Abstract
Background
It is well established that vicinal fluorines (RCHF-CHFR) prefer to adopt a gauche rather than an anti conformation when placed along aliphatic chains. This has been particularly recognised for 1,2-difluoroethane and extends to 2,3-difluorobutane and longer alkyl chains. It follows in these latter cases that if erythro and threo vicinal difluorinated stereoisomers are compared, they will adopt different overall conformations if the fluorines prefer to be gauche in each case. This concept is explored in this paper with erythro- and threo- diastereoisomers of 2,3-difluorosuccinates.
Results
A synthetic route to 2,3-difluorosuccinates has been developed through erythro- and threo- 1,2-difluoro-1,2-diphenylethane which involved the oxidation of the aryl rings to generate the corresponding 2,3-difluorosuccinic acids. Ester and amide derivatives of the erythro- and threo- 2,3-difluorosuccinic acids were then prepared. The solid and solution state conformation of these compounds was assessed by X-ray crystallography and NMR. Ab initio calculations were also carried out to model the conformation of erythro- and threo- 1,2-difluoro-1,2-diphenylethane as these differed from the 2,3-difluorosuccinates.
Conclusion
In general the overall chain conformations of the 2,3-difluorosuccinates diastereoisomers were found to be influenced by the fluorine gauche effect. The study highlights the prospects of utilising the vicinal difluorine motif (RCHF-CHFR) as a tool for influencing the conformation of performance organic molecules and particularly tuning conformation by selecting specific diastereoisomers (erythro or threo).

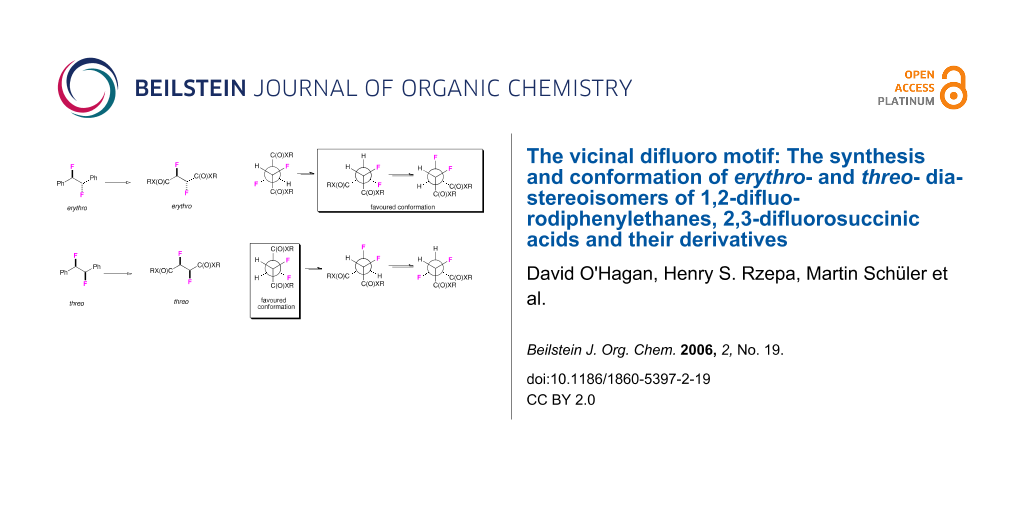
Graphical Abstract
Background
Of the 298,876 registered fluorinated structures in the Beilstein Chemical Database (for 2005) only 279 compounds contain a genuine vicinal difluoro motif-CHF-CHF- and only 12 crystal structures of this motif are deposited in the Cambridge Structure Data Base. The relatively rare presence of this motif may partly be attributed to the difficulty of their selective synthesis. It remains a synthetic challenge to prepare vicinal difluorocompounds efficiently and particularly in a stereoselective manner. There are attractive reasons to explore this motif. It is well known that the conformation of 1,2-difluoroethane is influenced by the fluorine gauche effect, where the fluorines prefer to be gauche rather than anti to each other [1]. This preference extends to 2,3-difluorobutane [2], and we have shown that erythro- and threo-9,10 difluorostearic acids have very different physical properties [3], the origin of which appears to lie in the different conformational preferences associated with the vicinal difluoro- motif for each diastereoisomer. Early synthetic methods to vicinal difluoro compounds have involved direct fluorination of alkenes with for eg. elemental fluorine (F2) [4] or XeF2 [5]. Such methods however are either difficult to carry out in a standard laboratory environment or they suffer from very poor stereoselectivity. The direct conversion of vicinal diols to vicinal difluorides has been explored with some success. For example both erythro and threo stereoisomers of dimethyl 2,3-difluorosuccinic acid were obtained either from methyl esters of the L-tartrate 1 or the meso-tartrate 1 by treatment with SF4/HF (Scheme 1) [6,7]. Conversion to the product erythro-2 proved efficient (97%) but that to threo-2 was poor (23%) due to competing elimination. The preparation of the erythro isomer of 2 is attractive on a large scale although SF4 has to be used with care and it is not amenable to reactions on a small scale. Our attempts to replace SF4 with DAST failed in trying to develop an analogous small scale laboratory process. Deoxofluor is finding use in the stereoselective conversion of vicinal diols to vicinal difluorocompounds and seems less prone to elimination than DAST [8]. In addition, Deoxofluor has been reported to be thermally more stable than related aminosulfur trifluoride reagents which allows the conversions to be carried out safely at elevated temperatures [9,10]. The stereoselective conversion of vicinal ditriflates to vicinal difluorides by treatment with TBAF has also been reported, particularly for the synthesis of 3,4-difluoropyrrolidine ring systems, and these reactions are finding currency in pharmaceutical products [11,12]. Schlosser et al. [13] have developed the most practical and straightforward method to access a variety of erythro- or threo- vicinal difluoro compounds in a diastereoselective manner, using either cis- or trans- epoxides 3 obtained directly from either the Z- or the E- alkenes. (Scheme 2). Ring opening of the epoxides 3 with HF-amine reagents generate the corresponding threo- and erythro- fluorohydrins 4 in largely a stereoselective manner. The resulting fluorohydrins 4 can then be converted to the erythro- or the threo- vicinal difluoro compounds 5 with reagents such as DAST [9,10] or Deoxofluor [8,14], although elimination products often compete with fluoride substitution depending on the nature of the substrate.
