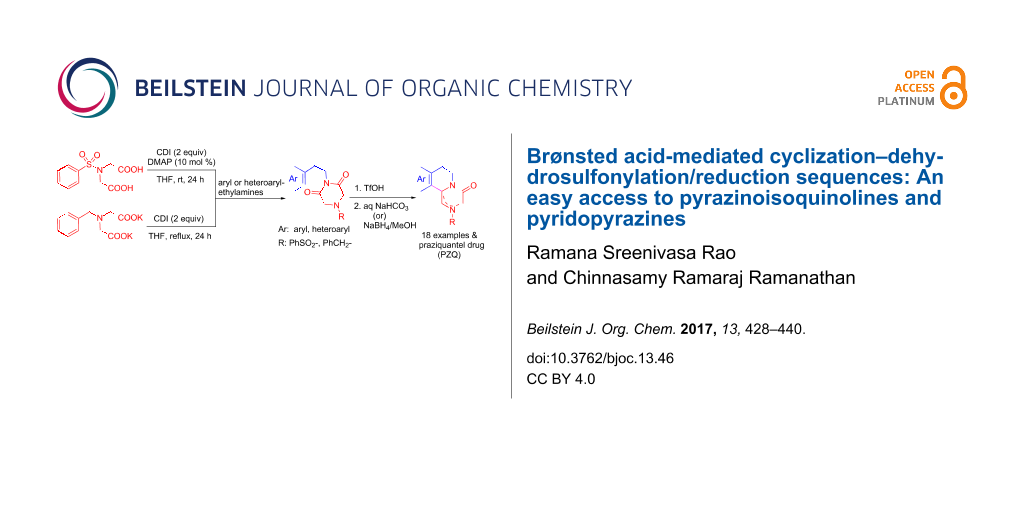
Abstract
An efficient and alternative synthetic approach has been developed to prepare various N-(arylethyl)piperazine-2,6-diones from 4-benzenesulfonyliminodiacetic acid and primary amines using carbonyldiimidazole in the presence of a catalytic amount of DMAP at ambient temperature. Piperazine-2,6-diones are successfully transformed to pharmaceutically useful pyridopyrazines or pyrazinoisoquinolines and ene-diamides via an imide carbonyl group activation strategy using a Brønsted acid. Subsequent dehydrosulfonylation reactions of the ene-diamides, in a one pot manner, smoothly transformed them to substituted pyrazinones. A concise synthesis of praziquantel (1) has also been achieved through this method.

Graphical Abstract
Introduction
Piperazine is an important structural core found in many biologically active natural products [1]. Piperazines are also useful intermediates in the synthesis of a variety of drug molecules having important biological activities such as anticancer [2,3], antifungal [4], amoebiasis, trypanosomiasis, bilharziasis [5,6], and schistosomiasis [7,8]. Some are cell adhesion inhibitors [9] brandykinin receptor antagonists [10,11] and chymase inhibitors [12]. Quinoxaline derivatives are known to act as aldose reductase (ALR2) inhibitors and are active towards chronic diabetic complications including neuropathy, nephropathy, cataracts and retinopathy [13,14]. On the other hand, piperazinone condensed tetrahydroisoquinolines (THIQs) are widespread in nature with interesting biological activities [15]. They are also found to be building blocks for the synthesis of many alkaloids (Figure 1) [16].
![[1860-5397-13-46-1]](/bjoc/content/figures/1860-5397-13-46-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected active pyrazinoisoquinolines, 2-oxopiperazines and aldose reductase inhibitors (ALR2).
Figure 1: Selected active pyrazinoisoquinolines, 2-oxopiperazines and aldose reductase inhibitors (ALR2).
To synthesize pyrazinoisoquinoline and its derivatives, various approaches such as the Ugi multicomponent reaction [17] amidoalkylation [18,19], N-acyliminium ion cyclization [20] and a radical cyclization [21] have been reported. To this end, recently, we have reported the activation of an imide carbonyl group with TfOH, for the synthesis of tetrahydroisoquinoline (THIQ) and tetrahydro-β-carboline (THBC) skeletons and related alkaloids [22-26].
The present study, describes the synthesis of 4-benzenesulfonylpiperazine-2,6-dione derivatives by using the combination of carbonyldiimidazole/4-dimethylaminopyridine (CDI/DMAP). Even though there are an adequate number of reports available in the literature for the synthesis of piperazine-2,6-dione derivatives [27]. To the best of our knowledge, this is the first report describing the synthesis of 4-benzenesulfonylpiperazine-2,6-diones at ambient temperature. The advantage of this methodology is that the more reactive and acid labile groups can be installed to the piperazine-2,6-diones at N-4 position, which would then be utilized for synthetically useful transformations to yield the corresponding products.
Further, these 4-benzenesulfonylpiperazine-2,6-diones were subjected to an imide carbonyl group activation strategy, to develop a practical approach to synthesize pyrazinoisoquinoline and pyridopyrazines via Brønsted acid assisted 6-exo-trig cyclization of arylethylpiperazine-2,6-diones.
Results and Discussion
Piperazine-2,6-diones are usually synthesized through an Ugi [30] multicomponent approach. The condensation of primary amines with benzyliminodiacetic acid at high temperatures or CDI/THF at reflux leads also to the formation of piperazine-2,6-diones (Scheme 1). Using CDI/THF under reflux conditions, the piperazine-2,6-dione 7a from 4-benzenesulfonyliminodiacetic acid was obtained only in 50% yield along with the formation of benzenesulfonic acid (Table 1, entry 1). The formation of benzenesulfonic acid may be due to the labile nature of the benzenesulfonyl protecting group. Therefore, the reactions were performed at temperatures below 70 °C and a higher yield of piperazine-2,6-dione 7a was observed at 60 °C (Table 1, entry 2). Further lowering of the reaction temperature did not improve the yield of 7a (Table 1, entry 3). Hence, there is a need to develop a method for the preparation of piperazine-2,6-dione derivatives from N-benzenesulfonyliminodiacetic acid at room temperature. After careful reviewing of reagents for amide bond formation, the reagent CDI proved to be a very successful reagent for the preparation of imides, amides, esters and thioesters. Therefore, the reaction was carried out with N-benzenesulfonyliminodiacetic acid, a primary amine and CDI (2 equiv) in THF, the corresponding piperazine-2,6-dione 7a was obtained in 21% yield at room temperature (Table 1, entry 4).
