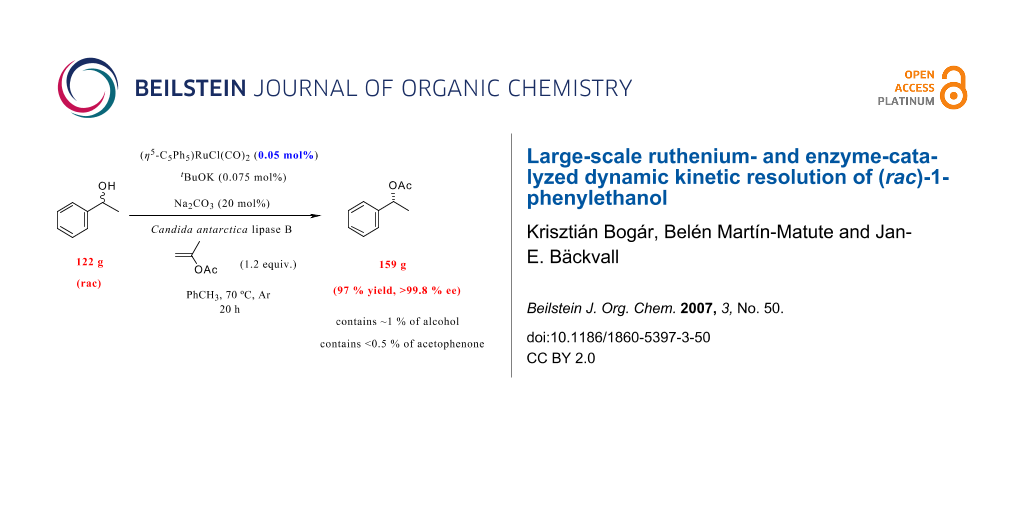
Abstract
The scale-up of the ruthenium- and enzyme-catalyzed dynamic kinetic resolution (DKR) of (rac)-1-phenylethanol (2) is addressed. The immobilized lipase Candida antarctica lipase B (CALB) was employed for the resolution, which shows high enantioselectivity in the transesterification. The ruthenium catalyst used, (η 5-C5Ph5)RuCl(CO)2 1, was shown to possess very high reactivity in the "in situ" redox racemization of 1-phenylethanol (2) in the presence of the immobilized enzyme, and could be used in 0.05 mol% with high efficiency. Commercially available isopropenyl acetate was employed as acylating agent in the lipase-catalyzed transesterifications, which makes the purification of the product very easy. In a successful large-scale DKR of 2, with 0.05 mol% of 1, (R)-1-phenylethanol acetate (3) was obtained in 159 g (97% yield) in excellent enantiomeric excess (99.8% ee).

Graphical Abstract
Background
Chiral alcohols are important synthetic intermediates and are structural elements in biologically active compounds and natural products. [1-4] A few methods have been developed for the enantioselective synthesis of chiral alcohols including catalytic asymmetric hydrogenation [5-14] and enantioselective hydride addition [15,16] of prochiral ketones, asymmetric dialkylzinc addition to aldehydes, [17,18] and asymmetric aldol reactions. [19-23] Biocatalysts have also been successfully employed in the enantioselective reduction of ketones.[24] The enzymatic resolution of racemic alcohols is a convenient alternative for preparing enantiomerically pure alcohols,[1,25-28] and recently this approach has been extended to dynamic kinetic resolution (DKR) by combining the enzymatic resolution with a metal-catalyzed racemization. [29-37] Today, many stable lipases are commercially available and they are frequently used in synthetic organic chemistry.[27]
In the course of the chemo-enzymatic synthesis of optically pure alcohols and esters, Candida antarctica lipase B (CALB) was found to be one of the most active and selective biocatalysts compared to other enzymes. Lipases do not need the presence of a cofactor and they can be employed in pure organic solvents.[38,39] CALB is immobilized on polyacrylate and this increases its thermostability and makes it easy to recover from the reaction mixture. Recently, we have developed highly efficient DKR protocols for secondary alcohols in which the traditional enzymatic resolution is combined with an "in situ" racemization of the substrate using a ruthenium(II) racemization catalyst (1) at ambient temperature (Figure 1).[32,34]
![[1860-5397-3-50-1]](/bjoc/content/figures/1860-5397-3-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Many 1-arylethanols are used as intermediates for the synthesis of pharmaceuticals and agrochemicals,[40,41] and they are therefore needed in enantiomerically pure form. Herein, we address some of the scale-up issues of the ruthenium- and enzyme-catalyzed DKR of (rac)-1-phenylethanol under mild conditions.