![[1860-5397-2-19-i1]](/bjoc/content/inline/1860-5397-2-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of vicinal dimethyl difluorosuccinates. The conversion of the tartrates 1 with SF4 and HF [6,7].
Scheme 1: Synthesis of vicinal dimethyl difluorosuccinates. The conversion of the tartrates 1 with SF4 and HF ...
![[1860-5397-2-19-i2]](/bjoc/content/inline/1860-5397-2-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Schlosser's route to vicinal erythro- or threo- difluoro alkanes 5 [13].
Scheme 2: Schlosser's route to vicinal erythro- or threo- difluoro alkanes 5 [13].
Vicinal difluoro compounds have been prepared by halo(bromo/iodo)fluorination of alkenes followed by halide substitution with silver fluoride [15]. The reaction has been applied to a variety of alkenes some of which (eg 6-9) are illustrated in Scheme 3.
![[1860-5397-2-19-i3]](/bjoc/content/inline/1860-5397-2-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Halofluorination of electron-rich alkenes with in situ fluoride displacement generates vicinal difluoro products. PPHF is Olah's reagent, pyridinium poly(hydrogen fluoride) [15].
Scheme 3: Halofluorination of electron-rich alkenes with in situ fluoride displacement generates vicinal difl...
We were interested in accessing diastereomerically pure samples of erythro- and threo- 2,3-difluorosuccinic acids 19. The preparation of stereoisomers of 2,3-difluorosuccinic acids, has involved conversions of tartaric acids (esters) [6,7], as described above in Scheme 1. Other approaches have involved the direct fluorination of fumaric acid [16] and the catalytic hydrogenation of 2,3-difluoromaleic acid [7], but these processes result in significant by-product formation and gave only poor yields of the desired products. Our alternative approach chose to explore the oxidation of the aromatic rings of erythro- and threo- diastereoisomers of 1,2-diphenyl-1,2-difluoroethane 13, exploiting the ability of the phenyl group to act as a latent carboxylic acid [17]. This paper describes these studies and we report the solid and solution state conformation of the erythro- and threo- diastereoisomers of 13 and the resultant 2,3-difluorosuccinic acid stereoisomers and some of their derivatives. Some of these results have recently been communicated [18]. The study suggests that the vicinal fluorine gauche effect can have a significant influence on the conformation of the 1,2-difluorosuccinates.
Results and Discussion
Synthesis of erythro- and threo- 1,2-diphenyl-1,2-difluoroethanes 13
Stilbene 9 is readily converted to its bromofluoro adduct by treatment with NBS and pyridine:HF following Olah's method [19] (Scheme 4).
![[1860-5397-2-19-i4]](/bjoc/content/inline/1860-5397-2-19-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Bromofluorination of stilbene [19].
Scheme 4: Bromofluorination of stilbene [19].
In our hands product 14 was generated with a diastereoselectivity of 94%. The predominant anti stereochemistry of 14 was established from the coupling constants of the olefin products obtained after a dehydrobromination reaction. The elimination of hydrogen bromide from such β-fluorobromides had been explored previously, and the reaction proceeds in a stereospecific manner to generate either E or Z fluoroalkene products [20]. Accordingly treatment of 14 with potassium tert-butoxide in a refluxing solution of hexane or pentane lead to the exclusive formation of the E-alkene 15 as judged by the 3JHF coupling constant of 21.1 Hz obtained from 19F-NMR. This is indicative of a stereospecific anti-elimination of hydrogen bromide from 14 to generate 15 with a cisoid relationship between H and F, rather than compound 16 which would have a trans relationship and a much larger 3JHF coupling constant (~30 Hz), and reinforces the stereochemical assignment made to 14 as illustrated in Scheme 5 [21].
![[1860-5397-2-19-i5]](/bjoc/content/inline/1860-5397-2-19-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Treatment of anti-14 with base generated the E-fluorostilbene 15 by an anti elimination mechanism.
Scheme 5: Treatment of anti-14 with base generated the E-fluorostilbene 15 by an anti elimination mechanism.
Substitution of the bromine in anti-14 with fluorine was accomplished by treatment with Ag(I)F in acetonitrile in the dark. Under these conditions, the substitution proceeds smoothly to erythro-13 but only in 56% de indicating a significant loss of stereochemical control during the reaction. The predominant stereochemical outcome of the fluorine substitution reaction suggests a double inversion mechanism as the major erythro-13 isomer must arise by replacement of the bromine of anti-14 by fluorine with an overall retention of configuration. Various examples of anchimeric assistance by phenyl groups have been reported [22] and in this case a carbocation is most reasonably generated which finds benzylic as well as anchimeric stabilisation via phenonium ring formation 18 with the β-phenyl group as illustrated in Scheme 6.
![[1860-5397-2-19-i6]](/bjoc/content/inline/1860-5397-2-19-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Hypothesis for the predominent retention of configuration during fluoride substitution via phenonium intermediate 18.
Scheme 6: Hypothesis for the predominent retention of configuration during fluoride substitution via phenoniu...
Isolation of the minor threo-13 isomer required careful chromatography. In order to improve the synthesis of threo-13 a reaction with cis-stilbene 17 was investigated. The one pot process with NBS, PPHF and Ag(I)F again proceeded smoothly however it also gave erythro-13 as the major product of the reaction, although with a reduced diastereoisomeric ratio (47% de) more suitable for threo- 13 isolation. The bias towards erythro- 13 in this case is clearly a result of internal rotation about the central carbon-carbon bond, to relieve a steric clash between the vicinal phenyl groups, after initial formation of an intermediate bromonium ion 18 as illustrated in Scheme 7.
