567–3570. doi:10.1016/0040-4039(88)85294-8
Return to citation in text: [1] -
Suzuki, K.; Maeta, H.; Matsumoto, T. Tetrahedron Lett. 1989, 30, 4853–4856. doi:10.1016/s0040-4039(01)80526-8
Return to citation in text: [1] -
Matsumoto, T.; Katsuki, M.; Suzuki, K. Tetrahedron Lett. 1989, 30, 833–836. doi:10.1016/s0040-4039(01)80628-6
Return to citation in text: [1] -
Kunz, H.; Sager, W. Helv. Chim. Acta 1985, 68, 283–287. doi:10.1002/hlca.19850680134
Return to citation in text: [1] -
Jona, H.; Mandai, H.; Chavasiri, W.; Takeuchi, K.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2002, 75, 291–309. doi:10.1246/bcsj.75.291
Return to citation in text: [1] -
Manabe, S.; Ito, Y. J. Org. Chem. 2013, 78, 4568–4572. doi:10.1021/jo400282x
Return to citation in text: [1] -
Ma, T.; Li, C.; Zhang, Z.-x.; Wang, Z.; Yu, L.; Xue, W. Synlett 2017, 28, 2633–2636. doi:10.1055/s-0036-1589121
Return to citation in text: [1] -
Sim, J.; Kim, S.-H.; Hur, J.; Lim, C.; Kim, H. S.; Suh, Y.-G. Asian J. Org. Chem. 2019, 8, 107–110. doi:10.1002/ajoc.201800582
Return to citation in text: [1] -
Sati, G. C.; Martin, J. L.; Xu, Y.; Malakar, T.; Zimmerman, P. M.; Montgomery, J. J. Am. Chem. Soc. 2020, 142, 7235–7242. doi:10.1021/jacs.0c03165
Return to citation in text: [1] -
Zeng, J.; Vedachalam, S.; Xiang, S.; Liu, X.-W. Org. Lett. 2011, 13, 42–45. doi:10.1021/ol102473k
Return to citation in text: [1] -
Okada, Y.; Asakura, N.; Bando, M.; Ashikaga, Y.; Yamada, H. J. Am. Chem. Soc. 2012, 134, 6940–6943. doi:10.1021/ja301480g
Return to citation in text: [1] -
Heuckendorff, M.; Pedersen, C. M.; Bols, M. J. Org. Chem. 2013, 78, 7234–7248. doi:10.1021/jo4012464
Return to citation in text: [1] -
Motoyama, A.; Arai, T.; Ikeuchi, K.; Aki, K.; Wakamori, S.; Yamada, H. Synthesis 2018, 50, 282–294. doi:10.1055/s-0036-1590927
Return to citation in text: [1] -
Pelletier, G.; Zwicker, A.; Allen, C. L.; Schepartz, A.; Miller, S. J. J. Am. Chem. Soc. 2016, 138, 3175–3182. doi:10.1021/jacs.5b13384
Return to citation in text: [1] -
Wadzinski, T. J.; Steinauer, A.; Hie, L.; Pelletier, G.; Schepartz, A.; Miller, S. J. Nat. Chem. 2018, 10, 644–652. doi:10.1038/s41557-018-0041-8
Return to citation in text: [1] -
Yang, Y.; Zhang, X.; Yu, B. Nat. Prod. Rep. 2015, 32, 1331–1355. doi:10.1039/c5np00033e
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2005, 61, 2947–2993. doi:10.1016/j.tet.2005.01.070
Return to citation in text: [1] -
Yang, Y.; Yu, B. Chem. Rev. 2017, 117, 12281–12356. doi:10.1021/acs.chemrev.7b00234
Return to citation in text: [1] -
Cardona, A.; Boutureira, O.; Castillón, S.; Díaz, Y.; Matheu, M. I. Green Chem. 2017, 19, 2687–2694. doi:10.1039/c7gc00722a
Return to citation in text: [1] -
Müller, H. Sulfur Dioxide. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012; Vol. 35, pp 73–118.