Results and Discussions
The DKR of 1-phenylethanol has been tested under different reaction conditions with the aim of decreasing the catalyst loading. When the reaction is performed on a 1 mmol scale, 4 mol% of ruthenium complex 1 is used for an efficient reaction.[32,34] We believe that the need of this high catalyst loading is due to a fast decomposition of the ruthenium active intermediates in the presence of small amounts of molecular oxygen. To test this hypothesis, the DKR of 1-phenylethanol under argon was compared to the DKR of 1-phenylethanol (2) under an oxygen atmosphere (Scheme 1).
![[1860-5397-3-50-i1]](/bjoc/content/inline/1860-5397-3-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: DKR of 1-phenylethanol under an Ar atmosphere (top) and DKR of 1-phenylethanol under an O2 atmosphere (bottom).
Scheme 1: DKR of 1-phenylethanol under an Ar atmosphere (top) and DKR of 1-phenylethanol under an O2 atmosphe...
While 98% of enantiomerically pure (>99% ee) (R)-1-phenylethanol acetate (3) was obtained in only 3 hours under an argon atmosphere, only 60% of 3 was produced after 16 hours under an oxygen atmosphere. We envisioned that running the reaction on a larger scale would allow a decrease in the catalyst loading, since decomposition of the catalyst due to the presence of traces of molecular oxygen may be avoided. The reaction in Scheme 1 (under argon) was therefore run on a larger scale and the results are summarized in Table 1. On a 10 mmol-scale, 1 mol% of Ru-catalyst 1 can be used to achieve 90% yield of acetate 3 (99% ee) after 18 h at room temperature (entry 1). A decrease of the catalyst loading to 0.5 mol% afforded 87% of enantiopure acetate, although a longer reaction time was required (42 h, entry 2). A significant improvement was obtained when the reaction was carried out at 40°C (entry 3). At this temperature, 0.5 mol% of catalyst 1 is sufficient to obtain a quantitative yield (99.5%) of the enantiomerically pure acetate 3 (>99% ee) within 18 h. The very high efficiency of the latter reaction suggests that the racemization catalyst loading can be further decreased, and that an increased reaction temperature may facilitate this decrease.
Table 1: Dynamic kinetic resolution of 1-phenylethanol (2) under different reaction conditions.a
| Entry | 2 (mmol) | [Ru] (mol%) | tBuOK (mol%) | CALB (mg/mmol) | Na2CO3 (mg/mmol) | T (°C) | Ratio 2/solv (w/w) | time | Yield of 3 (%) | ee of 3 (%)b |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10 | 1 | 1.5 | 2 | 50 | rt | 0.14 | 18 h | 90c | >99 |
| 2 | 10 | 0.5 | 0.75 | 2 | 25 | rt | 0.28 | 42 h | 87c | >99 |
| 3 | 10 | 0.5 | 0.75 | 2 | 25 | 40 | 0.28 | 18 h | 99.5c | >99 |
| 4 | 1000 | 0.05 | 0.075 | 0.5 | 21.2 | 70 | 0.94 | 20 h | 97d | >99 |
| 5 | 10000 | 0.01 | 0.015 | 0.1 | 3 | 70 | 20.1 | 21 d | 87d | 97 |
a. Toluene was used as solvent. b: ee measured by GLC; c: Yield measured by GLC; d: Isolated yield by distillation.
Indeed, a DKR reaction at 70°C on a 1 mol-scale employing only 0.05 mol% Ru-catalyst in a more concentrated reaction mixture afforded enantiopure acetate 3 in 97% yield after 20 h. Careful analysis of the enantiomeric purity of acetate 3 revealed that it was >99.8% ee. This result is remarkable since only a very low catalyst loading is employed and in addition, only 150 mL of toluene as the solvent is used to produce 159 g of enantiomerically pure 3. Finally, to prove the high efficiency of the racemization catalyst 1, DKR was carried out on a 10 mol-scale employing lower amount of metal- and biocatalyst in a more concentrated reaction mixture. After 21 days, enantiomerically enriched acetate 3 (97% ee) was obtained in 87% yield (corresponds to 1.43 kg). The remaining alcohol had an ee of 19%, demonstrating the high racemization ability activity of ruthenium complex 1.