![[1860-5397-2-19-i7]](/bjoc/content/inline/1860-5397-2-19-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Proposed C-C bond rotation during the preparation of 14 from cis-stilbene.
Scheme 7: Proposed C-C bond rotation during the preparation of 14 from cis-stilbene.
Erythro 13 was readily purified after several crystallisations whereas isolation of the threo isomer of 13 was more challenging. Partial separation of threo-13 was achieved by means of preparative thin layer chromatography. The enriched diasteroisomeric mixture could be crystallised to purity and crystals suitable for X-ray structure analysis were obtained (Figure 1). In the solid state erythro-13 adopts a conformation in which the phenyl substituents are anti to each other, with a Ph-C-C-Ph torsion angle of 180°. As a result the C-F bonds also align anti with respect to each other with a F-C-C-F torsion angle also close to 180°.
![[1860-5397-2-19-1]](/bjoc/content/figures/1860-5397-2-19-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystal structure of erythro-13.
Figure 1: Crystal structure of erythro-13.
A stereochemical mixture enriched in favour of threo-13 was crystallised to purity and a suitable crystal was used for X-ray structure analysis. The resultant structure is shown in Figure 2.
![[1860-5397-2-19-2]](/bjoc/content/figures/1860-5397-2-19-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The most obvious feature of this structure is the perhaps unexpected gauche relationship between the phenyl rings which places the fluorine atoms also gauche to each other. This superficially suggests that the fluorine gauche effect is over-riding any steric repulsion between the phenyl rings. To explore the significance of these solid state conformations further, NMR solution studies and ab initio analysis, exploring the preferred conformations for each of the diastereoisomers was carried out.
NMR studies on erythro- and threo-13
The most obvious feature in the 1H- and 19F- NMR spectra of the diastereoisomers of 13 is the coupling pattern from the AA'XX' spin system (Figure 3). Due to the chemical equivalence but magnetic non-equivalence of the F and H atoms a second-order spectrum is generated in each case.
![[1860-5397-2-19-3]](/bjoc/content/figures/1860-5397-2-19-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Expanded regions of the second order AA'XX' spin systems in the 1H-NMR (left) and 19F-NMR spectra (right) of erythro-13 (a), threo-13 (b) and the four individual coupling constants for the central 1H and 19F nuclei are given in (c).
Figure 3: Expanded regions of the second order AA'XX' spin systems in the 1H-NMR (left) and 19F-NMR spectra (...
Measuring of coupling constants from such second-order spectra has been described by Abrahams et. al. [23] although the analysis requires an intuitive fitting of values to specific coupling relationships. These deduced values are tabulated in Figure 3c. The large values of 45.2 & 47.2 Hz clearly correlate to the geminal 2JHF coupling, and the values of 15.2 & 14.1 Hz to the vicinal 3JHF coupling. The smaller coupling constant of 2.6 & 6.0 Hz most appropriately correlate to the 3JHH couplings, and thus, the value of -16.5 & -17.3 Hz is assigned to the vicinal 3JFF coupling. The 19F NMR spectrum can similarly be assigned in each case and reinforced these values. The magnitude of the different vicinal NMR coupling constants can be rationalised in terms of rotational isomerism of the individual diastereoisomers. Only the three staggered conformations for erythro- and threo- 13 are considered (Figure 4).
![[1860-5397-2-19-4]](/bjoc/content/figures/1860-5397-2-19-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: NMR coupling constants and calculated relative energies (kcalmol-1) of the staggered conformers of erythro- and threo- 13 calculated at the B3LYP//cc-pVTZ level. Relative energies (i) in the gas phase (ii) corrected for entropy and zero point energy differences and (iii) using a solvation model are reported. Calculated dipole moments {μ} are also given.
Figure 4: NMR coupling constants and calculated relative energies (kcalmol-1) of the staggered conformers of ...
It is not obvious from the NMR data which of a or b is the favoured solution conformation for the erythro isomer. We infer a significant contribution from rotamer b where the C-H bonds are gauche on the basis of the small 3JHH value (2.6 Hz), however the relatively small 3JHF value (15 Hz) suggests two C-H and C-F gauche relationships implying a contribution from rotamer a. Rotamer a most closely resembles the X-ray structure for erythro-13 shown in Figure 1. The situation is much clearer for threo-13. The relatively large 3JHH coupling constant (6.0 Hz) and the small 3JHF coupling constant (14 Hz) suggests a significant population of rotatmer d. This isomer has the vicinal C-H bonds anti to each other and both of the C-F/C-H and C-F/C-F bonds gauche. This is also the preferred conformation for this compound in the solid state (X-ray structure in Figure 2).
Conformational energy calculations on erythro and threo-13
Due to the ambiguous solution state study particularly for erythro-13, ab initio calculations were carried out at the B3LYP//cc-pVTZ level exploring absolute energies of the three staggered conformers of both erythro- and threo- 13 [24,25]. The geometries were optimized at this level for a gas phase model, and corrected for entropy and zero-point energy differences at this level. A separate solvation correction (chloroform) was applied using a continuum model (PCPM) and the larger cc-pV5Z basis set (using pVTZ geometries). Chloroform was studied in an attempt to relate the calculated values to the NMR solution conformations (vide infra). The relative energy data and dipole moments for each diastereoisomer are presented in Figure 4. The calculated conformations and energies can be viewed at http://www.ch.ic.ac.uk/rzepa/ohagan/ (see Supporting Information File 2).