Return to citation in text: [1] -
Burow, D. F. Liquid Sulfur Dioxide. In The Chemistry of Nonaqueous Solvents; Lagowski, J. J., Ed.; Academic Press: New York, NY, USA, 1970; Vol. 3, pp 137–185. doi:10.1016/b978-0-12-433803-6.50008-0
Return to citation in text: [1] -
Walden, P. Ber. Dtsch. Chem. Ges. 1902, 35, 2018–2031. doi:10.1002/cber.190203502153
Return to citation in text: [1] -
Ross, J.; Percy, J. H.; Brandt, R. L.; Gebhart, A. I.; Mitchell, J. E.; Yolles, S. Ind. Eng. Chem., Ind. Ed. 1942, 34, 924–926. doi:10.1021/ie50392a006
Return to citation in text: [1] -
Tokura, N. Synthesis 1971, 639–645. doi:10.1055/s-1971-21781
Return to citation in text: [1] -
Schmidt, D.; Leutbecher, H.; Conrad, J.; Klaiber, I.; Mika, S.; Greiner, G.; Beifuss, U. Synlett 2007, 1725–1729. doi:10.1055/s-2007-984506
Return to citation in text: [1] -
Lugiņina, J.; Uzuleņa, J.; Posevins, D.; Turks, M. Eur. J. Org. Chem. 2016, 1760–1771. doi:10.1002/ejoc.201600141
Return to citation in text: [1] -
Lugiņina, J.; Turks, M. Synlett 2017, 28, 939–943. doi:10.1055/s-0036-1588670
Return to citation in text: [1] -
Olah, G. A.; Donovan, D. J. J. Am. Chem. Soc. 1978, 100, 5163–5169. doi:10.1021/ja00484a043
Return to citation in text: [1] -
Feigel, M.; Kessler, H.; Leibfritz, D.; Walter, A. J. Am. Chem. Soc. 1979, 101, 1943–1950. doi:10.1021/ja00502a003
Return to citation in text: [1] -
Mayr, H.; Gorath, G.; Bauer, B. Angew. Chem., Int. Ed. Engl. 1994, 33, 788–789. doi:10.1002/anie.199407881
Return to citation in text: [1] -
Olah, G. A. Angew. Chem., Int. Ed. Engl. 1995, 34, 1393–1405. doi:10.1002/anie.199513931
Return to citation in text: [1] -
Soares, B. G. Prog. Polym. Sci. 1997, 22, 1397–1430. doi:10.1016/s0079-6700(96)00025-1
Return to citation in text: [1] -
Chénedé, A.; Fleming, I.; Salmon, R.; West, M. C. J. Organomet. Chem. 2003, 686, 84–93. doi:10.1016/s0022-328x(03)00548-5
Return to citation in text: [1] -
Posevins, D.; Suta, K.; Turks, M. Eur. J. Org. Chem. 2016, 1414–1419. doi:10.1002/ejoc.201600013
Return to citation in text: [1] [2] [3] -
Suta, K.; Turks, M. ACS Omega 2018, 3, 18065–18077. doi:10.1021/acsomega.8b01630
Return to citation in text: [1] -
Leškovskis, K.; Lugiņina, J.; Suta, K.; Turks, M. Key Eng. Mater. 2019, 800, 42–46. doi:10.4028/www.scientific.net/kem.800.42
Return to citation in text: [1] -
Leškovskis, K.; Gulbe, K.; Mishnev, A.; Turks, M. Tetrahedron Lett. 2020, 61, 152528. doi:10.1016/j.tetlet.2020.152528
Return to citation in text: [1] [2] -
Igarashi, K.; Honma, T.; Irisawa, J. Carbohydr. Res. 1970, 15, 329–337. doi:10.1016/s0008-6215(00)80449-8
Return to citation in text: [1] [2] -
Milanova, E.; Benoit, R. L. Can. J. Chem. 1977, 55, 2807–2812. doi:10.1139/v77-390
Return to citation in text: [1] -
Lee, K.-Y.; Kim, C.-S.; Kim, H.-G.; Cheong, M.-S.; Mukherjee, D. K.; Jung, K.-D. Bull. Korean Chem. Soc. 2010, 31, 1937–1940. doi:10.5012/bkcs.2010.31.7.1937