![[1860-5397-13-46-i1]](/bjoc/content/inline/1860-5397-13-46-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Comparison of previous reports with present work for piperazine-2,6-dione synthesis.
Scheme 1: Comparison of previous reports with present work for piperazine-2,6-dione synthesis.
Table 1: Standardization for the preparation of piperazine-2,6-dione.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-46-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Reagents | Equivalent | Temp (°C ) | Time (h) | Yield (%)b |
|---|---|---|---|---|---|
| 1 | CDI | 2 | 70 | 24 | 50 |
| 2 | CDI | 2 | 60 | 24 | 61 |
| 3 | CDI | 2 | 40 | 24 | 42 |
| 4 | CDI | 2 | rt | 48 | 21 |
| 5 | CDI/DMAP | 2/1 | rt | 24 | 35 |
| 6 | CDI/DMAP | 2/0.5 | rt | 24 | 50 |
| 7 | CDI/DMAP | 2/0.25 | rt | 24 | 57 |
| 8 | CDI/DMAP | 2/0.1 | rt | 24 | 72 |
aReaction conditions: amine 5a (1 mmol), benzenesulfonyl substituted iminodiacetic acid (1 mmol), THF (15 mL), rt. bYields of isolated products.
To facilitate the peptide bond formation DMAP has been used in conjuncture with coupling reagents such as DCC. We presumed that the use of DMAP along with CDI may facilitate the imide bond formation at room temperature. Hence, the reaction of 5a with N-benzenesulfonyliminodiacetic acid was carried out using CDI in the presence of DMAP (Table 1, entries 5–8). Piperazine-2,6-dione 7a was obtained in 72% yield at room temperature in the presence of 10 mol % of DMAP. Similarly, this strategy has been extended to couple various arylethylamines with N-benzenesulfonyliminodiacetic acid at room temperature to furnish expected imides 7a–h in good yields (Scheme 2).
![[1860-5397-13-46-i2]](/bjoc/content/inline/1860-5397-13-46-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Coupling of N-benzenesulfonyliminodiacetic acid with primary amines using CDI/DMAP.
Scheme 2: Coupling of N-benzenesulfonyliminodiacetic acid with primary amines using CDI/DMAP.
The successful development of this methodology for the synthesis of 1-arylethylpiperazine-2,6-diones intrigued us to examine them in 6-exo-trig cyclization using TfOH followed by reduction with NaBH4/MeOH to accomplish the synthesis of tetrahydropyrazinoisoquinoline. Surprisingly, the reaction of 7a furnished a mixture of ene-diamide 9a and substituted pyrazinone 10a in 90:10 ratio. Generally, the syntheses of these types of units are very limited in the literature [31]. Under controlled experimental conditions, in the absence of NaBH4/MeOH, the ene-diamide 9a was successfully generated in 90% yield using 4 equivalents of TfOH in 30 minutes from 7a.
While increasing the reaction time from 30 minutes to 2 h, the formation of biologically active pyrazinone 10a has been realized as a major product along with ene-diamide 9a. A literature survey revealed that sulfonamides are known to undergo hydrolysis in the presence of a Brønsted acid [32]. Sulfonamides also participate in amide hydrolysis with external nucleophiles such as phosphide anions [33] or phenyldimethylsilyllithium [34]. The combinations of thiophenol/K2CO3 [35] or NaOH/MeOH [36] are also known to hydrolyse sulfonamides. These methods lead to the formation of the corresponding free amines. Such desulfonylation of sulfonamides has been less utilized to make an unsaturated bond, for example, imine. Hence, attention has been paid to find suitable conditions for the formation of pyrazinones directly from piperazine-2,6-diones via cyclization followed by dehydrosulfonylation (Scheme 3).
![[1860-5397-13-46-i3]](/bjoc/content/inline/1860-5397-13-46-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of ene-diamides 9a–g and pyrazinones 10a–f.
Scheme 3: Formation of ene-diamides 9a–g and pyrazinones 10a–f.
Following extensive optimization, it was realized that after cyclization, addition of MeOH followed by reflux proved to be suitable conditions to generate the substituted pyrazinones. The formation of substituted pyrazinones through dehydrosulfonylation (1,2-elimination) may be facilitated by 3-factors, such as, (i) extended conjugation in pyrazinones, (ii) good leaving capacity of the benzenesulfonyl group and (iii) the presence of an acidic proton which is α to the amide carbonyl group. Hence, we believe that the selective elimination of the sulfonyl group in refluxing methanol would serve as a simple and novel alternate methodology to the reported oxidative strategy [37] for the syntheses of substituted pyrazinones. To demonstrate the generality of this procedure various methoxy/methyl substituted phenethyl and heterocyclic ethylpiperazine-2,6-diones 7a–g have been successfully converted to the substituted pyrazinones 10a–f in excellent yields as shown in Scheme 3. To elucidate the mechanism involved in the formation of substituted pyrazinones, the aliquot obtained from the reaction of 7c with TfOH followed by reflux in MeOH, was analyzed through ESI–HRMS technique. The appearance of peaks at m/z = 143.0030 and m/z = 159.0773 indicates the formation of benzenesulfinic acid and benzenesulfonic acid, respectively. The formation of benzenesulfinic acid could be explained through a desulfonylization reaction via a 1,2-elimination process (Scheme 4 and Figure 2).
![[1860-5397-13-46-i4]](/bjoc/content/inline/1860-5397-13-46-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism for the formation of substituted pyrazinones.
Scheme 4: Mechanism for the formation of substituted pyrazinones.
![[1860-5397-13-46-2]](/bjoc/content/figures/1860-5397-13-46-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: HRMS spectra of aliquot generated from cyclization reaction of 7c.
Figure 2: HRMS spectra of aliquot generated from cyclization reaction of 7c.
The structural evidence for cyclized compounds 9b and 10a was supported by single-crystal X-ray diffraction analysis (Figure 3) in addition to IR, NMR and HRMS data.
![[1860-5397-13-46-3]](/bjoc/content/figures/1860-5397-13-46-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP diagrams of compound 9b and 10a.
Figure 3: ORTEP diagrams of compound 9b and 10a.