Of the three staggered conformers of the erythro-13 isomer two are enantiomeric and have identical energies thus analysis of erythro-13 is reduced to a comparison of the energies of conformers a and b. Conformer a emerges as the more stable in the gas phase, with this stability originating predominantly from entropy and zero-point energy corrections (1.06 kcal/mol). This is also the conformer that most closely represents the X-ray structure (Figure 1). The solvent correction (which takes into account free energy differences associated with the solvent cavity, but does not allow for free energy differences arising from vibrational terms) does not alter the relative energies of a and b, despite a having a zero dipole moment and b having a relatively large value (3.5D) [26]. Although the more polar b should perhaps gain more from electrostatic solvation, it has a smaller solvent accessible surface area (239A2 vs 246 A2 for a) and these two appear to cancel in their overall effect on the relative energies. Our best estimate of the relative stability of a and b is about 1.0 kcal/mol in favour of the former as noted above. Thus structure a does not conform to a fluorine gauche effect and appears to be dominated by solvation of the trans relationship of the aryl rings and the zero dipole moment, although the smaller 3JHH coupling of 2.6 Hz and the slightly larger 3JHF coupling of 15 Hz in the NMR, measurement does suggest some contribution of conformer b in solution.
The threo-13 isomer has three distinct staggered conformations; c, d and e. Computationally, this requires modelling the subtle balance between the correlation effects due to gauche fluorine atoms and those due to gauche phenyl rings. In the gas phase (entropy and zero energy corrected) conformers c and d are iso-energetic. The dipole moments for these conformers vary significantly, with d > c > e. As with the erythro isomer, the greater solvation for d is partially offset by a smaller solvent-accessible surface (238 vs 247 Å 2 for c). Although d is slightly favoured in this model (by 0.11 kcal/mol), this is significantly smaller than the NMR estimate and may reflect a limitation of the solvation model. Taking all of the data together (theory, X-ray and NMR) conformer d appears to be the most favoured conformer for threo-13 with both the fluorine and the phenyl rings gauche, despite its larger dipole moment.
2,3-Difluorosuccinic acids 19
The synthesis of the 2,3-difluorosuccinic acid diastereoisomers 19 was explored by the oxidation of the aryl rings of 13 to carboxylic acids. Oxidative degradation of aromatic rings has been achieved by RuCl3/NaIO4 oxidation [17] however this method proved unsatisfactory in our hands and lead to poor conversions and a complex product mixture. As an alternative strategy ozonolysis in acetic acid, with a hydrogen peroxide work-up was explored [27,28], and this proved successful as illustrated in Scheme 8. For example, reaction of a 4:1 mixture of erythro- and threo- 13 led to the formation of 19 also in a 4:1 ratio of diastereoisomers. Erythro 2,3-difluorosuccinic acid 19 was obtained in a modest yield as a single stereoisomer from a stereochemically pure sample of the erythro 13. A crystal of erythro-19 suitable for X-ray analysis was obtained after sublimation, and the resultant structure is shown in Figure 5.
![[1860-5397-2-19-i8]](/bjoc/content/inline/1860-5397-2-19-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of erythro-19 via ozonolysis of erythro-13.
Scheme 8: Synthesis of erythro-19 via ozonolysis of erythro-13.
![[1860-5397-2-19-5]](/bjoc/content/figures/1860-5397-2-19-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the X-ray crystal structure of erythro-19 both of the carboxylic acid carbonyl oxygens adopt a syn periplanar conformation with respect to the C-F bonds. In the crystal packing, the carboxylate groups of two neighbouring molecules are hydrogen bonded and this clearly determines the three dimensional structure of the unit cell. The threo-19 diastereoisomer was prepared by a similar aryl oxidation reaction on a diastereomerically pure sample of threo-13 and this allowed crystallisation of a sample of racemic threo-19. The X-ray structure in Figure 6 shows the molecule in an extended chain conformation with both of the C-F bonds gauche to each other. One molecule of water is bound for every succinic acid molecule and this water clearly participates in hydrogen bonding to the carboxylic acid groups.
![[1860-5397-2-19-6]](/bjoc/content/figures/1860-5397-2-19-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
The major by-product of the ozonolysis reaction of 13 was the vicinal difluorophenylpropionic acid 20 as a mixture of stereoisomers. The compound was purified by esterification with methanol to generate esters 21. These diastereoisomers could be separated by chromatography and then hydrolysis was achieved under acidic conditions, followed by recrystallisation as illustrated in Scheme 9 to generate racemic, but diastereomerically pure samples of erythro- and threo- 20.
![[1860-5397-2-19-i9]](/bjoc/content/inline/1860-5397-2-19-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Strategy for the preparation of diastereoisomers of erythro- and threo- 20.
Scheme 9: Strategy for the preparation of diastereoisomers of erythro- and threo- 20.
The 3JHH coupling constants of esters 21 remain small (2.8-3.6 Hz) and indicate a gauche relationship between these vicinal hydrogens as summarised in Figure 7. It follows that in each case the fluorines will be predominantly gauche to each other.
![[1860-5397-2-19-7]](/bjoc/content/figures/1860-5397-2-19-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: NMR (CDCl3, RT) coupling constants of erythro- and threo- 2,3-difluoro-3-phenylpropionates 21.
Figure 7: NMR (CDCl3, RT) coupling constants of erythro- and threo- 2,3-difluoro-3-phenylpropionates 21.
The observed values for erythro- 21 report a maximal 3JHF coupling constant for the β-fluorine (28 Hz), but an intermediate one for the α-fluorine (20.8 Hz). This suggests a conformational preference for rotamer c, which has a gauche vicinal fluorine relationship, over a (Figure 8). For the threo- 21 isomer, there are two vicinal 3JHF couplings of similar and large magnitude (26.3 and 23.6 Hz) suggesting that rotamer d, with two trans 3JHF relationships and again with the fluorines gauche, is the most significant contributor to the solution conformation.