Conclusion
In this article, some of the scale-up issues in the ruthenium- and enzyme-catalyzed DKR of (rac)-1-phenylethanol were investigated. Under optimized reaction conditions, DKR of 1-phenylethanol (2) was performed delivering 159 g (97% yield) of enantiomerically pure (R)-1-phenylethanol acetate (3) in a short reaction time (20 h) using 0.05 mol% of Ru-catalyst 1, and small amounts of enzyme. The employed heterogeneous biocatalyst, Candida antarctica lipase B, catalyzes the transesterification with excellent selectivity in the presence of the homogeneous ruthenium racemization catalyst 1. Isopropenyl acetate seems to be an appropriate acyl donor, which makes the purification of the product acetate very easy via simple distillation.
Supporting Information
| Supporting Information File 1: Large-scale ruthenium- and enzyme-catalyzed dynamic kinetic resolution of (rac)-1-phenylethanol: Supporting Information. Experimental procedure of DKR of 2 on 1 mol scale and GLC chromatogram of this experiment. | ||
| Format: DOC | Size: 1.2 MB | Download |
References
-
Theil, F. Chem. Rev. 1995, 95, 2203–2227. doi:10.1021/cr00038a017
Return to citation in text: [1] [2] -
Cossy, J.; BouzBouz, S.; Pradaux, F.; Willis, C.; Bellosta, V. Synlett 2002, 1595–1606. doi:10.1055/s-2002-34233
Return to citation in text: [1] -
Chen, S.-L.; Hu, Q.-Y.; Loh, T.-P. Org. Lett. 2004, 6, 3365–3367. doi:10.1021/ol048608q
Return to citation in text: [1] -
Kamal, A.; Khanna, G. B. R.; Ramu, R. Tetrahedron: Asymmetry 2002, 13, 2039–2051. doi:10.1016/S0957-4166(02)00537-2
Return to citation in text: [1] -
Noyori, R. Asymmetric Catalysis in Organic Synthesis; Wiley: New York, 1994.
Return to citation in text: [1] -
Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis; Springer: Berlin, 1999; Vol. 6 and 29.
Return to citation in text: [1] -
Palmer, M. J.; Wills, M. Tetrahedron: Asymmetry 1999, 10, 2045–2061. doi:10.1016/S0957-4166(99)00216-5
Return to citation in text: [1] -
Wills, M.; Palmer, M.; Smith, A.; Kenny, J.; Walsgrove, T. Molecules 2000, 5, 4–18.
Return to citation in text: [1] -
Ojima, I., Ed. Catalytic Asymmetric Synthesis; Wiley: New York, 2000; Vol. 1 and 8.
Return to citation in text: [1] -
Bäckvall, J.-E. J. Organomet. Chem. 2002, 652, 105–111. doi:10.1016/S0022-328X(02)01316-5
Return to citation in text: [1] -
Nishimura, S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis; Wiley: New York, 2001.
Return to citation in text: [1] -
Gladiali, S.; Alberico, E. In Transition Metals for Organic Synthesis, 2nd ed.; Beller, M.; Bolm, C., Eds.; Wiley-VCH: Weinheim, 2004; Vol. 2, pp 145–166.
Return to citation in text: [1] -
Clapham, S. E.; Hadzovic, A.; Morris, R. H. Coord. Chem. Rev. 2004, 248, 2201–2237. doi:10.1016/j.ccr.2004.04.007
Return to citation in text: [1] -
Gladiali, S.; Alberico, E. Chem. Soc. Rev. 2006, 35, 226–236. doi:10.1039/b513396c
Return to citation in text: [1] -
Corey, E. J.; Helal, C. J. Angew. Chem., Int. Ed. 1998, 37, 1986–2012. doi:10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z
Return to citation in text: [1] -
Riant, O.; Mostefaï, N.; Courmarcel, J. Synthesis 2004, 2943–2958. doi:10.1055/s-2004-834932
and reference therein.
Return to citation in text: [1] -
Soai, K.; Niwa, S. Chem. Rev. 1992, 92, 833–856. doi:10.1021/cr00013a004
Return to citation in text: [1] -
Zhu, H. J.; Jiang, J. X.; Ren, J.; Yan, Y. M.; Pittman, C. U., Jr. Curr. Org. Synth. 2005, 2, 547–587. doi:10.2174/157017905774322677
Return to citation in text: [1] -
Nicolau, K. C.; Snyder, S. A. Classics in Total Synthesis II; Wiley-VCH: Weinheim, 2003; pp 31–74.