Return to citation in text: [1] -
Kumar, A.; McGrady, G. S.; Passmore, J.; Grein, F.; Decken, A. Z. Anorg. Allg. Chem. 2012, 638, 744–753. doi:10.1002/zaac.201100476
Return to citation in text: [1] -
Maulitz, A. H.; Boese, R.; Kuhn, N. J. Mol. Struct.: THEOCHEM 1995, 333, 227–232. doi:10.1016/0166-1280(94)03955-k
Return to citation in text: [1] -
Eisfeld, W.; Regitz, M. J. Am. Chem. Soc. 1996, 118, 11918–11926. doi:10.1021/ja961398v
Return to citation in text: [1] -
Christensen, H. M.; Oscarson, S.; Jensen, H. H. Carbohydr. Res. 2015, 408, 51–95. doi:10.1016/j.carres.2015.02.007
Return to citation in text: [1] -
Nielsen, M. M.; Qiao, Y.; Wang, Y.; Pedersen, C. M. Eur. J. Org. Chem. 2020, 140–144. doi:10.1002/ejoc.201901755
Return to citation in text: [1] [2] -
Jona, H.; Takeuchi, K.; Mukaiyama, T. Chem. Lett. 2000, 29, 1278–1279. doi:10.1246/cl.2000.1278
Return to citation in text: [1] -
Pinnick, H. W.; Bal, B. S.; Lajis, N. H. Tetrahedron Lett. 1978, 19, 4261–4262. doi:10.1016/s0040-4039(01)95196-2
Return to citation in text: [1] -
Morita, T.; Okamoto, Y.; Sakurai, H. Tetrahedron Lett. 1980, 21, 835–838. doi:10.1016/s0040-4039(00)71518-8
Return to citation in text: [1] -
Laurence, C.; Gal, J.-F. Lewis Basicity and Affinity Scales: Data and Measurement; John Wiley & Sons: Chichester, UK, 2010.
Return to citation in text: [1] [2] -
Olah, G. A.; Prakash, G. K. S.; Rasul, G. J. Am. Chem. Soc. 2008, 130, 9168–9172. doi:10.1021/ja802445s
Return to citation in text: [1] -
Dere, R. T.; Zhu, X. Org. Lett. 2008, 10, 4641–4644. doi:10.1021/ol8019555
Return to citation in text: [1] -
Bosco, M.; Rat, S.; Dupré, N.; Hasenknopf, B.; Lacôte, E.; Malacria, M.; Rémy, P.; Kovensky, J.; Thorimbert, S.; Wadouachi, A. ChemSusChem 2010, 3, 1249–1252. doi:10.1002/cssc.201000218
Return to citation in text: [1] -
Araki, Y.; Watanabe, K.; Kuan, F.-H.; Itoh, K.; Kobayashi, N.; Ishido, Y. Carbohydr. Res. 1984, 127, C5–C9. doi:10.1016/0008-6215(84)85121-6
Return to citation in text: [1] -
Jünnemann, J.; Lundt, I.; Thiem, J. Liebigs Ann. Chem. 1991, 759–764. doi:10.1002/jlac.1991199101130
Return to citation in text: [1] -
Schene, H.; Waldmann, H. Synthesis 1999, 1411–1422. doi:10.1055/s-1999-3651
Return to citation in text: [1] -
Toshima, K.; Kasumi, K.-i.; Matsumura, S. Synlett 1999, 813–815. doi:10.1055/s-1999-2742
Return to citation in text: [1] -
Hansen, T.; Elferink, H.; van Hengst, J. M. A.; Houthuijs, K. J.; Remmerswaal, W. A.; Kromm, A.; Berdern, G.; van der Vorm, S.; Rijs, A. M.; Overkleeft, H. S.; Filippov, D. V.; Rutjes, F. P. J. T.; van der Marel, G. A.; Martens, J.; Oomens, J.; Codée, J. D. C.; Boltje, T. J. Nat. Commun. 2020, 11, No. 2664. doi:10.1038/s41467-020-16362-x
Return to citation in text: [1] -
Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. doi:10.1021/ja00847a032
Return to citation in text: [1] -