![[1860-5397-2-19-8]](/bjoc/content/figures/1860-5397-2-19-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Newman projections showing the staggered conformations of erythro- and threo- 21.
Figure 8: Newman projections showing the staggered conformations of erythro- and threo- 21.
In the solid state structure of threo-21 in Figure 9, the C-F bonds adopt a gauche relationship and the phenyl and ester groups are anti to each other. This is consistent with the preference for rotamer d found in solution. Attempts to crystallise erythro-21 as its free carboxylic acid resulted only in the formation of amorphous material and thus a comparison of solution and solid state structures was not possible for this isomer.
![[1860-5397-2-19-9]](/bjoc/content/figures/1860-5397-2-19-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: X-ray structure of methyl threo- 21.
Figure 9: X-ray structure of methyl threo- 21.
Amides of 2,3-difluorosuccinic acid
It was an objective of this research to explore the conformational preferences of amides of 2,3-difluorosuccinamides, particularly as we have previously noticed a conformational presence in α-fluoroamides [29], where the C-F bond aligns anti and planar to the amide carbonyl as illustrated in Figure 10. This adds an additional conformational constraint to these amides with a barrier to rotation around the C(CO)-C(F) bond of around 7-8 kcal mol-1. The preference of the C-F bond in α-fluoroamides to align anti periplanar to the carbonyl bond can be rationalized in terms of C-F bond and amide dipole relaxation as well as N-H...F hydrogen bonding [30].
![[1860-5397-2-19-10]](/bjoc/content/figures/1860-5397-2-19-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: The preferred conformation of α-fluoroamides has the C-F and amide carbonyl anti-planar [29,30].
Figure 10: The preferred conformation of α-fluoroamides has the C-F and amide carbonyl anti-planar [29,30].
The solution and solid state structures of 2,3-difluorosuccinate benzylamides 22 have been evaluated. These compounds were prepared by a straightforward EDCI amide coupling between benzylamine and 2,3-difluorosuccinate 19 as shown in Scheme 10.
![[1860-5397-2-19-i10]](/bjoc/content/inline/1860-5397-2-19-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: The synthesis of stereoisomers of erythro- and threo- 22. These isomers could be separated by chromatography.
Scheme 10: The synthesis of stereoisomers of erythro- and threo- 22. These isomers could be separated by chrom...
The diasteroisomers of 22 were separated by silica gel chromatography and recrystallisation of each allowed their X-ray structures to be compared. The structure of erythro-22 is illustrated in Figure 11.
![[1860-5397-2-19-11]](/bjoc/content/figures/1860-5397-2-19-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: X-ray structure of erythro-22.
Figure 11: X-ray structure of erythro-22.
Erythro- 22 adopts an extended conformation of the main chain in which the C-F bonds are anti with respect to each other. In that conformation the large benzyl substituents point in opposite directions. The α-fluoroamide groups tend towards a syn-planar C-F...N-H conformation as it is typical for this functional group (Figure 10) with the C-F bonds only 23° off the plane. The carbonyls point in opposite directions and thus intramolecular hydrogen bonding is not possible. There is however strong intermolecular hydrogen bonding between the amide hydrogen and the carbonyl oxygen of adjacent molecules which is dominating the unit cell structure (Figure 12).
![[1860-5397-2-19-12]](/bjoc/content/figures/1860-5397-2-19-12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Crystal packing of erythro-22 clearly indicating intermolecular hydrogen bonding.
Figure 12: Crystal packing of erythro-22 clearly indicating intermolecular hydrogen bonding.
These intermolecular interactions apparently over-ride the stereoelectronic preference for the gauche arrangement of the C-F bonds, which is observed in solution (vide infra). So we conclude that the solid and solution state structures of erythro- 22 are quite different. By comparison with erythro-22, the crystal structure of threo- 22 in Figure 13 shows both C-F bonds perfectly syn planar with respect to the amide N-H bonds, consistent with the typical planar arrangement of the α-fluoroamide group (Figure 10). The vicinal fluorines are gauche to each other. In this case the solution and solid state structures appear to be much more similar.
![[1860-5397-2-19-13]](/bjoc/content/figures/1860-5397-2-19-13.svg?scale=2.0&max-width=1024&background=FFFFFF)
In a recent Communication [18] we have reported the synthesis of peptides with 2,3-difluorosuccinic acid cores and revealed that such compounds adopt different conformations as a consequence of either the erythro or threo vicinal fluorine stereochemistry. That study highlighted both the solution and solid state conformations of the erythro and threo diastereoisomers of the bis-(S)-phenylalanine amides 23 as shown in Figure 14. The solution and solid state structures reinforced each other and the two diastereoisomers of 23 had preferred conformations where the fluorine atoms were again gauche to each other. This however gave very different shapes to the backbone connectivity in each diastereoisomer as illustrated in Figure 14.
![[1860-5397-2-19-14]](/bjoc/content/figures/1860-5397-2-19-14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: The conformations of erythro- and threo- 23 are very different as a consequence of each conformation preserving a vicinal fluorine gauche relationship [18].
Figure 14: The conformations of erythro- and threo- 23 are very different as a consequence of each conformatio...
NMR studies of vicinal difluoro diastereoisomers
The vicinal difluorosuccinates again give rise to second order NMR spectra due to the chemical equivalence but magnetic non equivalence of the fluorine and CHF methine hydrogen atoms similar to Figure 3. A comparison of the 3JHH and 3JHF coupling constants is outlined for the vicinal difluorosuccinate diastereoisomers 19 – 22, 24 and the 1,2-difluoro-1,2-diphenylethanes 13 in Figure 15.