Return to citation in text: [1] -
Machajewski, T. D.; Wong, C.-H. Angew. Chem., Int. Ed. 2000, 39, 1352–1374. doi:10.1002/(SICI)1521-3773(20000417)39:8<1352::AID-ANIE1352>3.0.CO;2-J
Return to citation in text: [1] -
Sawamura, M.; Ito, Y.; Carreira, E. M. In Catalytic Asymmetric Synthesis, 2nd ed.; Ojima, I., Ed.; Wiley-VCH: New York, 2000; pp 493–541.
Return to citation in text: [1] -
Palomo, C.; Oiarbide, M.; García, J. M. Chem. Soc. Rev. 2004, 33, 65–75. doi:10.1039/b202901d
Return to citation in text: [1] -
Saito, S.; Yamamoto, H. Acc. Chem. Res. 2004, 37, 570–579. doi:10.1021/ar030064p
Return to citation in text: [1] -
Nakamura, K.; Matsuda, T. Curr. Org. Chem. 2006, 10, 1217–1246. doi:10.2174/138527206777698020
Return to citation in text: [1] -
Wong, C.-H.; Whitesides, G. M. Enzymes in Organic Chemistry; Pergamon: Oxford, 1994.
Return to citation in text: [1] -
Bornscheuer, U. T.; Kazlauskas, R. J. Hydrolases in Organic Synthesis, Regio- and Stereoselective biotransformations; Wiley: Weinheim, 1999.
Return to citation in text: [1] -
Faber, K. Biotransformations in Organic Chemistry, 5th ed.; Springer: Berlin, 2004.
Return to citation in text: [1] [2] -
Silverman, R. B. The Organic Chemistry of Enzyme-Catalyzed Reactions; Academic Press: San Diego, 2002.
Return to citation in text: [1] -
Pàmies, O.; Bäckvall, J.-E. Chem. Rev. 2003, 103, 3247–3261. doi:10.1021/cr020029g
Return to citation in text: [1] -
Kim, M.-J.; Anh, Y.; Park, J. Curr. Opin. Biotechnol. 2002, 13, 578–587. doi:10.1016/S0958-1669(02)00347-6
And erratum 2003, 14, 131.
Return to citation in text: [1] -
Martín-Matute, B.; Bäckvall, J.-E. Curr. Opin. Chem. Biol. 2007, 11, 226–232. doi:10.1016/j.cbpa.2007.01.724
Return to citation in text: [1] -
Martín-Matute, B.; Edin, M.; Bogár, K.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2004, 43, 6535–6539. doi:10.1002/anie.200461416
Return to citation in text: [1] [2] [3] -
Choi, J. H.; Choi, Y. K.; Kim, Y. H.; Park, E. S.; Kim, E. J.; Kim, M.-J.; Park, J. J. Org. Chem. 2004, 69, 1972–1977. doi:10.1021/jo0355799
Return to citation in text: [1] -
Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F. B.; Bäckvall, J.-E. J. Am. Chem. Soc. 2005, 127, 8817–8825. doi:10.1021/ja051576x
Return to citation in text: [1] [2] [3] -
Kim, N.; Ko, S.-B.; Kwon, M. S.; Kim, M.-J.; Park, J. Org. Lett. 2005, 7, 4523–4526. doi:10.1021/ol051889x
Return to citation in text: [1] -
Borén, L.; Martín-Matute, B.; Xu, Y.; Cordova, A.; Bäckvall, J.-E. Chem.–Eur. J. 2006, 12, 225–232. doi:10.1002/chem.200500758
Return to citation in text: [1] -
Ko, S.-B.; Baburaj, B.; Kim, M.-J.; Park, J. J. Org. Chem. 2007, 72, 6860–6864. doi:10.1021/jo071065o
Return to citation in text: [1] -
Klibanov, A. M. Acc. Chem. Res. 1990, 23, 114–120. doi:10.1021/ar00172a004
Return to citation in text: [1] -
Klibanov, A. M. Nature 2001, 409, 241–246. doi:10.1038/35051719
Return to citation in text: [1] -