Zhu, S.-Z.; Huang, Q.-C.; Wu, K. Inorg. Chem. 1994, 33, 4584–4585. doi:10.1021/ic00098a028
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
Return to citation in text: [1]
| 41. | Ross, J.; Percy, J. H.; Brandt, R. L.; Gebhart, A. I.; Mitchell, J. E.; Yolles, S. Ind. Eng. Chem., Ind. Ed. 1942, 34, 924–926. doi:10.1021/ie50392a006 |
| 42. | Tokura, N. Synthesis 1971, 639–645. doi:10.1055/s-1971-21781 |
| 43. | Schmidt, D.; Leutbecher, H.; Conrad, J.; Klaiber, I.; Mika, S.; Greiner, G.; Beifuss, U. Synlett 2007, 1725–1729. doi:10.1055/s-2007-984506 |
| 44. | Lugiņina, J.; Uzuleņa, J.; Posevins, D.; Turks, M. Eur. J. Org. Chem. 2016, 1760–1771. doi:10.1002/ejoc.201600141 |
| 45. | Lugiņina, J.; Turks, M. Synlett 2017, 28, 939–943. doi:10.1055/s-0036-1588670 |
| 46. | Olah, G. A.; Donovan, D. J. J. Am. Chem. Soc. 1978, 100, 5163–5169. doi:10.1021/ja00484a043 |
| 47. | Feigel, M.; Kessler, H.; Leibfritz, D.; Walter, A. J. Am. Chem. Soc. 1979, 101, 1943–1950. doi:10.1021/ja00502a003 |
| 48. | Mayr, H.; Gorath, G.; Bauer, B. Angew. Chem., Int. Ed. Engl. 1994, 33, 788–789. doi:10.1002/anie.199407881 |
| 49. | Olah, G. A. Angew. Chem., Int. Ed. Engl. 1995, 34, 1393–1405. doi:10.1002/anie.199513931 |
| 50. | Soares, B. G. Prog. Polym. Sci. 1997, 22, 1397–1430. doi:10.1016/s0079-6700(96)00025-1 |
| 51. | Chénedé, A.; Fleming, I.; Salmon, R.; West, M. C. J. Organomet. Chem. 2003, 686, 84–93. doi:10.1016/s0022-328x(03)00548-5 |
| 52. | Posevins, D.; Suta, K.; Turks, M. Eur. J. Org. Chem. 2016, 1414–1419. doi:10.1002/ejoc.201600013 |
| 53. | Suta, K.; Turks, M. ACS Omega 2018, 3, 18065–18077. doi:10.1021/acsomega.8b01630 |
| 54. | Leškovskis, K.; Lugiņina, J.; Suta, K.; Turks, M. Key Eng. Mater. 2019, 800, 42–46. doi:10.4028/www.scientific.net/kem.800.42 |
| 55. | Leškovskis, K.; Gulbe, K.; Mishnev, A.; Turks, M. Tetrahedron Lett. 2020, 61, 152528. doi:10.1016/j.tetlet.2020.152528 |
| 56. | Igarashi, K.; Honma, T.; Irisawa, J. Carbohydr. Res. 1970, 15, 329–337. doi:10.1016/s0008-6215(00)80449-8 |
| 56. | Igarashi, K.; Honma, T.; Irisawa, J. Carbohydr. Res. 1970, 15, 329–337. doi:10.1016/s0008-6215(00)80449-8 |
| 64. | Jona, H.; Takeuchi, K.; Mukaiyama, T. Chem. Lett. 2000, 29, 1278–1279. doi:10.1246/cl.2000.1278 |
| 65. | Pinnick, H. W.; Bal, B. S.; Lajis, N. H. Tetrahedron Lett. 1978, 19, 4261–4262. doi:10.1016/s0040-4039(01)95196-2 |
| 66. | Morita, T.; Okamoto, Y.; Sakurai, H. Tetrahedron Lett. 1980, 21, 835–838. doi:10.1016/s0040-4039(00)71518-8 |
| 63. | Nielsen, M. M.; Qiao, Y.; Wang, Y.; Pedersen, C. M. Eur. J. Org. Chem. 2020, 140–144. doi:10.1002/ejoc.201901755 |
| 63. | Nielsen, M. M.; Qiao, Y.; Wang, Y.; Pedersen, C. M. Eur. J. Org. Chem. 2020, 140–144. doi:10.1002/ejoc.201901755 |
| 61. | Eisfeld, W.; Regitz, M. J. Am. Chem. Soc. 1996, 118, 11918–11926. doi:10.1021/ja961398v |
| 62. | Christensen, H. M.; Oscarson, S.; Jensen, H. H. Carbohydr. Res. 2015, 408, 51–95. doi:10.1016/j.carres.2015.02.007 |