![[1860-5397-2-19-15]](/bjoc/content/figures/1860-5397-2-19-15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: 3JHF and 3JHH coupling constants for the erythro (yellow) and threo (blue) diastereoisomers of the 2,3-difluorosuccinates 19–22, 24 as well as 1,2-difluoro-1,2-diphenylethanes 13. NMR spectra were recorded in CDCl3 with the exception of 2,3-difluorosuccinic acid, which was measured in CD3CN. The coupling constants were determined from second order spectra.
Figure 15: 3JHF and 3JHH coupling constants for the erythro (yellow) and threo (blue) diastereoisomers of the ...
Interestingly, the 3JHF coupling constants are very similar to each other within each diastereoisomeric series and are essentially independent of the nature of the substituents attached to the carboxylate group. The only significant exception are the diastereoisomers of 1,2-difluoro-1,2-diphenylethanes 13 which have already been discussed in detail. For the 2,3-difluorosuccinate derivatives 19–22,24 the threo stereoisomers have larger 3JHF coupling constants than the erythro stereoisomers.
In order to interpret the data in Figure 15, it is again useful to consider the staggered conformations of each threo and erythro diastereoisomer as shown in Figure 16. Each rotational isomer has two 3JHF and two 3JHH coupling constants the overall magnitude of each being an average of the two.
![[1860-5397-2-19-16]](/bjoc/content/figures/1860-5397-2-19-16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 16: Newman projections of the three staggered conformations of the erythro and threo stereoisomers of the vicinaldifluoro compounds succinates.
Figure 16: Newman projections of the three staggered conformations of the erythro and threo stereoisomers of t...
The angular dependence of the 3JHF coupling constant is largely influenced by the electronegativity of the substituents adjacent to the coupling nuclei [31]. For related compounds, the full trans 3JHF coupling constant has been estimated to be approximately 32 Hz and the gauche 3JHF coupling constant is approximately 8 Hz [32]. With no conformational bias the average 3JHF coupling constants will be (16 Hz) for each of the diastereoisomers according to these values (Figure 17).
![[1860-5397-2-19-17]](/bjoc/content/figures/1860-5397-2-19-17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 17: The average coupling constant with no conformational bias. The limiting coupling constants Jg = 8 Hz and Jt = 32 Hz are estimated values.
Figure 17: The average coupling constant with no conformational bias. The limiting coupling constants Jg = 8 H...
The experimental 3JHF coupling constants are clearly different for the two diastereoiomeric series. The contributions of the different conformers can then be estimated from the observed 3JHF NMR coupling constants as illustrated in the equations in Figure 18.
![[1860-5397-2-19-18]](/bjoc/content/figures/1860-5397-2-19-18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 18: The observed 3JHF coupling constants are an average over the rotational isomers.
Figure 18: The observed 3JHF coupling constants are an average over the rotational isomers.
For the erythro diastereoisomer the enantiotopic conformers b (which will be equally populated), dominate the conformer profile. This is consistent with the observed average 3JHH values of (2–3 Hz) where in conformers b only H-H gauche relationships are found with no contributions from anti H-H couplings, which would raise this low value. The high value 3JHF of 32 Hz for the threo diastereoisomers is essentially a maximum value for a trans coupling constant indicating the dominant contribution from conformer c. This is also consistent with the observed average 3JHH values of (2–3 Hz) where in conformer c there are only H-H gauche relationships.
In overview the dominant conformers in each diastereoisomer series have structures which accommodate gauche relationships between the C-F bonds and these results suggest that the fluorine "gauche effect" is influencing the preferred conformations in solution. It is notable that the coupling constants for the 1,2-difluoro-1,2-diphenylethanes isomers 13 are different in the series and do not conform to the ratios described above.
Conclusion
In this paper we have described the synthesis and comparative structures of a series of diastereoisomers of vicinal difluoro compounds, which were generated by converting stilbenes to 1,2-difluoro,1-2-diphenylethanes 13 and then oxidation of the aryl rings to generate 2,3-difluorosuccinic acids and their derivatives. The preparative methods allowed the preparation of individual erythro or threo diastereoisomers. The tendency of the vicinal fluorines to adopt predominant gauche conformations in solution emerges from an analysis of vicinal 3JHH and 3JHF coupling constants of these molecules and reinforces earlier studies on the conformation of vicinal difluoro compounds. This is in line with the well described fluorine gauche effect. The only exception to this was found for the threo stereoisomer of 1,2-difluoro-1,2-diphenylethanes 13, where all of the data (ab initio, NMR and X-ray) did not converge on a consensus structure. It emerges from this study that the stereoselective incorporation of vicinal fluorines can be used to influence the conformation of organic molecules. This is an attractive tool for the design of performance molecules in areas as diverse as pharmaceutical and medicinal chemistry research to materials science.
Experimental details for the preparation and characterisation of compounds 13, 14, 15, 19, 21, 22 and 24 are given in Supporting Information File 1.