Hansen, K. B.; Chilenski, J. R.; Desmond, R.; Devine, P. N.; Grabovski, E. J. J.; Heid, R.; Kubryk, M.; Mathre, D. J.; Varsolona, R. Tetrahedron: Asymmetry 2003, 14, 3581–3587. doi:10.1016/j.tetasy.2003.08.043
Return to citation in text: [1] -
Badone, D.; Guzzi, U. Bioorg. Med. Chem. Lett. 1994, 4, 1921–1924. doi:10.1016/S0960-894X(01)80535-7
Return to citation in text: [1]
| 1. | Theil, F. Chem. Rev. 1995, 95, 2203–2227. doi:10.1021/cr00038a017 |
| 2. | Cossy, J.; BouzBouz, S.; Pradaux, F.; Willis, C.; Bellosta, V. Synlett 2002, 1595–1606. doi:10.1055/s-2002-34233 |
| 3. | Chen, S.-L.; Hu, Q.-Y.; Loh, T.-P. Org. Lett. 2004, 6, 3365–3367. doi:10.1021/ol048608q |
| 4. | Kamal, A.; Khanna, G. B. R.; Ramu, R. Tetrahedron: Asymmetry 2002, 13, 2039–2051. doi:10.1016/S0957-4166(02)00537-2 |
| 19. | Nicolau, K. C.; Snyder, S. A. Classics in Total Synthesis II; Wiley-VCH: Weinheim, 2003; pp 31–74. |
| 20. | Machajewski, T. D.; Wong, C.-H. Angew. Chem., Int. Ed. 2000, 39, 1352–1374. doi:10.1002/(SICI)1521-3773(20000417)39:8<1352::AID-ANIE1352>3.0.CO;2-J |
| 21. | Sawamura, M.; Ito, Y.; Carreira, E. M. In Catalytic Asymmetric Synthesis, 2nd ed.; Ojima, I., Ed.; Wiley-VCH: New York, 2000; pp 493–541. |
| 22. | Palomo, C.; Oiarbide, M.; García, J. M. Chem. Soc. Rev. 2004, 33, 65–75. doi:10.1039/b202901d |
| 23. | Saito, S.; Yamamoto, H. Acc. Chem. Res. 2004, 37, 570–579. doi:10.1021/ar030064p |
| 17. | Soai, K.; Niwa, S. Chem. Rev. 1992, 92, 833–856. doi:10.1021/cr00013a004 |
| 18. | Zhu, H. J.; Jiang, J. X.; Ren, J.; Yan, Y. M.; Pittman, C. U., Jr. Curr. Org. Synth. 2005, 2, 547–587. doi:10.2174/157017905774322677 |
| 15. | Corey, E. J.; Helal, C. J. Angew. Chem., Int. Ed. 1998, 37, 1986–2012. doi:10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z |
| 16. |
Riant, O.; Mostefaï, N.; Courmarcel, J. Synthesis 2004, 2943–2958. doi:10.1055/s-2004-834932
and reference therein. |
| 32. | Martín-Matute, B.; Edin, M.; Bogár, K.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2004, 43, 6535–6539. doi:10.1002/anie.200461416 |
| 34. | Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F. B.; Bäckvall, J.-E. J. Am. Chem. Soc. 2005, 127, 8817–8825. doi:10.1021/ja051576x |
| 5. | Noyori, R. Asymmetric Catalysis in Organic Synthesis; Wiley: New York, 1994. |
| 6. | Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis; Springer: Berlin, 1999; Vol. 6 and 29. |
| 7. | Palmer, M. J.; Wills, M. Tetrahedron: Asymmetry 1999, 10, 2045–2061. doi:10.1016/S0957-4166(99)00216-5 |
| 8. | Wills, M.; Palmer, M.; Smith, A.; Kenny, J.; Walsgrove, T. Molecules 2000, 5, 4–18. |
| 9. | Ojima, I., Ed. Catalytic Asymmetric Synthesis; Wiley: New York, 2000; Vol. 1 and 8. |
| 10. | Bäckvall, J.-E. J. Organomet. Chem. 2002, 652, 105–111. doi:10.1016/S0022-328X(02)01316-5 |
| 11. | Nishimura, S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis; Wiley: New York, 2001. |
| 12. | Gladiali, S.; Alberico, E. In Transition Metals for Organic Synthesis, 2nd ed.; Beller, M.; Bolm, C., Eds.; Wiley-VCH: Weinheim, 2004; Vol. 2, pp 145–166. |