| 57. | Milanova, E.; Benoit, R. L. Can. J. Chem. 1977, 55, 2807–2812. doi:10.1139/v77-390 |
| 58. | Lee, K.-Y.; Kim, C.-S.; Kim, H.-G.; Cheong, M.-S.; Mukherjee, D. K.; Jung, K.-D. Bull. Korean Chem. Soc. 2010, 31, 1937–1940. doi:10.5012/bkcs.2010.31.7.1937 |
| 59. | Kumar, A.; McGrady, G. S.; Passmore, J.; Grein, F.; Decken, A. Z. Anorg. Allg. Chem. 2012, 638, 744–753. doi:10.1002/zaac.201100476 |
| 60. | Maulitz, A. H.; Boese, R.; Kuhn, N. J. Mol. Struct.: THEOCHEM 1995, 333, 227–232. doi:10.1016/0166-1280(94)03955-k |
| 55. | Leškovskis, K.; Gulbe, K.; Mishnev, A.; Turks, M. Tetrahedron Lett. 2020, 61, 152528. doi:10.1016/j.tetlet.2020.152528 |
| 67. | Laurence, C.; Gal, J.-F. Lewis Basicity and Affinity Scales: Data and Measurement; John Wiley & Sons: Chichester, UK, 2010. |
| 52. | Posevins, D.; Suta, K.; Turks, M. Eur. J. Org. Chem. 2016, 1414–1419. doi:10.1002/ejoc.201600013 |
| 75. | Hansen, T.; Elferink, H.; van Hengst, J. M. A.; Houthuijs, K. J.; Remmerswaal, W. A.; Kromm, A.; Berdern, G.; van der Vorm, S.; Rijs, A. M.; Overkleeft, H. S.; Filippov, D. V.; Rutjes, F. P. J. T.; van der Marel, G. A.; Martens, J.; Oomens, J.; Codée, J. D. C.; Boltje, T. J. Nat. Commun. 2020, 11, No. 2664. doi:10.1038/s41467-020-16362-x |
| 67. | Laurence, C.; Gal, J.-F. Lewis Basicity and Affinity Scales: Data and Measurement; John Wiley & Sons: Chichester, UK, 2010. |
| 71. | Araki, Y.; Watanabe, K.; Kuan, F.-H.; Itoh, K.; Kobayashi, N.; Ishido, Y. Carbohydr. Res. 1984, 127, C5–C9. doi:10.1016/0008-6215(84)85121-6 |
| 72. | Jünnemann, J.; Lundt, I.; Thiem, J. Liebigs Ann. Chem. 1991, 759–764. doi:10.1002/jlac.1991199101130 |
| 73. | Schene, H.; Waldmann, H. Synthesis 1999, 1411–1422. doi:10.1055/s-1999-3651 |
| 74. | Toshima, K.; Kasumi, K.-i.; Matsumura, S. Synlett 1999, 813–815. doi:10.1055/s-1999-2742 |
| 3. | Demchenko, A. V., Ed. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527621644 |
| 69. | Dere, R. T.; Zhu, X. Org. Lett. 2008, 10, 4641–4644. doi:10.1021/ol8019555 |
| 70. | Bosco, M.; Rat, S.; Dupré, N.; Hasenknopf, B.; Lacôte, E.; Malacria, M.; Rémy, P.; Kovensky, J.; Thorimbert, S.; Wadouachi, A. ChemSusChem 2010, 3, 1249–1252. doi:10.1002/cssc.201000218 |
| 52. | Posevins, D.; Suta, K.; Turks, M. Eur. J. Org. Chem. 2016, 1414–1419. doi:10.1002/ejoc.201600013 |
| 68. | Olah, G. A.; Prakash, G. K. S.; Rasul, G. J. Am. Chem. Soc. 2008, 130, 9168–9172. doi:10.1021/ja802445s |
| 76. | Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. doi:10.1021/ja00847a032 |
| 77. | Zhu, S.-Z.; Huang, Q.-C.; Wu, K. Inorg. Chem. 1994, 33, 4584–4585. doi:10.1021/ic00098a028 |
| 1. | Bennett, C. S., Ed. Selective Glycosylations: Synthetic Methods and Catalysts; Wiley-VCH: Weinheim, Germany, 2017. doi:10.1002/9783527696239 |