References
-
Craig, N. C.; Chen, A.; Suh, K. H.; Klee, S.; Mellau, G. C.; Winnewisser, B. P.; Winnewisser, M. J. Am. Chem. Soc. 1997, 119, 4789–4790. doi:10.1021/ja963819e
Return to citation in text: [1] -
Angelini, G.; Gavuzzo, E.; Segre, A. L.; Speranza, M. J. Phys. Chem. 1990, 94, 8762–8766. doi:10.1021/j100388a004
Return to citation in text: [1] -
Tavasli, M.; O'Hagan, D.; Pearson, C.; Petty, M. C. Chem. Commun. 2002, 1226–1227. doi:10.1039/b202891c
Return to citation in text: [1] -
Merritt, R. F. J. Am. Chem. Soc. 1967, 89, 609–612. doi:10.1021/ja00979a025
Return to citation in text: [1] -
Chia, T.; Yang, N. C.; Chernick, C. L. J. Am. Chem. Soc. 1964, 86, 5021–5022. doi:10.1021/ja01076a069
Return to citation in text: [1] -
Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7
Return to citation in text: [1] [2] [3] -
Hudlicky, M. J. Fluorine Chem. 1983, 23, 241–259. doi:10.1016/S0022-1139(00)85130-1
Return to citation in text: [1] [2] [3] [4] -
Singh, R. P.; Shreeve, J. M. J. Fluorine Chem. 2002, 116, 23–26. doi:10.1016/S0022-1139(02)00065-9
Return to citation in text: [1] [2] -
Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M. Chem. Commun. 1999, 215–216. doi:10.1039/a808517j
Return to citation in text: [1] [2] -
Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048–7054. doi:10.1021/jo990566+
Return to citation in text: [1] [2] -
Hulin, B.; Cabral, S.; Lopaze, M. G.; Van Volkenburg, M. A.; Andrews, K. M.; Parker, J. C. Bioorg. Med. Chem. Lett. 2005, 15, 4770–4773. doi:10.1016/j.bmcl.2005.07.026
Return to citation in text: [1] -
Caldwell, C. G.; Chen, P.; He, J.; Parmee, R. E.; Leiting, B.; Marsilio, F.; Patel, R. A.; Wu, J. K.; Eiermann, G. J.; Petrov, A.; He, H.; Lyons, K. A.; Thornberry, N. A.; Weber, A. E. Bioorg. Med. Chem. Lett. 2004, 14, 1265–1268. doi:10.1016/j.bmcl.2003.12.040
Return to citation in text: [1] -
Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3
Return to citation in text: [1] [2] -
Lal, G. S.; Labach, E.; Evans, A. J. Org. Chem. 2000, 65, 4830–4832. doi:10.1021/jo000020j
Return to citation in text: [1] -
Olah, G. J. Org. Chem. 1979, 44, 3872–3881. doi:10.1021/jo01336a027
Return to citation in text: [1] [2] -
Rozen, S. J. Org. Chem. 1986, 51, 3607–3611. doi:10.1021/jo00369a011
Return to citation in text: [1] -
Norsikian, S. Chem.–Eur. J. 1999, 5, 2055–2068. doi:10.1002/(SICI)1521-3765(19990702)5:7<2055::AID-CHEM2055>3.0.CO;2-9
Return to citation in text: [1] [2] -
Schüler, M.; O'Hagan, D.; Slawin, A. M. Z. Chem. Commun. 2005, 4324–4326. doi:10.1039/b506010a
Return to citation in text: [1] [2] [3] -
Olah, G.; Nojima Keres, M. I. Synthesis 1973, 780–783. doi:10.1055/s-1973-22298
Return to citation in text: [1] [2] -
Ernet, E.; Haufe, G. Synthesis 1997, 953–956. doi:10.1055/s-1997-1279
Return to citation in text: [1] -
Barton, D. H. R. J. Chem. Soc., Perkin Trans. 1 1974, 739–742. doi:10.1039/p19740000739
Return to citation in text: [1] -
Harwood, L. Polar Rearrangements; Oxford Chemistry Primers; Oxford University Press: Tokyo, 1992.
Return to citation in text: [1] -
Abraham, R. J.; Loftus, P. Tetrahedron 1977, 33, 1227–1234. doi:10.1016/0040-4020(77)80419-5
Return to citation in text: [1] -
Gaussian03, Revision C.02; Gaussian, Inc.: Wallingford, CT, 2004.
Return to citation in text: [1] -
Kendall, R. A.; Dunning, T. H., Jr.; Harrison, R. J. J. Chem. Phys. 1992, 96, 6796–6806. doi:10.1063/1.462569
Return to citation in text: [1] -
Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995–2001. doi:10.1021/jp9716997
Return to citation in text: [1] -
Aoyama, T. J. Chem. Soc., Perkin Trans. 1 1995, 15, 1905–1912. doi:10.1039/p19950001905
Return to citation in text: [1] -
Nakajima, M.; Tomioka, K.; Koga, K. Tetrahedron 1993, 49, 9735–9750. doi:10.1016/S0040-4020(01)80176-9
Return to citation in text: [1] -
Banks, J. W.; Batsanov, A. S.; Howard, J. A. K.; O'Hagan, D.; Rzepa, H. S.; Martin-Santamaria, S. J. Chem. Soc., Perkin Trans. 2 1999, 2409–2411. doi:10.1039/a907452j
Return to citation in text: [1] [2] -
Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899–926. doi:10.1021/cr00088a005
Return to citation in text: [1] [2] -
Abraham, R. J.; Cavalli, L. Mol. Phys. 1965, 9, 67. doi:10.1080/00268976500100091
Return to citation in text: [1] -
Ihrig, A. M.; Smith, S. L. J. Am. Chem. Soc. 1972, 94, 34–41. doi:10.1021/ja00756a007
Return to citation in text: [1]
| 24. | Gaussian03, Revision C.02; Gaussian, Inc.: Wallingford, CT, 2004. |
| 25. | Kendall, R. A.; Dunning, T. H., Jr.; Harrison, R. J. J. Chem. Phys. 1992, 96, 6796–6806. doi:10.1063/1.462569 |
| 26. | Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995–2001. doi:10.1021/jp9716997 |