To show the synthetic utility of substituted pyrazinones, we introduce a phenyl group at the C-3 position in pyrazinone derivative 10a. To begin with, the phenyl group was proposed to introduce at the 3-position in substituted pyrazinone via regioselective bromination followed by Suzuki coupling with phenylboronic acid. Accordingly, the bromination of 10a was successfully carried out to furnish regioisomers 12a and 12b in 82% yield upon treatment with bromodimethylsulfonium bromide (BDMS) [38] in dichloromethane at 0 °C to room temperature. The conventional Suzuki coupling of the regioisomers 12a and 12b with phenylboronic acid furnished the corresponding arylated products 13a and 13b in excellent yield (Scheme 5).
![[1860-5397-13-46-i5]](/bjoc/content/inline/1860-5397-13-46-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 3-phenylpyrazinone.
Scheme 5: Synthesis of 3-phenylpyrazinone.
To our dismay, the imide 7h did not participate in the cyclization reaction in the presence of TfOH at room temperature to generate a potential precursor for the synthesis of praziquantel. To realize the cyclization, the imide 7h was treated with TfOH under neat conditions at 70 °C, quite unfortunately to witness the decomposition of imide 7h. This may be due to the labile nature of the benzenesulfonyl group in 7h. To avoid this decomposition problem, the amino group in iminodiacetic acid was protected as a N-benzyl group instead of a N-benzenesulfonyl group. Accordingly, the potassium salt of N-benzyliminodiacetic acid was synthesized following the reported procedure [39]. The N-protected N-phenethylpiperazine-2,6-dione 8h was formed, while treating the potassium salt of N-benzyliminodiacetic acid with phenethylamine in presence of CDI in THF under reflux conditions. Similarly, this strategy has been extended to couple arylethylamines with the potassium salt of N-benzyliminodiacetic acid in THF at reflux to furnish the expected imides 8a–c, 8h, and 8i in good yields (Scheme 6).
![[1860-5397-13-46-i6]](/bjoc/content/inline/1860-5397-13-46-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of 4-N-benzyl-1-N-(aryl/heteroarylethyl)piperazine-2,6-dione.
Scheme 6: Synthesis of 4-N-benzyl-1-N-(aryl/heteroarylethyl)piperazine-2,6-dione.
The imide 8h was subjected to the cyclization reaction conditions using 6 equivalents of TfOH under neat conditions at 70 °C followed by reduction using NaBH4/MeOH at room temperature, which successfully furnished N-benzylpraziquantel 11h in 75% yield. The electron rich phenethyl group containing imides 8a–c, electron rich heteroaryl ethyl imide 8i, smoothly delivered the cyclized products 11a–c and 11i in excellent yields through the cyclization followed by the reduction sequence at room temperature (Scheme 7).
![[1860-5397-13-46-i7]](/bjoc/content/inline/1860-5397-13-46-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Cyclization of pyrazinoisoquinolines.
Scheme 7: Cyclization of pyrazinoisoquinolines.
The synthesis of praziquantel from intermediate 11h was accomplished through N-debenzylation at 80 °C under 1 atmosphere of H2 in the presence of Pd/C [40]. The present synthetic protocol involves the debenzylation reaction under milder conditions using Pd(OH)2 on charcoal/H2 at room temperature followed by coupling the secondary amine with cyclohexanecarboxylic acid using CDI. Praziquantel (1) was obtained in 80% overall yield from 11h (Scheme 8).
![[1860-5397-13-46-i8]](/bjoc/content/inline/1860-5397-13-46-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of the drug praziquantel 1.
Scheme 8: Synthesis of the drug praziquantel 1.
Conclusion
In conclusion, an efficient and alternative synthetic approach has been developed to prepare various N-(arylethyl)piperazine-2,6-diones. Brønsted acid assisted 6-exo-trig cyclization of piperazine-2,6-dione derivatives of aryl/heteroarylethylamines through carbonyl group activation to assemble the pyridoisoquinoline and pyrazinoisoquinoline derivatives have been demonstrated. The ene-diamides furnished the substituted pyrazinones through an acid–mediated dehydrosulfonylation in methanol. This strategy can be adopted to develop value added pyrazinone-based potential precursors useful to synthesize various drug targets. Further, the synthetic utility of pyrazinoisoquinoline was exemplified by the successful synthesis of fused tetrahydroisoquinoline drug molecule, praziquantel. Extending this strategy towards the stereoselective reduction of pyrazinones/ene-diamides is under investigation in our laboratory.
Experimental
General procedure for the synthesis of 4-benzenesulfonylpiperazinone-2,6-diones 7a–h
An oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, benzenesulfonyliminodiacetic acid (1 mmol), carbonyldiimidazole (2 mmol) and 10 mol % DMAP and dry THF (25 mL). Stirring was continued at room temperature to provide a clear solution. To this mixture was added aryl and heteroarylethylamine (5a–h) (1 mmol) and the resulting solution was stirred at room temperature for 24 h (monitored by TLC). The solution was concentrated to dryness using a rotary evaporator under reduced pressure. The crude reaction product was purified by chromatography on a short silica gel column using ethyl acetate/hexane, (30:70) as eluent to afford 7a–h in pure form.
1-(3,4-Dimethoxyphenethyl)-4-(phenylsulfonyl)piperazine-2,6-dione (7a)
300 mg, (72% yield) colorless solid; mp 138–139 °C; IR (KBr, cm−1): 3002, 2937, 2835, 1686, 1515, 1447, 1391, 1270, 1237, 1171, 1114, 1026, 960, 852, 811, 811, 690, 668, 572, 525; 1H NMR (400 MHz, CDCl3) δ 7.79–7.78 (m, 2H), 7.66–7.62 (m, 1H), 7.59–7.55 (m, 2H), 7.67 (d, J = 7.3 Hz, 1H), 6.69–6.66 (m, 2H), 4.11 (s, 4H), 3.86 (s, 3H), 3.84 (s, 3H), 3.6–3.65 (m, 2H), 2.43–2.47 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.4, 149.0, 147.9, 135.6, 134.2, 130.0, 127.7, 120.9, 112.1, 111.4, 56.0, 48.9, 40.1, 33.4; HRMS–ESI (m/z): [M + H] calcd for C20H22N2O6S, 419.1277; found, 419.1268.