| 1. | Bennett, C. S., Ed. Selective Glycosylations: Synthetic Methods and Catalysts; Wiley-VCH: Weinheim, Germany, 2017. doi:10.1002/9783527696239 |
| 2. | Fraser-Reid, B. O.; Tatsuta, K.; Thiem, J.; Coté, G. L.; Flitsch, S.; Ito, Y.; Kondo, H.; Nishimura, S.-i.; Yu, B., Eds. Glycoscience: Chemistry and Chemical Biology; Springer: Berlin, Germany, 2008. |
| 3. | Demchenko, A. V., Ed. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527621644 |
| 4. | Nielsen, M. M.; Pedersen, C. M. Chem. Rev. 2018, 118, 8285–8358. doi:10.1021/acs.chemrev.8b00144 |
| 8. | Shimizu, M.; Togo, H.; Yokoyama, M. Synthesis 1998, 799–822. doi:10.1055/s-1998-2070 |
| 9. | Williams, S. J.; Withers, S. G. Carbohydr. Res. 2000, 327, 27–46. doi:10.1016/s0008-6215(00)00041-0 |
| 26. | Sim, J.; Kim, S.-H.; Hur, J.; Lim, C.; Kim, H. S.; Suh, Y.-G. Asian J. Org. Chem. 2019, 8, 107–110. doi:10.1002/ajoc.201800582 |
| 7. | Shoda, S.-I.; Kulkarni, S. S.; Gervay-Hague, J. Glycoside Synthesis from Anomeric Halides. In Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Demchenko, A. V., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 29–93. doi:10.1002/9783527621644.ch2 |
| 27. | Sati, G. C.; Martin, J. L.; Xu, Y.; Malakar, T.; Zimmerman, P. M.; Montgomery, J. J. Am. Chem. Soc. 2020, 142, 7235–7242. doi:10.1021/jacs.0c03165 |
| 6. | Mukaiyama, T.; Murai, Y.; Shoda, S.-i. Chem. Lett. 1981, 10, 431–432. doi:10.1246/cl.1981.431 |
| 5. | Yokoyama, M. Carbohydr. Res. 2000, 327, 5–14. doi:10.1016/s0008-6215(99)00324-9 |
| 25. | Ma, T.; Li, C.; Zhang, Z.-x.; Wang, Z.; Yu, L.; Xue, W. Synlett 2017, 28, 2633–2636. doi:10.1055/s-0036-1589121 |
| 6. | Mukaiyama, T.; Murai, Y.; Shoda, S.-i. Chem. Lett. 1981, 10, 431–432. doi:10.1246/cl.1981.431 |
| 18. | Mukaiyama, T.; Maeshima, H.; Jona, H. Chem. Lett. 2001, 30, 388–389. doi:10.1246/cl.2001.388 |
| 22. | Kunz, H.; Sager, W. Helv. Chim. Acta 1985, 68, 283–287. doi:10.1002/hlca.19850680134 |
| 7. | Shoda, S.-I.; Kulkarni, S. S.; Gervay-Hague, J. Glycoside Synthesis from Anomeric Halides. In Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Demchenko, A. V., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 29–93. doi:10.1002/9783527621644.ch2 |
| 14. | Toshima, K. Carbohydr. Res. 2000, 327, 15–26. doi:10.1016/s0008-6215(99)00325-0 |
| 15. | Toshima, K. Glycosyl Halides. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid, B. O.; Tatsuta, K.; Thiem, J.; Coté, G. L.; Flitsch, S.; Ito, Y.; Kondo, H.; Nishimura, S.-i.; Yu, B., Eds.; Springer: Berlin, Germany, 2008; pp 429–449. |
| 16. | Mukaiyama, T.; Jona, H. Proc. Jpn. Acad., Ser. B 2002, 78, 73–83. doi:10.2183/pjab.78.73 |
| 17. | Mukaiyama, T. Angew. Chem., Int. Ed. 2004, 43, 5590–5614. doi:10.1002/anie.200300641 |
| 23. | Jona, H.; Mandai, H.; Chavasiri, W.; Takeuchi, K.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2002, 75, 291–309. doi:10.1246/bcsj.75.291 |
| 12. | Pearson, R. G. J. Am. Chem. Soc. 1963, 85, 3533–3539. doi:10.1021/ja00905a001 |