| 17. | Norsikian, S. Chem.–Eur. J. 1999, 5, 2055–2068. doi:10.1002/(SICI)1521-3765(19990702)5:7<2055::AID-CHEM2055>3.0.CO;2-9 |
| 1. | Craig, N. C.; Chen, A.; Suh, K. H.; Klee, S.; Mellau, G. C.; Winnewisser, B. P.; Winnewisser, M. J. Am. Chem. Soc. 1997, 119, 4789–4790. doi:10.1021/ja963819e |
| 5. | Chia, T.; Yang, N. C.; Chernick, C. L. J. Am. Chem. Soc. 1964, 86, 5021–5022. doi:10.1021/ja01076a069 |
| 31. | Abraham, R. J.; Cavalli, L. Mol. Phys. 1965, 9, 67. doi:10.1080/00268976500100091 |
| 32. | Ihrig, A. M.; Smith, S. L. J. Am. Chem. Soc. 1972, 94, 34–41. doi:10.1021/ja00756a007 |
| 3. | Tavasli, M.; O'Hagan, D.; Pearson, C.; Petty, M. C. Chem. Commun. 2002, 1226–1227. doi:10.1039/b202891c |
| 6. | Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7 |
| 7. | Hudlicky, M. J. Fluorine Chem. 1983, 23, 241–259. doi:10.1016/S0022-1139(00)85130-1 |
| 18. | Schüler, M.; O'Hagan, D.; Slawin, A. M. Z. Chem. Commun. 2005, 4324–4326. doi:10.1039/b506010a |
| 2. | Angelini, G.; Gavuzzo, E.; Segre, A. L.; Speranza, M. J. Phys. Chem. 1990, 94, 8762–8766. doi:10.1021/j100388a004 |
| 13. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3 |
| 18. | Schüler, M.; O'Hagan, D.; Slawin, A. M. Z. Chem. Commun. 2005, 4324–4326. doi:10.1039/b506010a |
| 11. | Hulin, B.; Cabral, S.; Lopaze, M. G.; Van Volkenburg, M. A.; Andrews, K. M.; Parker, J. C. Bioorg. Med. Chem. Lett. 2005, 15, 4770–4773. doi:10.1016/j.bmcl.2005.07.026 |
| 12. | Caldwell, C. G.; Chen, P.; He, J.; Parmee, R. E.; Leiting, B.; Marsilio, F.; Patel, R. A.; Wu, J. K.; Eiermann, G. J.; Petrov, A.; He, H.; Lyons, K. A.; Thornberry, N. A.; Weber, A. E. Bioorg. Med. Chem. Lett. 2004, 14, 1265–1268. doi:10.1016/j.bmcl.2003.12.040 |
| 9. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M. Chem. Commun. 1999, 215–216. doi:10.1039/a808517j |
| 10. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048–7054. doi:10.1021/jo990566+ |
| 30. | Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899–926. doi:10.1021/cr00088a005 |
| 9. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M. Chem. Commun. 1999, 215–216. doi:10.1039/a808517j |
| 10. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048–7054. doi:10.1021/jo990566+ |
| 8. | Singh, R. P.; Shreeve, J. M. J. Fluorine Chem. 2002, 116, 23–26. doi:10.1016/S0022-1139(02)00065-9 |
| 14. | Lal, G. S.; Labach, E.; Evans, A. J. Org. Chem. 2000, 65, 4830–4832. doi:10.1021/jo000020j |
| 29. | Banks, J. W.; Batsanov, A. S.; Howard, J. A. K.; O'Hagan, D.; Rzepa, H. S.; Martin-Santamaria, S. J. Chem. Soc., Perkin Trans. 2 1999, 2409–2411. doi:10.1039/a907452j |
| 30. | Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899–926. doi:10.1021/cr00088a005 |
| 8. | Singh, R. P.; Shreeve, J. M. J. Fluorine Chem. 2002, 116, 23–26. doi:10.1016/S0022-1139(02)00065-9 |
| 27. | Aoyama, T. J. Chem. Soc., Perkin Trans. 1 1995, 15, 1905–1912. doi:10.1039/p19950001905 |
| 28. | Nakajima, M.; Tomioka, K.; Koga, K. Tetrahedron 1993, 49, 9735–9750. doi:10.1016/S0040-4020(01)80176-9 |
| 6. | Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7 |
| 7. | Hudlicky, M. J. Fluorine Chem. 1983, 23, 241–259. doi:10.1016/S0022-1139(00)85130-1 |
| 13. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875–2881. doi:10.1016/S0040-4020(88)90023-3 |
| 29. | Banks, J. W.; Batsanov, A. S.; Howard, J. A. K.; O'Hagan, D.; Rzepa, H. S.; Martin-Santamaria, S. J. Chem. Soc., Perkin Trans. 2 1999, 2409–2411. doi:10.1039/a907452j |
| 7. | Hudlicky, M. J. Fluorine Chem. 1983, 23, 241–259. doi:10.1016/S0022-1139(00)85130-1 |
| 6. | Burmakov, A. I.; Motnyak, L. A.; Kunshenko, B. V.; Alexeeva, L. A.; Yagupolskii, L. M. J. Fluorine Chem. 1981, 19, 151–161. doi:10.1016/S0022-1139(00)81331-7 |
| 7. | Hudlicky, M. J. Fluorine Chem. 1983, 23, 241–259. doi:10.1016/S0022-1139(00)85130-1 |
| 22. | Harwood, L. Polar Rearrangements; Oxford Chemistry Primers; Oxford University Press: Tokyo, 1992. |
| 23. | Abraham, R. J.; Loftus, P. Tetrahedron 1977, 33, 1227–1234. doi:10.1016/0040-4020(77)80419-5 |
| 21. | Barton, D. H. R. J. Chem. Soc., Perkin Trans. 1 1974, 739–742. doi:10.1039/p19740000739 |
| 19. | Olah, G.; Nojima Keres, M. I. Synthesis 1973, 780–783. doi:10.1055/s-1973-22298 |
| 19. | Olah, G.; Nojima Keres, M. I. Synthesis 1973, 780–783. doi:10.1055/s-1973-22298 |
| 17. | Norsikian, S. Chem.–Eur. J. 1999, 5, 2055–2068. doi:10.1002/(SICI)1521-3765(19990702)5:7<2055::AID-CHEM2055>3.0.CO;2-9 |
| 18. | Schüler, M.; O'Hagan, D.; Slawin, A. M. Z. Chem. Commun. 2005, 4324–4326. doi:10.1039/b506010a |
© 2006 O'Hagan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)