General procedure for synthesis of 4-benzylpiperazinone-2,6-diones 8a–c, 8h, 8i
An oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, the potassium salt of benzyliminodiacetic acid (1 mmol) and carbonyldiimidazole (2 mmol) were added to dry THF (25 mL). The mixture was heated at refluxed for 10 min to obtain a clear solution. To this was added the aryl and heteroarylethylamines (5a–c, 5h, 5i) (1 mmol) and the resulting solution was heated at reflux for 24 h, then allowed to cool to room temperature. The solution was concentrated to dryness using a rotary evaporator under reduced pressure. The crude product was purified on a short silica gel column chromatography using ethyl acetate/hexane (2:3) as eluent to afford 8a–c, 8h, and 8i in pure form.
4-Benzyl-1-(3,4-dimethoxyphenethyl)piperazine-2,6-dione (8a)
250 mg, (68% yield) yellow liquid; IR (KBr, cm−1): 2950, 2853, 1734, 1683, 1594, 1514, 1453, 1348, 1268, 1156, 1028, 806, 755, 704, 631; 1H NMR (400 MHz, CDCl3) δ 7.29–7.26 (m, 1H), 7.24–7.21 (m, 2H), 7.19–7.16 (m, 2H), 6.70 (s, 2H), 6.68 (s, 1H), 3.94–3.86 (m, 2H), 3.79 (s, 3H), 3.76 (s, 3H), 3.49 (s, 2H), 3.29 (s, 4H), 2.72-2.68 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.8, 148.9, 147.7, 135.4, 130.8, 129.1, 128.7, 129.0, 121.0, 112.2, 111.3, 60.7, 56.3, 55.9, 40.4, 33.5; HRMS–ESI (m/z): [M + H] calcd for C21H24N2O4, 369.1814; found, 369.1808.
General procedure for the cyclization of 4-benzenesulfonylpiperazinone-2,6-diones 7a–g
Analogous as described in [26] an oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, imide 7a–g (0.5 mmol), and dry dichloromethane (15 mL), and the resulting solution was cooled to 0 °C (by using ice). To this solution was added TfOH (0.2 mL, 2 mmol) with stirring. After 30 min, the reaction mixture was quenched with aqueous NaHCO3. The organic layer was separated, and the aqueous layer was extracted with dichloromethane (2 × 15 mL). The combined organic extracts were washed with brine solution, dried over anhydrous Na2SO4 and filtered. The solution was concentrated to dryness by using a rotary evaporator. The dried crude product was purified by chromatography on a short silica gel chromatography column using ethyl acetate/hexane (1:1) as eluent to afford the 9a–g in pure form.
9,10-Dimethoxy-2-(phenylsulfonyl)-2,3,6,7-tetrahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (9a)
360 mg, (90% yield) colorless liquid; IR (KBr, cm−1): 2927, 2854, 1679, 1513, 1406, 1389, 1355, 1272, 1209, 1163, 1030, 992, 726, 576; 1H NMR (400 MHz, CDCl3) δ 7.82–7.79 (m, 2H), 7.59–7.55 (m, 1H), 7.51–7.47 (m, 2H), 6.92 (s, 1H), 6.58 (d, J = 7.6 Hz, 2H), 4.18 (s, 2H), 3.91 (s, 3H), 3.86 (s, 3H), 3.48–3.45 (m, 2H), 2.58–2.55 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0, 149.9, 148.6, 137.0, 133.6, 129.3, 127.3, 127.0, 119.9, 110.9, 106.4, 104.0, 56.4, 56.0, 48.3, 38.1, 28.0; HRMS–ESI (m/z): [M + H] calcd for C20H20N2O5S, 401.1171; found, 401.1156.
General procedure for the synthesis of pyrazinones 10a–f
Similar as described in [26] an oven-dried two-neck round-bottomed flask that had septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, imide 7a–f (0.5 mmol), and dry dichloromethane (15 mL), and the resulting solution was cooled to 0 °C. To this solution was added TfOH (0.2 mL, 2 mmol) with stirring. After the stipulated time, the contents were warmed to room temperature and methanol (25 mL) was added to the crude reaction mixture, the contents were refluxed for 1 h and then the solvent was evaporated to dryness under reduced pressure. The solid residue was dissolved in water and the aqueous layer was extracted with dichloromethane (2 × 15 mL). The combined organic extracts were washed with brine solution, dried over anhydrous Na2SO4 and filtered. The solution was concentrated to dryness by using a rotary evaporator. The dried compound was purified through a short silica gel column chromatography using ethyl acetate/hexane mixture (1:1) as eluent to afford the 10a–f in pure form.
9,10-Dimethoxy-6,7-dihydro-4H-pyrazino[2,1-a]isoquinolin-4-one (10a)
240 mg, (93% yield) yellow solid; mp 132–133 °C; IR (KBr, cm−1): 2926, 2850, 1680, 1514, 1354, 1162, 1030, 993, 732; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.74 (s, 1H), 7.17 (s, 1H), 6.74 (s, 1H), 4.23–4.17 (m, 2H), 3.94 (s, 3H), 3.93 (s, 3H), 3.04–2.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.8, 151.7, 148.9, 145.3, 135.5, 129.0, 119.8, 118.7, 110.8, 107.7, 56.3, 56.2, 38.7, 27.0; HRMS–ESI (m/z): [M + H] calcd for C14H14N2O3, 259.1083; found, 259.1073.
General procedure for the cyclization of 4-benzylpiperazine-2,6-diones 8a–c, 8h, 8i
Similar as described in [26] an oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, imide 8a–c, 8h, 8i (0.5 mmol), and dry dichloromethane (10 mL), and the resulting solution was cooled to 0 °C (by using ice). To this solution was added TfOH (0.2 mL, 2 mmol) with stirring. After 6 h the reaction mixture was diluted with methanol (2.5 mL) followed by portion wise addition of NaBH4 (2 mmol). The solution was stirred until the color disappeared (Additional NaBH4 and MeOH were used if the color persisted for a long time). The solution was evaporated to dryness under reduced pressure. The residue was dissolved in water and extracted with dichloromethane (2 × 20 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The solvent was evaporated under vacuum, and the crude product was purified by silica gel column chromatography using ethyl acetate/hexane (1:1) as eluent to afford the 11a–c, 11h, and 11i in pure form.