| 13. | Pearson, R. G.; Songstad, J. J. Am. Chem. Soc. 1967, 89, 1827–1836. doi:10.1021/ja00984a014 |
| 10. | Kaeothip, S.; Demchenko, A. V. Carbohydr. Res. 2011, 346, 1371–1388. doi:10.1016/j.carres.2011.05.004 |
| 11. | Kulkarni, S. S.; Wang, C.-C.; Sabbavarapu, N. M.; Podilapu, A. R.; Liao, P.-H.; Hung, S.-C. Chem. Rev. 2018, 118, 8025–8104. doi:10.1021/acs.chemrev.8b00036 |
| 19. | Matsumoto, T.; Maeta, H.; Suzuki, K.; Tsuchihashi, l. G.-i. Tetrahedron Lett. 1988, 29, 3567–3570. doi:10.1016/0040-4039(88)85294-8 |
| 20. | Suzuki, K.; Maeta, H.; Matsumoto, T. Tetrahedron Lett. 1989, 30, 4853–4856. doi:10.1016/s0040-4039(01)80526-8 |
| 21. | Matsumoto, T.; Katsuki, M.; Suzuki, K. Tetrahedron Lett. 1989, 30, 833–836. doi:10.1016/s0040-4039(01)80628-6 |
| 32. | Pelletier, G.; Zwicker, A.; Allen, C. L.; Schepartz, A.; Miller, S. J. J. Am. Chem. Soc. 2016, 138, 3175–3182. doi:10.1021/jacs.5b13384 |
| 33. | Wadzinski, T. J.; Steinauer, A.; Hie, L.; Pelletier, G.; Schepartz, A.; Miller, S. J. Nat. Chem. 2018, 10, 644–652. doi:10.1038/s41557-018-0041-8 |
| 28. | Zeng, J.; Vedachalam, S.; Xiang, S.; Liu, X.-W. Org. Lett. 2011, 13, 42–45. doi:10.1021/ol102473k |
| 29. | Okada, Y.; Asakura, N.; Bando, M.; Ashikaga, Y.; Yamada, H. J. Am. Chem. Soc. 2012, 134, 6940–6943. doi:10.1021/ja301480g |
| 30. | Heuckendorff, M.; Pedersen, C. M.; Bols, M. J. Org. Chem. 2013, 78, 7234–7248. doi:10.1021/jo4012464 |
| 31. | Motoyama, A.; Arai, T.; Ikeuchi, K.; Aki, K.; Wakamori, S.; Yamada, H. Synthesis 2018, 50, 282–294. doi:10.1055/s-0036-1590927 |
| 39. | Burow, D. F. Liquid Sulfur Dioxide. In The Chemistry of Nonaqueous Solvents; Lagowski, J. J., Ed.; Academic Press: New York, NY, USA, 1970; Vol. 3, pp 137–185. doi:10.1016/b978-0-12-433803-6.50008-0 |
| 40. | Walden, P. Ber. Dtsch. Chem. Ges. 1902, 35, 2018–2031. doi:10.1002/cber.190203502153 |
| 37. | Cardona, A.; Boutureira, O.; Castillón, S.; Díaz, Y.; Matheu, M. I. Green Chem. 2017, 19, 2687–2694. doi:10.1039/c7gc00722a |
| 38. | Müller, H. Sulfur Dioxide. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012; Vol. 35, pp 73–118. |
| 1. | Bennett, C. S., Ed. Selective Glycosylations: Synthetic Methods and Catalysts; Wiley-VCH: Weinheim, Germany, 2017. doi:10.1002/9783527696239 |
| 1. | Bennett, C. S., Ed. Selective Glycosylations: Synthetic Methods and Catalysts; Wiley-VCH: Weinheim, Germany, 2017. doi:10.1002/9783527696239 |
| 4. | Nielsen, M. M.; Pedersen, C. M. Chem. Rev. 2018, 118, 8285–8358. doi:10.1021/acs.chemrev.8b00144 |
| 34. | Yang, Y.; Zhang, X.; Yu, B. Nat. Prod. Rep. 2015, 32, 1331–1355. doi:10.1039/c5np00033e |
| 35. | Pellissier, H. Tetrahedron 2005, 61, 2947–2993. doi:10.1016/j.tet.2005.01.070 |
| 36. | Yang, Y.; Yu, B. Chem. Rev. 2017, 117, 12281–12356. doi:10.1021/acs.chemrev.7b00234 |
© 2021 Gulbe et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)