| 13. | Clapham, S. E.; Hadzovic, A.; Morris, R. H. Coord. Chem. Rev. 2004, 248, 2201–2237. doi:10.1016/j.ccr.2004.04.007 |
| 14. | Gladiali, S.; Alberico, E. Chem. Soc. Rev. 2006, 35, 226–236. doi:10.1039/b513396c |
| 27. | Faber, K. Biotransformations in Organic Chemistry, 5th ed.; Springer: Berlin, 2004. |
| 32. | Martín-Matute, B.; Edin, M.; Bogár, K.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2004, 43, 6535–6539. doi:10.1002/anie.200461416 |
| 34. | Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F. B.; Bäckvall, J.-E. J. Am. Chem. Soc. 2005, 127, 8817–8825. doi:10.1021/ja051576x |
| 29. | Pàmies, O.; Bäckvall, J.-E. Chem. Rev. 2003, 103, 3247–3261. doi:10.1021/cr020029g |
| 30. |
Kim, M.-J.; Anh, Y.; Park, J. Curr. Opin. Biotechnol. 2002, 13, 578–587. doi:10.1016/S0958-1669(02)00347-6
And erratum 2003, 14, 131. |
| 31. | Martín-Matute, B.; Bäckvall, J.-E. Curr. Opin. Chem. Biol. 2007, 11, 226–232. doi:10.1016/j.cbpa.2007.01.724 |
| 32. | Martín-Matute, B.; Edin, M.; Bogár, K.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2004, 43, 6535–6539. doi:10.1002/anie.200461416 |
| 33. | Choi, J. H.; Choi, Y. K.; Kim, Y. H.; Park, E. S.; Kim, E. J.; Kim, M.-J.; Park, J. J. Org. Chem. 2004, 69, 1972–1977. doi:10.1021/jo0355799 |
| 34. | Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F. B.; Bäckvall, J.-E. J. Am. Chem. Soc. 2005, 127, 8817–8825. doi:10.1021/ja051576x |
| 35. | Kim, N.; Ko, S.-B.; Kwon, M. S.; Kim, M.-J.; Park, J. Org. Lett. 2005, 7, 4523–4526. doi:10.1021/ol051889x |
| 36. | Borén, L.; Martín-Matute, B.; Xu, Y.; Cordova, A.; Bäckvall, J.-E. Chem.–Eur. J. 2006, 12, 225–232. doi:10.1002/chem.200500758 |
| 37. | Ko, S.-B.; Baburaj, B.; Kim, M.-J.; Park, J. J. Org. Chem. 2007, 72, 6860–6864. doi:10.1021/jo071065o |
| 40. | Hansen, K. B.; Chilenski, J. R.; Desmond, R.; Devine, P. N.; Grabovski, E. J. J.; Heid, R.; Kubryk, M.; Mathre, D. J.; Varsolona, R. Tetrahedron: Asymmetry 2003, 14, 3581–3587. doi:10.1016/j.tetasy.2003.08.043 |
| 41. | Badone, D.; Guzzi, U. Bioorg. Med. Chem. Lett. 1994, 4, 1921–1924. doi:10.1016/S0960-894X(01)80535-7 |
| 1. | Theil, F. Chem. Rev. 1995, 95, 2203–2227. doi:10.1021/cr00038a017 |
| 25. | Wong, C.-H.; Whitesides, G. M. Enzymes in Organic Chemistry; Pergamon: Oxford, 1994. |
| 26. | Bornscheuer, U. T.; Kazlauskas, R. J. Hydrolases in Organic Synthesis, Regio- and Stereoselective biotransformations; Wiley: Weinheim, 1999. |
| 27. | Faber, K. Biotransformations in Organic Chemistry, 5th ed.; Springer: Berlin, 2004. |
| 28. | Silverman, R. B. The Organic Chemistry of Enzyme-Catalyzed Reactions; Academic Press: San Diego, 2002. |
| 24. | Nakamura, K.; Matsuda, T. Curr. Org. Chem. 2006, 10, 1217–1246. doi:10.2174/138527206777698020 |
| 38. | Klibanov, A. M. Acc. Chem. Res. 1990, 23, 114–120. doi:10.1021/ar00172a004 |
| 39. | Klibanov, A. M. Nature 2001, 409, 241–246. doi:10.1038/35051719 |
© 2007 Bogár et al; licensee Beilstein-Institut
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)