2-Benzyl-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (11h)
219 mg, (75% yield) yellow liquid; IR (KBr, cm−1): 2926, 2824, 1646, 1517, 1457, 1258, 1147, 1033, 744; 1H NMR (400 MHz, CDCl3) δ 7.28–7.23 (m, 4H), 7.24–7.16 (m, 1H), 7.11–7.08 (m, 2H), 7.07–7.04 (m, 1H), 6.94–6.92 (m, 1H), 4.78 (dd, J = 12.0, 8.0 Hz, 1H), 4.70–4.65 (m, 1H), 3.55–3.51 (m, 2H), 3.50–3.41 (m, 2H), 2.91–2.87 (m, 2H), 2.85–2.77 (m, 2H), 2.67–2.62 (m, 1H), 2.30–1.96 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 166.7, 136.5, 134.9, 132.2, 129.3, 129.2,128.6, 127.7, 127.1, 126.6, 124.7, 61.6, 57.0, 55.7, 55.5, 39.0, 28.7, 20.9; HRMS–ESI (m/z): [M + H] calcd for C19H20N2O, 293.1654; found, 293.1730.
Procedure for the synthesis 9,10-dimethoxy-3-phenyl-6,7-dihydro-4H-pyrazino[2,1-a]isoquinolin-4-one (13)
An oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask charged with a stir bar, 3,4-dimethoxypyrazinone 10a (1.0 mmol) and CH2Cl2 (5 mL) at 0–5 °C, bromodimethylsulfonium bromide (BDMS) (1.0 mmol, 0.223 g) was added and stirred at room temperature. After 24 h, the reaction mixture was washed with water (2 × 10 mL) and extracted with CH2Cl2 (2 × 10 mL). The organic layers were combined and dried over anhydrous Na2SO4. Solvents were removed by evaporation in a rotary evaporator. The crude reaction mixture was purified through silica gel column chromatography using ethyl acetate/hexane, 30:70 as eluent to afford 12a and 12b in the ratio 79:21.
The regioisomers of bromopyrazinone (12a and 12b) (1.0 mmol), bis(triphenylphosphine)palladium(II) chloride (10 mol %, 7 mg) in dimethylformamide (2 mL) was added phenylboronic acid (122 mg) and 2 M aqueous sodium carbonate (0.8 mL) under a nitrogen atmosphere. The reaction mixture was heated to 90 °C and stirred for 2 h. The reaction mixture was quenched with water and the mixture was extracted with ethyl acetate. The organic phase was separated, dried over anhydrous Na2SO4, filtered and concentrated. The crude reaction mixture was purified through a short silica gel column chromatography using ethyl acetate/hexane 10:90 as eluent and afforded 13a and 13b in the ratio of 92:8.
3-Bromo-9,10-dimethoxy-6,7-dihydro-4H-pyrazino[2,1-a]isoquinolin-4-one (12a)
275 mg, (82% yield) orange yellow liquid; IR (KBr, cm−1): 2924, 2856, 1727, 1511, 1456, 1362, 1272, 845; 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1H), 7.13 (s, 1H), 6.74 (d, J = 9.6 Hz, 1H), 4.26 (t, J = 6.4 Hz, 2H), 3.94 (s, 6H), 2.96 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 152.7, 152.0, 149.0, 137.9, 136.0, 128.8, 118.5, 118.3, 110.9, 107.6, 56.4, 56.3, 40.9, 27.2; HRMS–ESI (m/z): [M + H] calcd for C14H13BrN2O3, 337.0188; found, 337.0171.
9,10-Dimethoxy-3-phenyl-6,7-dihydro-4H-pyrazino[2,1-a]isoquinolin-4-one (13a)
290 mg, (87% yield) yellow liquid ; IR (KBr, cm−1): 2924, 2856, 1728, 1595, 1512, 1458, 1216; 1H NMR (400 MHz, CDCl3) δ 8.35–8.30 (m, 2H), 7.89 (s, 1H), 7.46–7.39 (m, 3H), 7.22 (s, 1H), 6.76 (s, 1H), 4.30 (t, J = 6.4 Hz, 2H), 3.95 (s, 3H), 3.94 (s, 3H), 2.98 (t, J = 6.4 Hz 2H); 13C NMR (100 MHz, CDCl3) δ 154.9, 151.5, 149.1, 148.9, 136.6, 134.6, 129.5, 128.8, 128.1, 119.5, 119.3, 110.8, 107.5, 56.4, 56.2, 39.2, 27.3; HRMS–ESI (m/z): [M + H] calcd for C20H18N2O3, 335.1396; found, 335.1383.
Procedure for the synthesis of praziquantel [2-(cyclohexanecarbonyl)-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one]
An oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, 2-benzyl-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (292 mg, 1 mmol), palladium hydroxide on charcoal (30 mg) and ethyl acetate (15 mL). The reaction flask was degassed and filled with hydrogen gas twice through a rubber bladder and the contents were stirred under hydrogen atmosphere. After 24 h, the reaction mixture was filtered through a small bed of celite and washed with ethyl acetate (2 × 10 mL). The solvents were removed by evaporation using a rotary evaporator. The crude product was used in the next step without further purification
An oven-dried two-neck round-bottom flask that had a septum in the side arm was cooled to room temperature under a steady stream of nitrogen. The flask was charged with a stir bar, cyclohexanecarboxylic acid (0.14 mL, 1.1 mmol) and carbonyldiimidazole (194 mg, 1.2 mmol) and dry THF (10 mL). The contents were stirred to obtain a clear solution. To this solution was added dropwise a solution of 1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (200 mg, 2.32 mmol) in dry THF and the resulting solution was stirred at room temperature for 24 h. The solution was concentrated to dryness using a rotary evaporator. The crude reaction mixture was purified by a short silica gel column chromatography using ethyl acetate/hexane (10:90) as eluent to furnished praziquantel (1) in 249 mg, (80% yield) as a white solid; mp 129–131 °C; IR (KBr, cm−1): 2928, 2856, 1660, 1452, 1211, 1039, 755, 656; 1H NMR (400 MHz, CDCl3) δ 7.21–7.13 (m, 4H), 5.08 (dd, J = 13.2, 2.4 Hz, 1H), 4.79–4.72 (m, 2H), 4.39 (d, J = 17.2 Hz, 1H), 4.0 (d, J = 13.2 Hz, 1H), 2.91–2.69 (m, 4H), 2.40 (t, J = 11.6 Hz, 1H), 1.73–1.65 (m, 5H), 1.53–1.43 (m, 2H), 1.23-1.17 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 174.9, 164.6, 134.6, 132.6, 129.2, 127.4, 126.9, 125.4, 54.9, 48.9, 45.1, 42.8, 40.7, 39.1, 29.1, 28.9, 28.8, 28.6, 25.7, 25.6, 25.3, 20.8; HRMS–ESI (m/z): [M + H] calcd for C19H24N2O2, 313.1916; found, 313.1905.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 3.6 MB | Download |
Acknowledgements
We thank DST and CSIR, New Delhi for financial support. RSR thank UGC, New Delhi for SRF. We gratefully acknowledge the Department of Chemistry, Pondicherry University for HRMS data (DST-FIST Sponsored) and IR data; CIF, Pondicherry University for NMR data. We acknowledge IPLS programme, Pondicherry University.
References
-
Rathi, A. K.; Syed, R.; Shin, H.-S.; Patel, R. V. Expert Opin. Ther. Pat. 2016, 26, 777–797. doi:10.1080/13543776.2016.1189902
Return to citation in text: [1] -
Chetan, B.; Bunha, M.; Jagrat, M.; Sinha, B. N.; Saiko, P.; Graser, G.; Szekeres, T.; Raman, G.; Rajendran, P.; Moorthy, D.; Basu, A.; Jayaprakash, V. Bioorg. Med. Chem. Lett. 2010, 20, 3906–3910. doi:10.1016/j.bmcl.2010.05.020
Return to citation in text: [1] -
Adamczyk-Woźniak, A.; Czerwińska, K.; Madura, I. D.; Matuszewska, A.; Sporzyński, A.; Żubrowska-Zembrzuska, A. New J. Chem. 2015, 39, 4308–4315. doi:10.1039/C5NJ00084J
Return to citation in text: [1] -
Tang, H.; Zheng, C.; Lv, J.; Wu, J.; Li, Y.; Yang, H.; Fu, B.; Li, C.; Zhou, Y.; Zhu, J. Bioorg. Med. Chem. Lett. 2010, 20, 979–982. doi:10.1016/j.bmcl.2009.12.050
Return to citation in text: [1] -
Samuelson, J.; Ayala, P.; Orozco, E.; Wirth, D. Mol. Biochem. Parasitol. 1990, 38, 281–290. doi:10.1016/0166-6851(90)90031-G
Return to citation in text: [1] -
Flickinger, C. J. Exp. Cell Res. 1972, 74, 541–546. doi:10.1016/0014-4827(72)90414-4
Return to citation in text: [1] -
Steinmann, P.; Keiser, J.; Bos, R.; Tanner, M.; Utzinger, J. Lancet Infect. Dis. 2006, 6, 411–425. doi:10.1016/S1473-3099(06)70521-7
Return to citation in text: [1] -
Caffrey, C. R. Curr. Opin. Chem. Biol. 2007, 11, 433–439. doi:10.1016/j.cbpa.2007.05.031
Return to citation in text: [1] -
Lewis, I.; Rohde, B.; Mengus, M.; Weetall, M.; Maida, S.; Hugo, R.; Lake, P. Mol. Diversity 2000, 5, 61–73. doi:10.1023/A:1013990624215
Return to citation in text: [1] -
Askew, B. C., Jr.; Aya, T.; Biswas, K.; Cai, G.; Chen, J. J.; Han, N.; Liu, Q.; Nguyen, T.; Nishimura, N.; Nomak, R.; Peterkin, T.; Qian, W.; Yang, K.; Yuan, C. C.; Zhu, J.; D’amico, D. C.; Nguyen, T.; Qian, W. 1,2,3,4-Tetrahydropyrazin-2-yl acetamides and their use as bradykinin antagonists for the treatment of inflammation related disorders. WO Patent WO019975, Feb 23, 2006.
Return to citation in text: [1] -
Aya, T.; Cai, G.; Chen, J. J.; D’amico, D. C.; Nguyen, T.; Qian, W. Triazoles and their use as bradykinin b1 receptor antagonists. WO Patent WO044355, April 27, 2006.
Return to citation in text: [1] -
Matsumoto, E. Novel thiazine or pyrazine derivatives. WO Patent WO0107419, Feb 1, 2001.
Return to citation in text: [1] -
Kao, Y.-L.; Donaghue, K.; Chan, A.; Knight, J.; Silink, M. Diabetes 1999, 48, 1338–1340. doi:10.2337/diabetes.48.6.1338
Return to citation in text: [1] -
Alexiou, P.; Pegklidou, K.; Chatzopoulou, M.; Nicolaou, I.; Demopoulos, V. J. Curr. Med. Chem. 2009, 16, 734–752. doi:10.2174/092986709787458362
Return to citation in text: [1] -
Caldwell, J. J.; Veillard, N.; Collins, I. Tetrahedron 2012, 68, 9713–9728. doi:10.1016/j.tet.2012.09.039
Return to citation in text: [1] -
Kölzer, M.; Weitzel, K.; Göringer, H. U.; Thines, E.; Opatz, T. ChemMedChem 2010, 5, 1456–1464. doi:10.1002/cmdc.201000230
Return to citation in text: [1] -
Cao, H.; Liu, H.; Dömling, A. Chem. – Eur. J. 2010, 16, 12296–12298. doi:10.1002/chem.201002046
Return to citation in text: [1] -
Kim, C. S.; Min, D. Y. Arch. Pharmacal Res. 1998, 21, 744–748. doi:10.1007/BF02976769
Return to citation in text: [1] -
Guglielmo, S.; Cortese, D.; Vottero, F.; Rolando, B.; Kommer, V. P.; Williams, D. L.; Fruttero, R.; Gasco, A. Eur. J. Med. Chem. 2014, 84, 135–145. doi:10.1016/j.ejmech.2014.07.007
Return to citation in text: [1] -
Kim, J. H.; Lee, Y. S.; Park, H.; Kim, C. S. Tetrahedron 1998, 54, 7395–7400. doi:10.1016/S0040-4020(98)00401-3
Return to citation in text: [1] -
Todd, M. H.; Ndubaku, C.; Bartlett, P. A. J. Org. Chem. 2002, 67, 3985–3988. doi:10.1021/jo010990m
Return to citation in text: [1] -
Selvakumar, J.; Makriyannis, A.; Ramanathan, C. R. Org. Biomol. Chem. 2010, 2, 4056–4058. doi:10.1039/c0ob00269k
Return to citation in text: [1] -
Selvakumar, J.; Ramanathan, C. R. Org. Biomol. Chem. 2011, 9, 7643–7646. doi:10.1039/c1ob06349a
Return to citation in text: [1] -
Mangalaraj, S.; Selvakumar, J.; Ramanathan, C. R. J. Chem. Sci. 2015, 127, 811–819. doi:10.1007/s12039-015-0836-8
Return to citation in text: [1] -
Mangalaraj, S.; Ramanathan, C. R. RSC Adv. 2012, 2, 12665–12669. doi:10.1039/c2ra21734a
Return to citation in text: [1] -
Selvakumar, J.; Rao, R. S.; Srinivasapriyan, V.; Marutheeswaran, S.; Ramanathan, C. R. Eur. J. Org. Chem. 2015, 2175–2188. doi:10.1002/ejoc.201403617
Return to citation in text: [1] [2] [3] [4] -
Henry, D. W. J. Heterocycl. Chem. 1966, 3, 503–511. doi:10.1002/jhet.5570030423
Return to citation in text: [1] -
Brewer, M. D.; Burgess, M. N.; Dorgan, R. J. J.; Elliott, R. L.; Mamalis, P.; Manger, B. R.; Webster, R. A. B. J. Med. Chem. 1989, 32, 2058–2062. doi:10.1021/jm00129a007
-
Kruse, C. G.; Troost, J. J.; Cohen-Fernandes, P.; van der Linden, H.; van Loon, J. D. Recl. Trav. Chim. Pays-Bas 1988, 107, 303–309. doi:10.1002/recl.19881070402
-
Ugi, I.; Demharter, A.; Hörl, W.; Schmid, T. Tetrahedron 1996, 52, 11657–11664. doi:10.1016/0040-4020(96)00647-3
Return to citation in text: [1] -
Lee, S.-C.; Park, S. B. J. Comb. Chem. 2007, 9, 828–835. doi:10.1021/cc0700492
Return to citation in text: [1] -
Snyder, H. R.; Heckert, R. E. J. Am. Chem. Soc. 1952, 74, 2006–2009. doi:10.1021/ja01128a041
Return to citation in text: [1] -
Yoshida, S.; Igawa, K.; Tomooka, K. J. Am. Chem. Soc. 2012, 134, 19358–19361. doi:10.1021/ja309642r
Return to citation in text: [1] -
Fleming, I.; Frackenpohl, J.; Ila, H. J. Chem. Soc., Perkin Trans. 1 1998, 1229–1236. doi:10.1039/a709116h
Return to citation in text: [1] -
Dar'in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014
Return to citation in text: [1] -
Yadav, A. K.; Yadav, S. K. S.; Siddiqui, I.; Peruncheralathan, S.; Ila, H.; Junjappa, H. Synlett 2008, 2674–2680. doi:10.1055/s-0028-1083529
Return to citation in text: [1] -
Kiec'-Kononowicz, K. Arch. Pharm. 1989, 322, 795–799. doi:10.1002/ardp.19893221104
Return to citation in text: [1] -
Khan, A. T.; Ali, M. A.; Goswami, P.; Choudhury, L. H. J. Org. Chem. 2006, 71, 8961–8963. doi:10.1021/jo061501r
Return to citation in text: [1] -
Merz, A.; Schropp, R.; Dötterl, E. Synthesis 1995, 795–800. doi:10.1055/s-1995-3993
Return to citation in text: [1] -
Yuste, F.; Pallás, Y.; Barrios, H.; Ortíz, B.; Sánchez-Obregón, R. J. Heterocycl. Chem. 1986, 23, 189–190. doi:10.1002/jhet.5570230139
Return to citation in text: [1]
| 40. | Yuste, F.; Pallás, Y.; Barrios, H.; Ortíz, B.; Sánchez-Obregón, R. J. Heterocycl. Chem. 1986, 23, 189–190. doi:10.1002/jhet.5570230139 |
| 26. | Selvakumar, J.; Rao, R. S.; Srinivasapriyan, V.; Marutheeswaran, S.; Ramanathan, C. R. Eur. J. Org. Chem. 2015, 2175–2188. doi:10.1002/ejoc.201403617 |
| 26. | Selvakumar, J.; Rao, R. S.; Srinivasapriyan, V.; Marutheeswaran, S.; Ramanathan, C. R. Eur. J. Org. Chem. 2015, 2175–2188. doi:10.1002/ejoc.201403617 |
| 1. | Rathi, A. K.; Syed, R.; Shin, H.-S.; Patel, R. V. Expert Opin. Ther. Pat. 2016, 26, 777–797. doi:10.1080/13543776.2016.1189902 |
| 7. | Steinmann, P.; Keiser, J.; Bos, R.; Tanner, M.; Utzinger, J. Lancet Infect. Dis. 2006, 6, 411–425. doi:10.1016/S1473-3099(06)70521-7 |
| 8. | Caffrey, C. R. Curr. Opin. Chem. Biol. 2007, 11, 433–439. doi:10.1016/j.cbpa.2007.05.031 |
| 21. | Todd, M. H.; Ndubaku, C.; Bartlett, P. A. J. Org. Chem. 2002, 67, 3985–3988. doi:10.1021/jo010990m |
| 5. | Samuelson, J.; Ayala, P.; Orozco, E.; Wirth, D. Mol. Biochem. Parasitol. 1990, 38, 281–290. doi:10.1016/0166-6851(90)90031-G |
| 6. | Flickinger, C. J. Exp. Cell Res. 1972, 74, 541–546. doi:10.1016/0014-4827(72)90414-4 |
| 22. | Selvakumar, J.; Makriyannis, A.; Ramanathan, C. R. Org. Biomol. Chem. 2010, 2, 4056–4058. doi:10.1039/c0ob00269k |
| 23. | Selvakumar, J.; Ramanathan, C. R. Org. Biomol. Chem. 2011, 9, 7643–7646. doi:10.1039/c1ob06349a |
| 24. | Mangalaraj, S.; Selvakumar, J.; Ramanathan, C. R. J. Chem. Sci. 2015, 127, 811–819. doi:10.1007/s12039-015-0836-8 |
| 25. | Mangalaraj, S.; Ramanathan, C. R. RSC Adv. 2012, 2, 12665–12669. doi:10.1039/c2ra21734a |
| 26. | Selvakumar, J.; Rao, R. S.; Srinivasapriyan, V.; Marutheeswaran, S.; Ramanathan, C. R. Eur. J. Org. Chem. 2015, 2175–2188. doi:10.1002/ejoc.201403617 |
| 4. | Tang, H.; Zheng, C.; Lv, J.; Wu, J.; Li, Y.; Yang, H.; Fu, B.; Li, C.; Zhou, Y.; Zhu, J. Bioorg. Med. Chem. Lett. 2010, 20, 979–982. doi:10.1016/j.bmcl.2009.12.050 |
| 18. | Kim, C. S.; Min, D. Y. Arch. Pharmacal Res. 1998, 21, 744–748. doi:10.1007/BF02976769 |
| 19. | Guglielmo, S.; Cortese, D.; Vottero, F.; Rolando, B.; Kommer, V. P.; Williams, D. L.; Fruttero, R.; Gasco, A. Eur. J. Med. Chem. 2014, 84, 135–145. doi:10.1016/j.ejmech.2014.07.007 |
| 2. | Chetan, B.; Bunha, M.; Jagrat, M.; Sinha, B. N.; Saiko, P.; Graser, G.; Szekeres, T.; Raman, G.; Rajendran, P.; Moorthy, D.; Basu, A.; Jayaprakash, V. Bioorg. Med. Chem. Lett. 2010, 20, 3906–3910. doi:10.1016/j.bmcl.2010.05.020 |
| 3. | Adamczyk-Woźniak, A.; Czerwińska, K.; Madura, I. D.; Matuszewska, A.; Sporzyński, A.; Żubrowska-Zembrzuska, A. New J. Chem. 2015, 39, 4308–4315. doi:10.1039/C5NJ00084J |
| 20. | Kim, J. H.; Lee, Y. S.; Park, H.; Kim, C. S. Tetrahedron 1998, 54, 7395–7400. doi:10.1016/S0040-4020(98)00401-3 |
| 13. | Kao, Y.-L.; Donaghue, K.; Chan, A.; Knight, J.; Silink, M. Diabetes 1999, 48, 1338–1340. doi:10.2337/diabetes.48.6.1338 |
| 14. | Alexiou, P.; Pegklidou, K.; Chatzopoulou, M.; Nicolaou, I.; Demopoulos, V. J. Curr. Med. Chem. 2009, 16, 734–752. doi:10.2174/092986709787458362 |
| 16. | Kölzer, M.; Weitzel, K.; Göringer, H. U.; Thines, E.; Opatz, T. ChemMedChem 2010, 5, 1456–1464. doi:10.1002/cmdc.201000230 |
| 12. | Matsumoto, E. Novel thiazine or pyrazine derivatives. WO Patent WO0107419, Feb 1, 2001. |
| 17. | Cao, H.; Liu, H.; Dömling, A. Chem. – Eur. J. 2010, 16, 12296–12298. doi:10.1002/chem.201002046 |
| 10. | Askew, B. C., Jr.; Aya, T.; Biswas, K.; Cai, G.; Chen, J. J.; Han, N.; Liu, Q.; Nguyen, T.; Nishimura, N.; Nomak, R.; Peterkin, T.; Qian, W.; Yang, K.; Yuan, C. C.; Zhu, J.; D’amico, D. C.; Nguyen, T.; Qian, W. 1,2,3,4-Tetrahydropyrazin-2-yl acetamides and their use as bradykinin antagonists for the treatment of inflammation related disorders. WO Patent WO019975, Feb 23, 2006. |
| 11. | Aya, T.; Cai, G.; Chen, J. J.; D’amico, D. C.; Nguyen, T.; Qian, W. Triazoles and their use as bradykinin b1 receptor antagonists. WO Patent WO044355, April 27, 2006. |
| 26. | Selvakumar, J.; Rao, R. S.; Srinivasapriyan, V.; Marutheeswaran, S.; Ramanathan, C. R. Eur. J. Org. Chem. 2015, 2175–2188. doi:10.1002/ejoc.201403617 |
| 9. | Lewis, I.; Rohde, B.; Mengus, M.; Weetall, M.; Maida, S.; Hugo, R.; Lake, P. Mol. Diversity 2000, 5, 61–73. doi:10.1023/A:1013990624215 |
| 15. | Caldwell, J. J.; Veillard, N.; Collins, I. Tetrahedron 2012, 68, 9713–9728. doi:10.1016/j.tet.2012.09.039 |
| 31. | Lee, S.-C.; Park, S. B. J. Comb. Chem. 2007, 9, 828–835. doi:10.1021/cc0700492 |
| 27. | Henry, D. W. J. Heterocycl. Chem. 1966, 3, 503–511. doi:10.1002/jhet.5570030423 |
| 30. | Ugi, I.; Demharter, A.; Hörl, W.; Schmid, T. Tetrahedron 1996, 52, 11657–11664. doi:10.1016/0040-4020(96)00647-3 |
| 38. | Khan, A. T.; Ali, M. A.; Goswami, P.; Choudhury, L. H. J. Org. Chem. 2006, 71, 8961–8963. doi:10.1021/jo061501r |
| 39. | Merz, A.; Schropp, R.; Dötterl, E. Synthesis 1995, 795–800. doi:10.1055/s-1995-3993 |
| 36. | Yadav, A. K.; Yadav, S. K. S.; Siddiqui, I.; Peruncheralathan, S.; Ila, H.; Junjappa, H. Synlett 2008, 2674–2680. doi:10.1055/s-0028-1083529 |
| 37. | Kiec'-Kononowicz, K. Arch. Pharm. 1989, 322, 795–799. doi:10.1002/ardp.19893221104 |
| 34. | Fleming, I.; Frackenpohl, J.; Ila, H. J. Chem. Soc., Perkin Trans. 1 1998, 1229–1236. doi:10.1039/a709116h |
| 35. | Dar'in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930–3933. doi:10.1021/acs.orglett.5b02014 |
| 32. | Snyder, H. R.; Heckert, R. E. J. Am. Chem. Soc. 1952, 74, 2006–2009. doi:10.1021/ja01128a041 |
| 33. | Yoshida, S.; Igawa, K.; Tomooka, K. J. Am. Chem. Soc. 2012, 134, 19358–19361. doi:10.1021/ja309642r |
© 2017 Rao and Ramanathan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)